
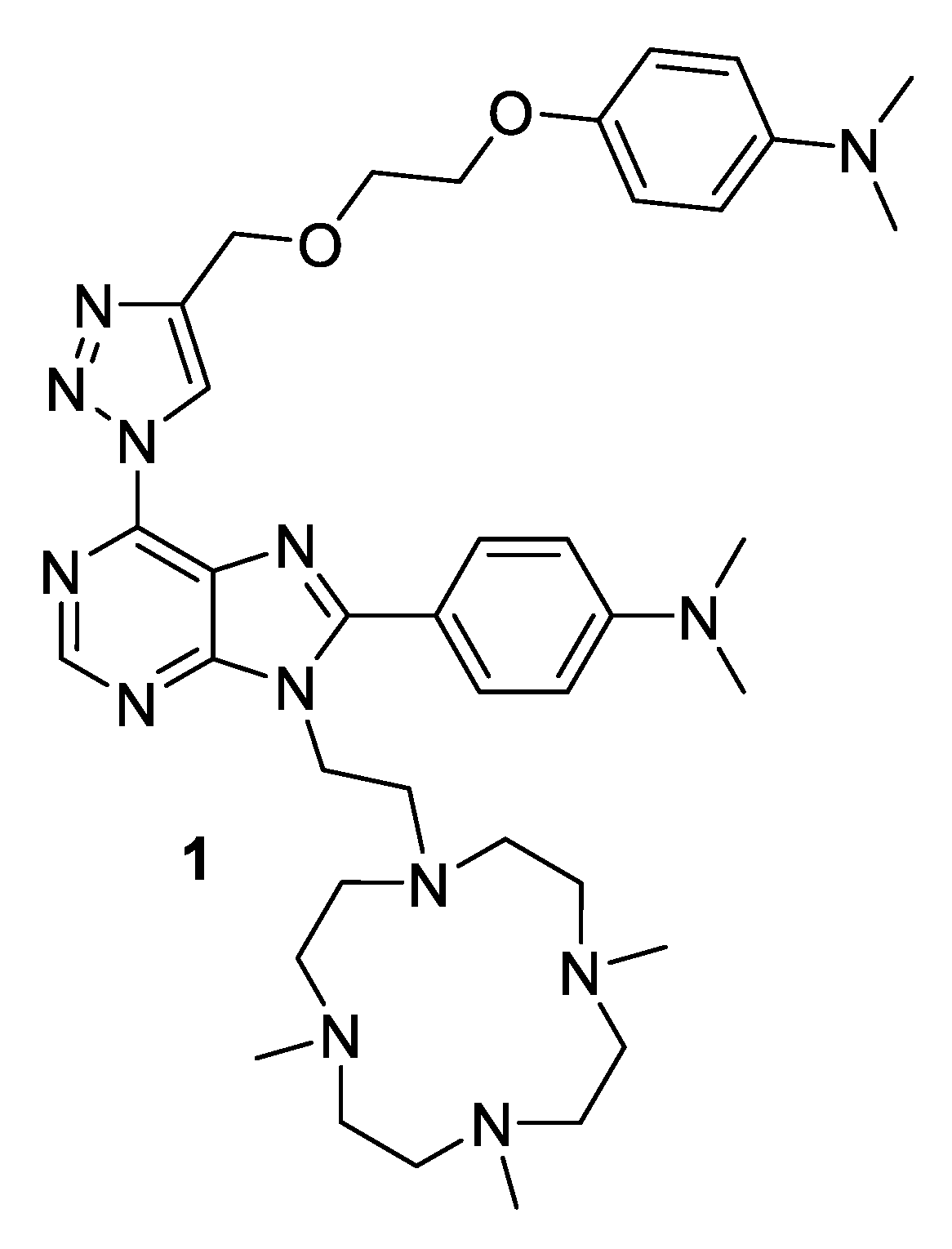
Synthesis of Purine-1,4,7,10-Tetraazacyclododecane Conjugate and Its Complexation Modes with Copper(II)


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
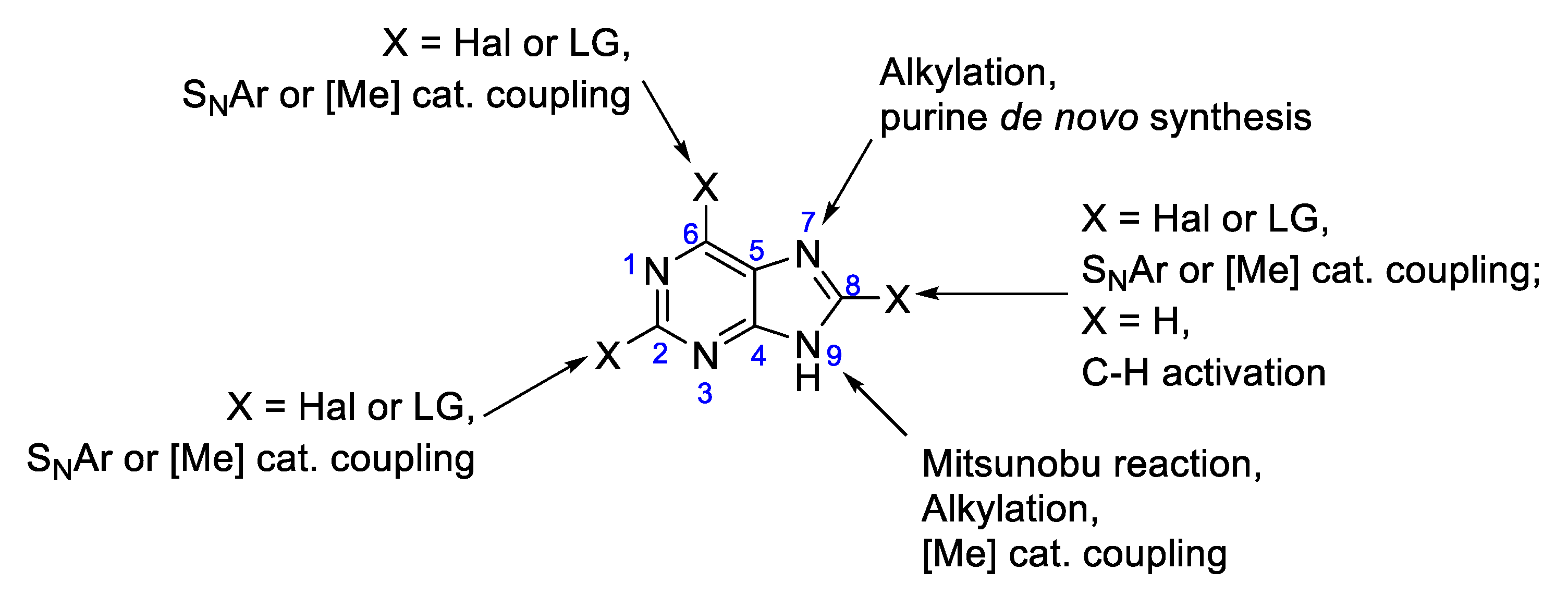
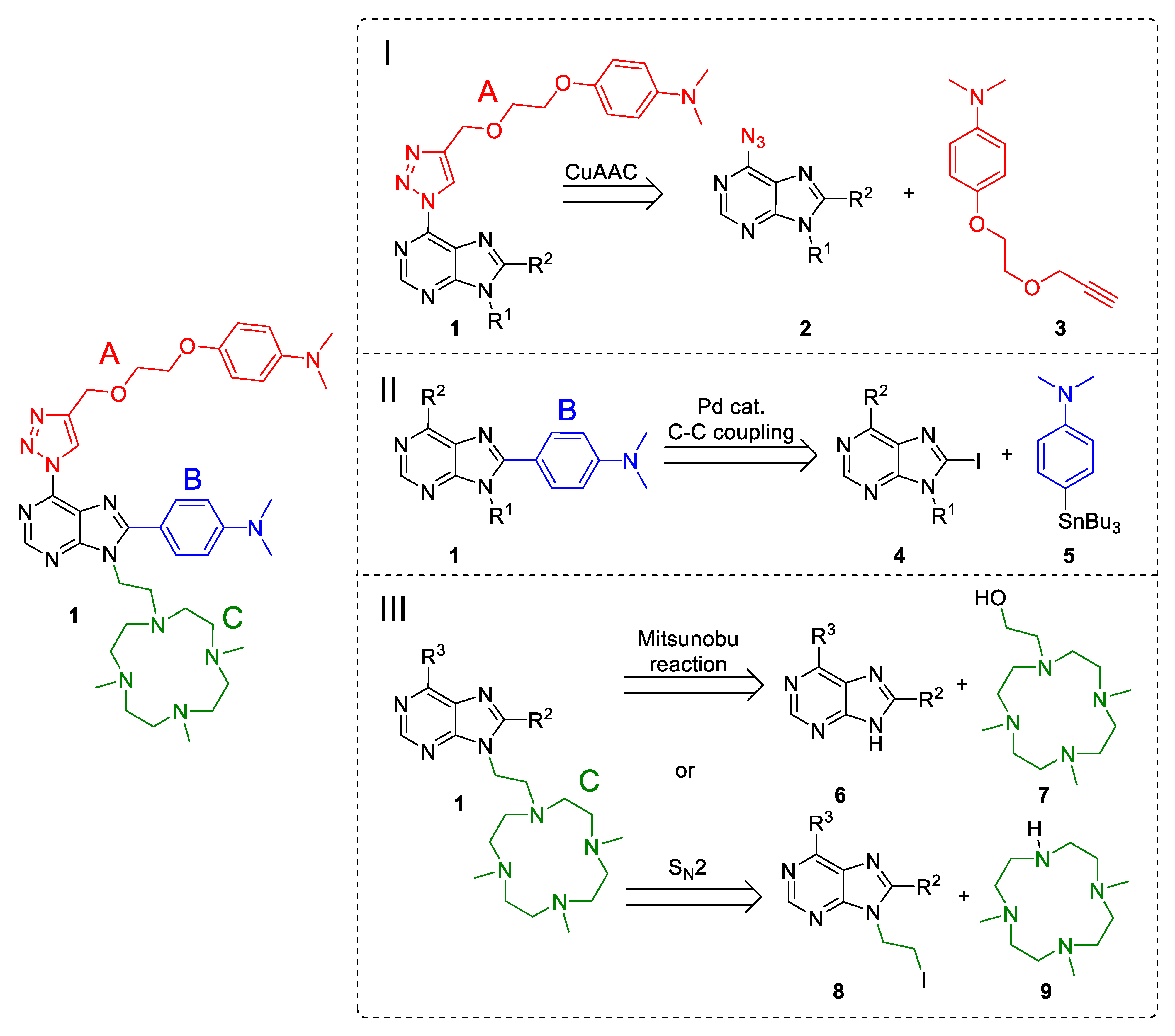
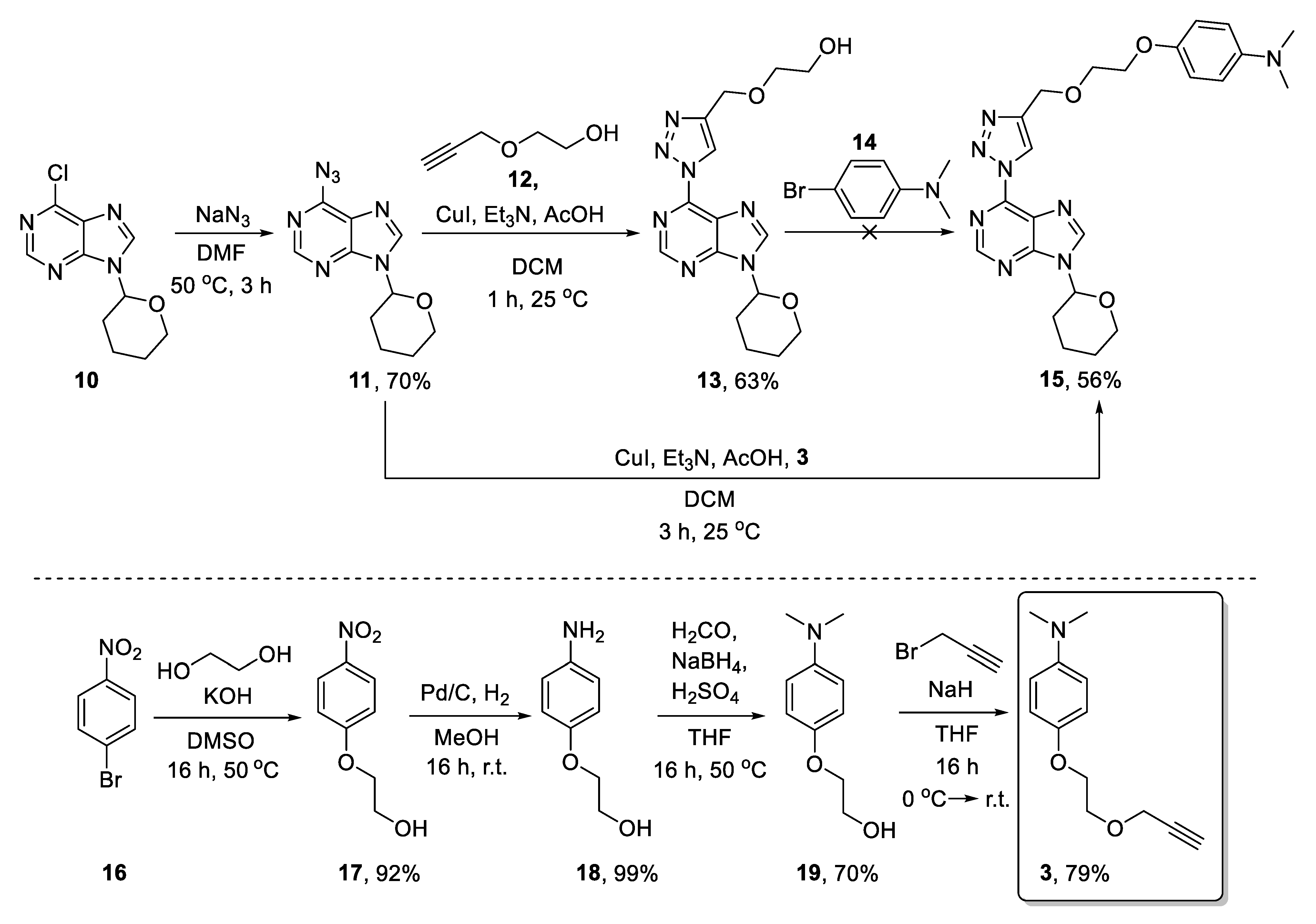
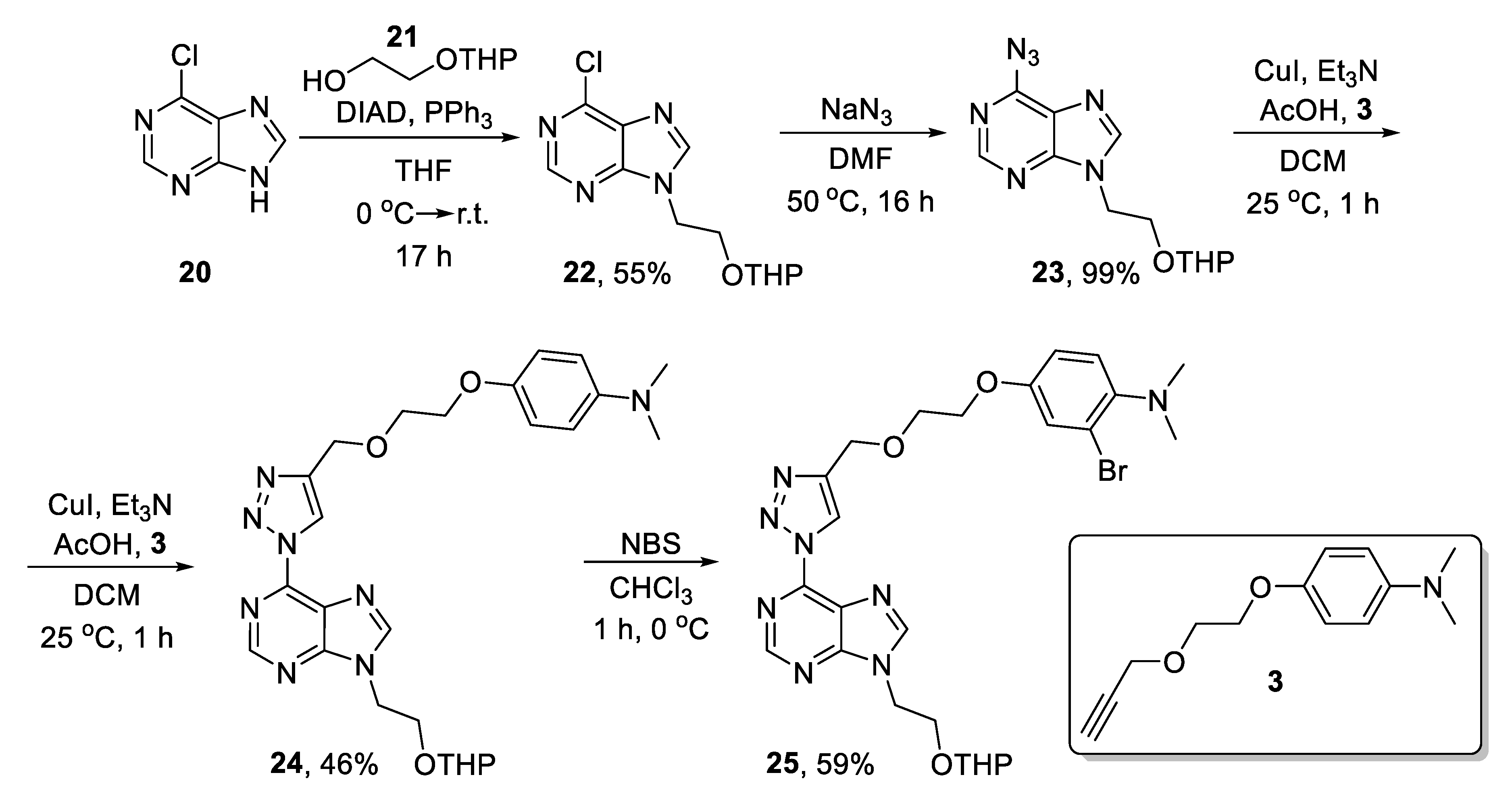
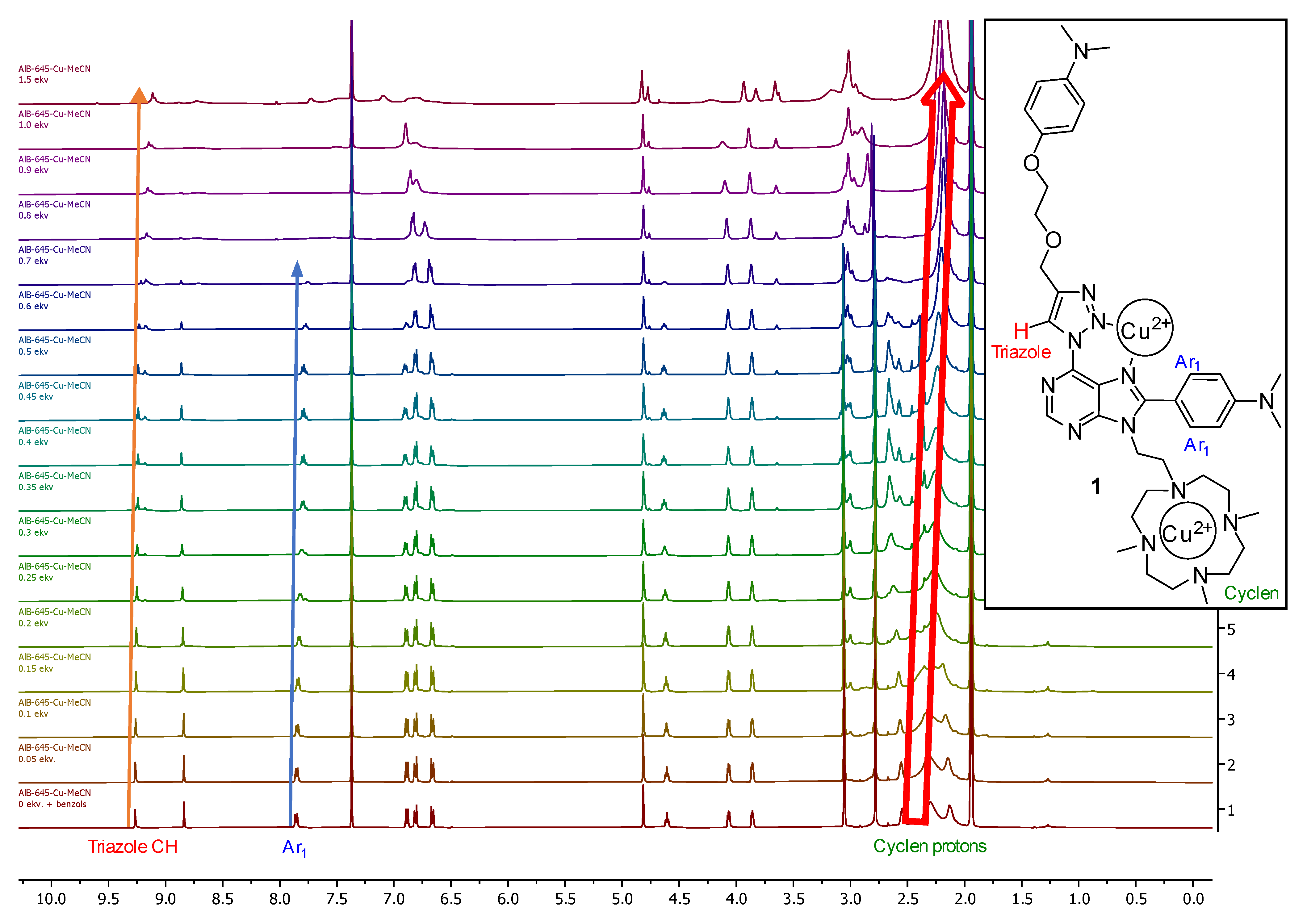
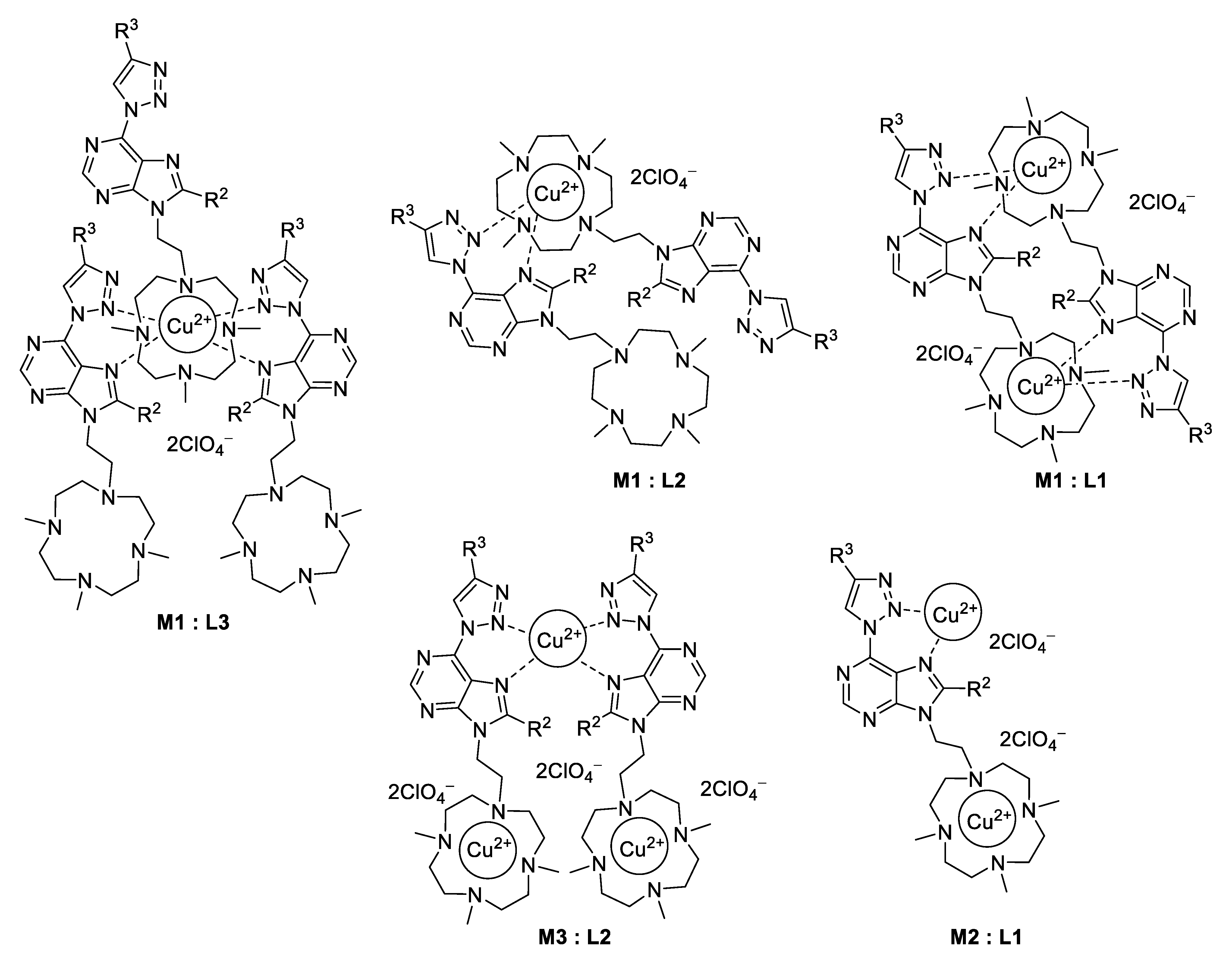
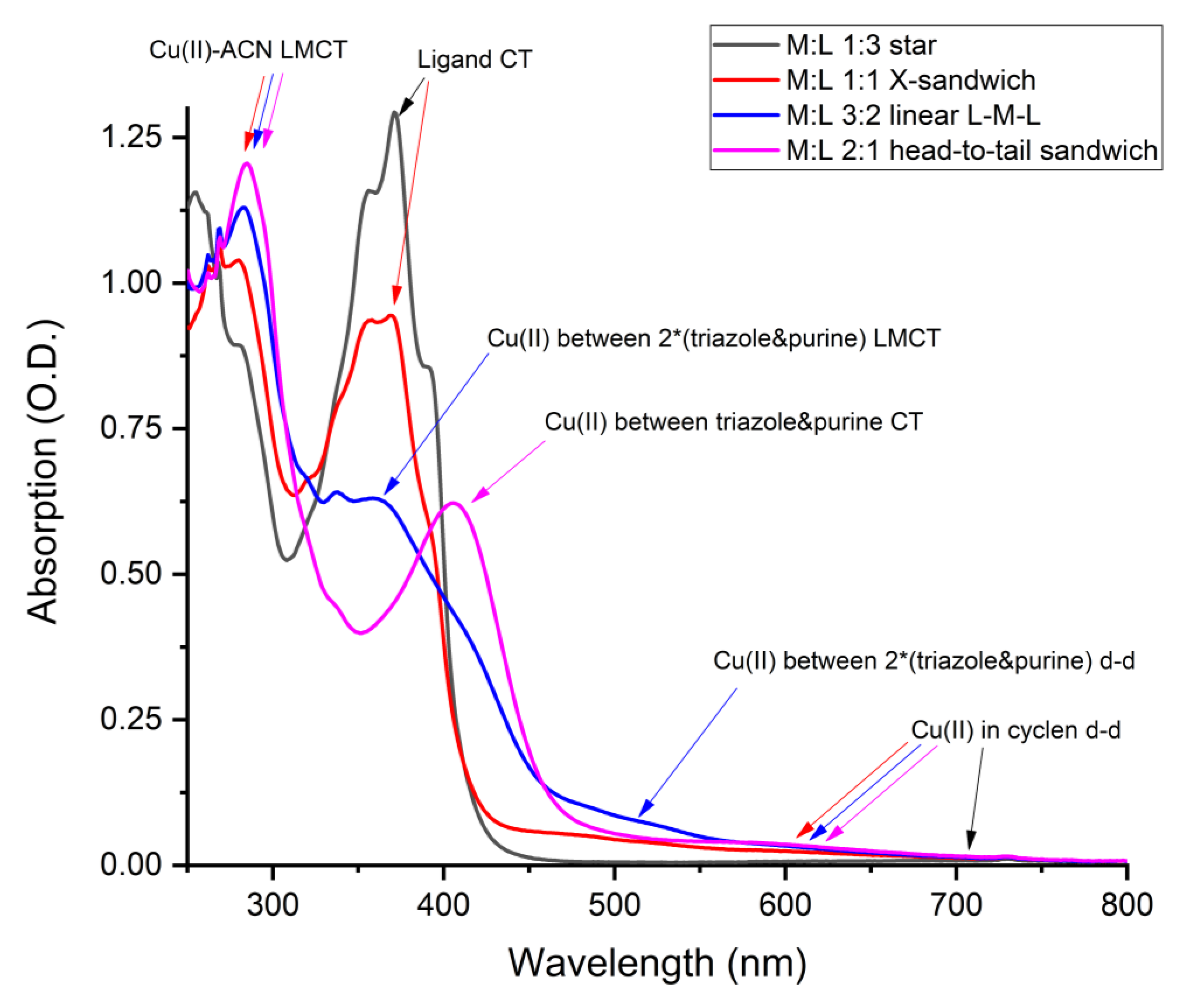
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Characterization Data for Products
3.2.1. 2-(4-Nitrophenoxy)ethan-1-ol (17)
3.2.2. 2-(4-Aminophenoxy)ethan-1-ol (18)
3.2.3. 2-(4-(Dimethylamino)phenoxy)ethan-1-ol (19)
3.2.4. N,N-Dimethyl-4-(2-(prop-2-yn-1-yloxy)ethoxy)aniline (3)
3.2.5. 6-Azido-9-(Tetrahydro-2H-pyran-2-yl)-9H-purine (11)
3.2.6. 2-({1-[9-(Tetrahydro-2H-pyran-2-yl)-9H-purin-6-yl]-1H-1,2,3-triazol-4-yl}methoxy)ethan-1-ol (13)
3.2.7. N,N-Dimethyl-4-[2-({1-[9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-yl]-1H-1,2,3-triazol-4-yl}methoxy)ethoxy]aniline (15)
3.2.8. 6-Chloro-9-{2-[(Tetrahydro-2H-pyran-2-yl)oxy]ethyl}-9H-purine (22)
3.2.9. 6-Azido-9-{2-[(Tetrahydro-2H-pyran-2-yl)oxy]ethyl}-9H-purine (23)
3.2.10. N,N-Dimethyl-4-(2-{[1-(9-{2-[(Tetrahydro-2H-pyran-2-yl)oxy]ethyl}-9H-purin-6-yl)-1H-1,2,3-triazol-4-yl]methoxy}ethoxy)aniline (24)
3.2.11. 2-Bromo-N,N-Dimethyl-4-{2-[(1-(9-{2-[(Tetrahydro-2H-pyran-2-yl)oxy]ethyl}-9H-purin-6-yl)-1H-1,2,3-triazol-4-yl)methoxy]ethoxy}aniline (25)
3.2.12. 6-Chloro-8-iodo-9-{2-[(Tetrahydro-2H-pyran-2-yl)oxy]ethyl}-9H-purine (26)
3.2.13. 4-(6-Chloro-9-{2-[(Tetrahydro-2H-pyran-2-yl)oxy]ethyl}-9H-purin-8-yl)-N,N-Dimethylaniline (27)
3.2.14. 4-(6-Azido-9-{2-[(Tetrahydro-2H-pyran-2-yl)oxy]ethyl}-9H-purin-8-yl)-N,N-Dimethylaniline (28)
3.2.15. 4-[6-(4-({2-[4-(Dimethylamino)phenoxy]ethoxy)methyl}-1H-1,2,3-triazol-1-yl)-9-{2-[(Tetrahydro-2H-pyran-2-yl)oxy]ethyl}-9H-purin-8-yl]-N,N-Dimethylaniline (29)
3.2.16. 2-{6-[4-({2-[4-(Dimethylamino)phenoxy]ethoxy}methyl)-1H-1,2,3-triazol-1-yl]-8-[4-(Dimethylamino)phenyl]-9H-purin-9-yl}ethan-1-ol (30)
3.2.17. 2-{6-[4-({2-[4-(Dimethylamino)phenoxy]ethoxy}methyl)-1H-1,2,3-triazol-1-yl]-8-[4-(Dimethylamino)phenyl]-9H-purin-9-yl}ethyl Methanesulfonate (31)
3.2.18. 4-(6-(4-((2-(4-(Dimethylamino)phenoxy)ethoxy)methyl)-1H-1,2,3-triazol-1-yl)-9-(2-(4,7,10-trimethyl-1,4,7,10-tetraazacyclododecan-1-yl)ethyl)-9H-purin-8-yl)-N,N-Dimethylaniline (1)
3.2.19. 4-(6-(4-((2-(4-(Dimethylamino)phenoxy)ethoxy)methyl)-1H-1,2,3-triazol-1-yl)-9-vinyl-9H-purin-8-yl)-N,N-Dimethylaniline (32)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Müller, C.E.; Thorand, M.; Qurishi, R.; Diekmann, M.; Jacobson, K.A.; Padgett, W.L.; Daly, J.W. Imidazo[2,1-i]Purin-5-Ones and Related Tricyclic Water-Soluble Purine Derivatives: Potent A2A- and A3-Adenosine Receptor Antagonistst. J. Med. Chem. 2002, 45, 3440–3450. [Google Scholar] [CrossRef] [PubMed]
- Al Jaroudi, W.; Iskandrian, A.E. Regadenoson: A New Myocardial Stress Agent. J. Am. Coll. Cardiol. 2009, 54, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.; Lu, X.; Hollenbaugh, J.A.; Cho, J.H.; Amblard, F.; Schinazi, R.F. Metabolism, Biochemical Actions, and Chemical Synthesis of Anticancer Nucleosides, Nucleotides, and Base Analogs. Chem. Rev. 2016, 116, 14379–14455. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.B. Enzymology of Purine and Pyrimidine Antimetabolites Used in the Treatment of Cancer. Chem. Rev. 2009, 109, 2880–2893. [Google Scholar] [CrossRef]
- Xu, W.; Chan, K.M.; Kool, E.T. Fluorescent Nucleobases as Tools for Studying DNA and RNA. Nat. Chem. 2017, 9, 1043–1055. [Google Scholar] [CrossRef]
- Sinkeldam, R.W.; Greco, N.J.; Tor, Y. Fluorescent Analogs of Biomolecular Building Blocks: Design, Properties, and Applications. Chem. Rev. 2010, 110, 2579–2619. [Google Scholar] [CrossRef]
- Dumas, A.; Luedtke, N.W. Site-Specific Control of N7-Metal Coordination in DNA by a Fluorescent Purine Derivative. Chem.-A Eur. J. 2012, 18, 245–254. [Google Scholar] [CrossRef]
- Omumi, A.; McLaughlin, C.K.; Ben-Israel, D.; Manderville, R.A. Application of a Fluorescent C-Linked Phenolic Purine Adduct for Selective N7-Metalation of DNA. J. Phys. Chem. B 2012, 116, 6158–6165. [Google Scholar] [CrossRef]
- Sun, K.M.; McLaughlin, C.K.; Lantero, D.R.; Manderville, R.A. Biomarkers for Phenol Carcinogen Exposure Act as pH-Sensing Fluorescent Probes. J. Am. Chem. Soc. 2007, 129, 1894–1895. [Google Scholar] [CrossRef]
- Venkatesh, V.; Shukla, A.; Sivakumar, S.; Verma, S. Purine-Stabilized Green Fluorescent Gold Nanoclusters for Cell Nuclei Imaging Applications. ACS Appl. Mater. Interfaces 2014, 6, 2185–2191. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Zhang, H.; Xuan, X.; Xie, M.; Xia, S.; Qu, G.; Guo, H. Nucleoside-Based Ultrasensitive Fluorescent Probe for the Dual-Mode Imaging of Microviscosity in Living Cells. Anal. Chem. 2016, 88, 5554–5560. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yao, J.; Zhan, L.; Gong, S.; Ma, D.; Yang, C. Purine-Based Thermally Activated Delayed Fluorescence Emitters for Efficient Organic Light-Emitting Diodes. Dye. Pigment. 2020, 180, 108437. [Google Scholar] [CrossRef]
- Kong, B.; Joshi, T.; Belousoff, M.J.; Tor, Y.; Graham, B.; Spiccia, L. Neomycin B-Cyclen Conjugates and Their Zn(II) Complexes as RNA-Binding Agents. J. Inorg. Biochem. 2016, 162, 334–342. [Google Scholar] [CrossRef]
- Abdollahi, M.; Hosseini, A. Hydrogen Sulfide. In Encyclopedia of Toxicology, 3rd ed.; Wexler, P., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 971–974. ISBN 9780123864550. [Google Scholar]
- Lima, L.M.P.; Esteban-Gómez, D.; Delgado, R.; Platas-Iglesias, C.; Tripier, R. Monopicolinate Cyclen and Cyclam Derivatives for Stable Copper(II) Complexation. Inorg. Chem. 2012, 51, 6916–6927. [Google Scholar] [CrossRef]
- Delgado, R.; Félix, V.; Lima, L.M.P.; Price, D.W. Metal Complexes of Cyclen and Cyclam Derivatives Useful for Medical Applications: A Discussion Based on Thermodynamic Stability Constants and Structural Data. Dalt. Trans. 2007, 2734–2745. [Google Scholar] [CrossRef]
- Paderni, D.; Voccia, M.; Macedi, E.; Formica, M.; Giorgi, L.; Caporaso, L.; Fusi, V. A Combined Solid State, Solution and DFT Study of a Dimethyl-Cyclen-Pd(II) Complex. Dalt. Trans. 2024, 53, 14300–14314. [Google Scholar] [CrossRef]
- Verma, A.; Bhuvanesh, N.; Reibenspies, J.; Tayade, S.B.; Kumbhar, A.S.; Bretosh, K.; Sutter, J.P.; Sunkari, S.S. Structurally Characterised New Twisted Conformer for Cyclen, Controlled by Metal Ion Complexation as Seen in NiII and CuII complexes with Halides and Pseudohalides. CrystEngComm 2022, 24, 119–131. [Google Scholar] [CrossRef]
- Harmand, L.; Cadet, S.; Kauffmann, B.; Scarpantonio, L.; Batat, P.; Jonusauskas, G.; McClenaghan, N.D.; Lastécouères, D.; Vincent, J.M. Copper Catalyst Activation Driven by Photoinduced Electron Transfer: A Prototype Photolatent Click Catalyst. Angew. Chemie-Int. Ed. 2012, 51, 7137–7141. [Google Scholar] [CrossRef]
- Zaķis, J.M.; Ozols, K.; Novosjolova, I.; Vilšķērsts, R.; Mishnev, A.; Turks, M. Sulfonyl Group Dance: A Tool for the Synthesis of 6-Azido-2-Sulfonylpurine Derivatives. J. Org. Chem. 2020, 85, 4753–4771. [Google Scholar] [CrossRef]
- Frieden, M.; Aviñó, A.; Eritja, R. Convenient Synthesis of 8-Amino-2′-Deoxyadenosine. Nucleosides Nucleotides Nucleic Acids 2003, 22, 193–202. [Google Scholar] [CrossRef]
- Cīrule, D.; Novosjolova, I.; Bizdēna, Ē.; Turks, M. 1,2,3-Triazoles as Leaving Groups: SNAr Reactions of 2,6-Bistriazolylpurines with O- And C-Nucleophiles. Beilstein J. Org. Chem. 2021, 17, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.M.; Xin, P.Y.; Niu, H.Y.; Wang, D.C.; Jiang, Y.; Qu, G.R. Microwave Irradiated C6-Functionalization of 6-Chloropurine Nucleosides with Various Mild Nucleophiles under Solvent-Free Conditions. Green Chem. 2010, 12, 2131–2134. [Google Scholar] [CrossRef]
- Borrmann, T.; Abdelrahman, A.; Volpini, R.; Lambertucci, C.; Alksnis, E.; Gorzalka, S.; Knospe, M.; Schiedel, A.C.; Cristalli, G.; Müller, C.E. Structure-Activity Relationships of Adenine and Deazaadenine Derivatives as Ligands for Adenine Receptors, a New Purinergic Receptor Family. J. Med. Chem. 2009, 52, 5974–5989. [Google Scholar] [CrossRef]
- Singh, B.; Diaz-Gonzalez, R.; Ceballos-Perez, G.; Rojas-Barros, D.I.; Gunaganti, N.; Gillingwater, K.; Martinez-Martinez, M.S.; Manzano, P.; Navarro, M.; Pollastri, M.P. Medicinal Chemistry Optimization of a Diaminopurine Chemotype: Toward a Lead for Trypanosoma Brucei Inhibitors. J. Med. Chem. 2020, 63, 9912–9927. [Google Scholar] [CrossRef]
- Malínková, V.; Řezníčková, E.; Jorda, R.; Gucký, T.; Kryštof, V. Trisubstituted Purine Inhibitors of PDGFRα and Their Antileukemic Activity in the Human Eosinophilic Cell Line EOL-1. Bioorg. Med. Chem. 2017, 25, 6523–6535. [Google Scholar] [CrossRef]
- Novosjolova, I.; Bizdēna, Ē.; Turks, M. Synthesis and Applications of Azolylpurine and Azolylpurine Nucleoside Derivatives. Eur. J. Org. Chem. 2015, 2015, 3629–3649. [Google Scholar] [CrossRef]
- Kurimoto, A.; Ogino, T.; Ichii, S.; Isobe, Y.; Tobe, M.; Ogita, H.; Takaku, H.; Sajiki, H.; Hirota, K.; Kawakami, H. Synthesis and Evaluation of 2-Substituted 8-Hydroxyadenines as Potent Interferon Inducers with Improved Oral Bioavailabilities. Bioorg. Med. Chem. 2004, 12, 1091–1099. [Google Scholar] [CrossRef]
- Buchanan, H.S.; Pauff, S.M.; Kosmidis, T.D.; Taladriz-Sender, A.; Rutherford, O.I.; Hatit, M.Z.C.; Fenner, S.; Watson, A.J.B.; Burley, G.A. Modular, Step-Efficient Palladium-Catalyzed Cross-Coupling Strategy to Access C6-Heteroaryl 2-Aminopurine Ribonucleosides. Org. Lett. 2017, 19, 3759–3762. [Google Scholar] [CrossRef]
- Havelková, M.; Dvořák, D.; Hocek, M. The Suzuki-Miyaura Cross-Coupling Reactions of 2-, 6- or 8-Halopurines with Boronic Acids Leading to 2-, 6- or 8-Aryl- and -Alkenylpurine Derivatives. Synthesis 2001, 11, 1704–1710. [Google Scholar] [CrossRef]
- Strouse, J.J.; Ješelnik, M.; Arterburn, J.B. Applications of Boronic Acids in Selective C-C and C-N Arylation of Purines. Acta Chim. Slov. 2005, 52, 187–199. [Google Scholar] [CrossRef]
- Wang, D.C.; Niu, H.Y.; Qu, G.R.; Liang, L.; Wei, X.J.; Zhang, Y.; Guo, H.M. Nickel-Catalyzed Negishi Cross-Couplings of 6-Chloropurines with Organozinc Halides at Room Temperature. Org. Biomol. Chem. 2011, 9, 7663–7666. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S.B.; Stevenson, T.M.; Citineni, J.R.; Nyzam, V.; Knochel, P. Preparation and Reactions of New Zincated Nitrogen-Containing Heterocycles. Tetrahedron 1997, 53, 7237–7254. [Google Scholar] [CrossRef]
- Nolsøe, J.M.J.; Gundersen, L.L.; Rise, F. Regiochemistry in the Pd-Mediated Coupling between 6,8-Dihalopurines and Organometallic Reagents. Acta Chem. Scand. 1999, 53, 366–372. [Google Scholar]
- Gundersen, L.L.; Langli, G.; Rise, F. Regioselective Pd-Mediated Coupling between 2,6-Dichloropurines and Organometallic Reagents. Tetrahedron Lett. 1995, 36, 1945–1948. [Google Scholar] [CrossRef]
- Bag, S.S.; Jana, S.; Kasula, M. Sonogashira Cross-Coupling: Alkyne-Modified Nucleosides and Their Applications. In Palladium-Catalyzed Modification of Nucleosides, Nucleotides and Oligonucleotides; Kapdi, A.R., Maiti, D., Sanghvi, Y.S., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 75–146. ISBN 9780128112922. [Google Scholar]
- Ibrahim, N.; Chevot, F.; Legraverend, M. Regioselective Sonogashira Cross-Coupling Reactions of 6-Chloro-2,8-diiodo-9-THP-9H-purine with Alkyne Derivatives. Tetrahedron Lett. 2011, 52, 305–307. [Google Scholar]
- Bilbao, N.; Vázquez-González, V.; Aranda, M.T.; González-Rodríguez, D. Synthesis of 5-/8-Halogenated or Ethynylated Lipophilic Nucleobases as Potential Synthetic Intermediates for Supramolecular Chemistry. Eur. J. Org. Chem. 2015, 2015, 7160–7175. [Google Scholar] [CrossRef]
- Hocek, M.; Pohl, R. Regioselectivity in Cross-Coupling Reactions of 2,6,8-Trichloro-9-(Tetrahydropyran-2-Yl)Purine: Synthesis of 2,6,8-Trisubstituted Purine Bases. Synthesis 2004, 36, 2869–2876. [Google Scholar] [CrossRef]
- Hocek, M.; Hocková, D.; Dvořáková, H. Dichotomy in Regioselective Cross-Coupling Reactions of 6,8-Dichloropurines with Phenylboronic Acid and Methylmagnesium Chloride: Synthesis of 6,8-Di-Substituted Purines. Synthesis 2004, 6, 889–894. [Google Scholar] [CrossRef]
- Mahajan, T.R.; Ytre-Arne, M.E.; Strøm-Andersen, P.; Dalhus, B.; Gundersen, L.L. Synthetic Routes to N-9 Alkylated 8-Oxoguanines; Weak Inhibitors of the Human DNA Glycosylase OGG1. Molecules 2015, 20, 15944–15965. [Google Scholar] [CrossRef]
- Abdoli, M.; Mirjafary, Z.; Saeidian, H.; Kakanejadifard, A. New Developments in Direct Functionalization of C-H and N-H Bonds of Purine Bases via Metal Catalyzed Cross-Coupling Reactions. RSC Adv. 2015, 5, 44371–44389. [Google Scholar] [CrossRef]
- Mooney, D.T.; Moore, P.R.; Lee, A.L. Direct Minisci-Type C-H Amidation of Purine Bases. Org. Lett. 2022, 24, 8008–8013. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.F.; Ulven, T. Direct N9-Arylation of Purines with Aryl Halide. Chem. Commun. 2014, 50, 4997–4999. [Google Scholar] [CrossRef]
- Chen, S.; Graceffa, R.F.; Boezio, A.A. Direct, Regioselective N-Alkylation of 1,3-Azoles. Org. Lett. 2016, 18, 16–19. [Google Scholar] [CrossRef]
- Tranová, L.; Stýskala, J. Study of the N7 Regioselective Glycosylation of 6-Chloropurine and 2,6-Dichloropurine with Tin and Titanium Tetrachloride. J. Org. Chem. 2021, 86, 13265–13275. [Google Scholar] [CrossRef]
- Nevrlka, F.; Bědroň, A.; Valenta, M.; Tranová, L.; Stýskala, J. Study of Direct N7 Regioselective Tert-Alkylation of 6-Substituted Purines and Their Modification at Position C6 through O, S, N, and C Substituents. ACS Omega 2024, 9, 17368–17378. [Google Scholar] [CrossRef]
- Sebris, A.; Novosjolova, I.; Turks, M. Synthesis of 7-Arylpurines from Substituted Pyrimidines. Synthesis 2022, 54, 5529–5539. [Google Scholar] [CrossRef]
- Bollier, M.; Klupsch, F.; Six, P.; Dubuquoy, L.; Azaroual, N.; Millet, R.; Leleu-Chavain, N. One- or Two-Step Synthesis of C-8 and N-9 Substituted Purines. J. Org. Chem. 2018, 83, 422–430. [Google Scholar] [CrossRef]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Taqui Khan, M.; Krishnamoorthy, C.R. Interaction of Metal Ions with Monosubstittuted Purines. J. Inorg. Nucl. Chem. 1971, 33, 1417–1425. [Google Scholar] [CrossRef]
- Gao, S.H.; Xie, M.S.; Wang, H.X.; Niu, H.Y.; Qu, G.R.; Guo, H.M. Highly Selective Detection of Hg2+ Ion by Push-Pull-Type Purine Nucleoside-Based Fluorescent Sensor. Tetrahedron 2014, 70, 4929–4933. [Google Scholar] [CrossRef]
- Li, J.P.; Wang, H.X.; Wang, H.X.; Xie, M.S.; Qu, G.R.; Niu, H.Y.; Guo, H.M. Push-Pull-Type Purine Nucleoside-Based Fluorescent Sensors for the Selective Detection of Pd2+ in Aqueous Buffer. Eur. J. Org. Chem. 2014, 2014, 2225–2230. [Google Scholar] [CrossRef]
- Chen, X.; Mao, Y.; Wang, A.; Lu, L.; Shao, Q.; Jiang, C.; Lu, H. Synthesis and Application of Purine-Based Fluorescence Probe for Continuous Recognition of Cu2+ and Glyphosate. Spectrochim. Acta-Part A Mol. Biomol. Spectrosc. 2024, 304, 123291. [Google Scholar] [CrossRef]
- Tao, X.; Mao, Y.; Alam, S.; Wang, A.; Qi, X.; Zheng, S.; Jiang, C.; Chen, S.Y.; Lu, H. Sensitive Fluorescence Detection of Glyphosate and Glufosinate Ammonium Pesticides by Purine-Hydrazone-Cu2+ Complex. Spectrochim. Acta-Part A Mol. Biomol. Spectrosc. 2024, 314, 124226. [Google Scholar] [CrossRef]
- Sadowska, P.; Jankowski, W.; Bregier-Jarzębowska, R.; Pietrzyk, P.; Jastrząb, R. Deciphering the Impact of Nucleosides and Nucleotides on Copper Ion and Dopamine Coordination Dynamics. Int. J. Mol. Sci. 2024, 25, 9137. [Google Scholar] [CrossRef]
- Schino, I.; Cantore, M.; de Candia, M.; Altomare, C.D.; Maria, C.; Barros, J.; Cachatra, V.; Calado, P.; Shimizu, K.; Freitas, A.A.; et al. Exploring Mannosylpurines as Copper Chelators and Cholinesterase Inhibitors with Potential for Alzheimer’s Disease. Pharmaceuticals 2023, 16, 54. [Google Scholar] [CrossRef]
- Xu, H.; Chen, W.; Ju, L.; Lu, H. A Purine Based Fluorescent Chemosensor for the Selective and Sole Detection of Al3+ and Its Practical Applications in Test Strips and Bio-Imaging. Spectrochim. Acta-Part A Mol. Biomol. Spectrosc. 2021, 247, 119074. [Google Scholar] [CrossRef]
- Jovaisaite, J.; Cīrule, D.; Jeminejs, A.; Novosjolova, I.; Turks, M.; Baronas, P.; Komskis, R.; Tumkevicius, S.; Jonusauskas, G.; Jursenas, S. Proof of Principle of a Purine D-A-D′ Ligand Based Ratiometric Chemical Sensor Harnessing Complexation Induced Intermolecular PET. Phys. Chem. Chem. Phys. 2020, 22, 26502–26508. [Google Scholar] [CrossRef]
- Tomczyk, M.D.; Kuźnik, N.; Walczak, K. Cyclen-Based Artificial Nucleases: Three Decades of Development (1989–2022). Part a–Hydrolysis of Phosphate Esters. Coord. Chem. Rev. 2023, 481, 215047. [Google Scholar] [CrossRef]
- Gałęzowska, J.; Boratyński, P.J.; Kowalczyk, R.; Lipke, K.; Czapor-Irzabek, H. Copper(II) Complexes of Chiral 1,2,3-Triazole Biheterocyclic ‘Click’ Ligands Equipped in Cinchona Alkaloid Moiety. Polyhedron 2017, 121, 1–8. [Google Scholar] [CrossRef]
- Cheney, G.E.; Freiser, H.; Fernando, Q.; Freiser, H. Metal Complexes of Purine and Some of Its Derivatives. J. Am. Chem. Soc. 1959, 81, 2611–2615. [Google Scholar] [CrossRef]
- Petrenko, Y.P.; Piasta, K.; Khomenko, D.M.; Doroshchuk, R.O.; Shova, S.; Novitchi, G.; Toporivska, Y.; Gumienna-Kontecka, E.; Martins, L.M.D.R.S.; Lampeka, R.D. An Investigation of Two Copper(II) Complexes with a Triazole Derivative as a Ligand: Magnetic and Catalytic Properties. RSC Adv. 2021, 11, 23442–23449. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Toro, I.; Domínguez-Martín, A.; Choquesillo-Lazarte, D.; Vílchez-Rodríguez, E.; González-Pérez, J.M.; Castiñeiras, A.; Niclós-Gutiérrez, J. Lights and Shadows in the Challenge of Binding Acyclovir, a Synthetic Purine-like Nucleoside with Antiviral Activity, at an Apical-Distal Coordination Site in Copper(II)-Polyamine Chelates. J. Inorg. Biochem. 2015, 148, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Yuan, H.Q.; Kikuchi, T.; Fujisawa, I.; Aoki, K. Metal Ion Interactions with Nucleobases in the Tetradentate 1,4,7,10-Tetraazacyclododecane (Cyclen)-Ligand System: Crystal Structures of [Cu(Cyclen)(Adeninato)]·ClO4·2H2O, [{Cu(Cyclen)}2(Hypoxanthinato)]·(ClO4)3, [Cu(Cyclen)(Theophyllinato)]3·(ClO4)3·2H2O, and [Cu(cyclen)(theophyllinato)]3·(ClO4)3·2H2O, and [Cu(cyclen)(xanthinato)]·(0.7ClO4)3·(0.3ClO4)3·3H2O·(0.5H2O)3. J. Mol. Struct. 2010, 966, 92–101. [Google Scholar] [CrossRef]
- Šišuļins, A.; Bucevičius, J.; Tseng, Y.T.; Novosjolova, I.; Traskovskis, K.; Bizdēna, Ē.; Chang, H.T.; Tumkevičius, S.; Turks, M. Synthesis and Fluorescent Properties of N(9)-Alkylated 2-Amino-6-Triazolylpurines and 7-Deazapurines. Beilstein J. Org. Chem. 2019, 15, 474–489. [Google Scholar] [CrossRef]
- Sebris, A.; Traskovskis, K.; Novosjolova, I.; Turks, M. Synthesis and Photophysical Properties of 2-Azolyl-6-Piperidinylpurines. Chem. Heterocycl. Compd. 2021, 57, 560–567. [Google Scholar] [CrossRef]
- Sessler, J.L.; Sathiosatham, M.; Doerr, K.; Lynch, V.V.; Abboud, K.A. A G-Quartet Formed in the Absence of a Templating Metal Cation: A New 8-(N,N-Dimethylaniline)Guanosine Derivative. Angew. Chem. Int. Ed. Engl. 2000, 39, 1300–1303. [Google Scholar] [CrossRef]
- Liu, T.; Zhan, W.; Wang, Y.; Zhang, L.; Yang, B.; Dong, X.; Hu, Y. Structure-Based Design, Synthesis and Biological Evaluation of Diphenylmethylamine Derivatives as Novel Akt1 Inhibitors. Eur. J. Med. Chem. 2014, 73, 167–176. [Google Scholar] [CrossRef]
- Selmani, A.; Serpier, F.; Darses, S. From Tetrahydrofurans to Tetrahydrobenzo[d]Oxepines via a Regioselective Control of Alkyne Insertion in Rhodium-Catalyzed Arylative Cyclization. J. Org. Chem. 2019, 84, 4566–4574. [Google Scholar] [CrossRef]
- Zhao, D.; Wu, N.; Zhang, S.; Xi, P.; Su, X.; Lan, J.; You, J. Synthesis of Phenol, Aromatic Ether, and Benzofuran Derivatives by Copper-Catalyzed Hydroxylation of Aryl Halides. Angew. Chemie-Int. Ed. 2009, 48, 8729–8732. [Google Scholar] [CrossRef]
- Altman, R.A.; Shafir, A.; Choi, A.; Lichtor, P.A.; Buchwald, S.L. An Improved Cu-Based Catalyst System for the Reactions of Alcohols with Aryl Halides. J. Org. Chem. 2008, 73, 284–286. [Google Scholar] [CrossRef]
- Niu, J.; Guo, P.; Kang, J.; Li, Z.; Xu, J.; Hu, S. Copper(I)-Catalyzed Aryl Bromides to Form Intermolecular and Intramolecular Carbon-Oxygen Bonds. J. Org. Chem. 2009, 74, 5075–5078. [Google Scholar] [CrossRef] [PubMed]
- Kreibich, M.; Petrović, D.; Brückner, R. Mechanistic Studies of the Deslongchamps Annulation. J. Org. Chem. 2018, 83, 1116–1133. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, M.; Konkol, M.; Del Rosal, I.; Poteau, R.; Maron, L.; Okuda, J. Rare-Earth Metal Alkyl and Hydride Complexes Stabilized by a Cyclen-Derived [NNNN] Macrocyclic Ancillary Ligand. J. Am. Chem. Soc. 2008, 130, 6920–6921. [Google Scholar] [CrossRef]
- Mereshchenko, A.S.; Olshin, P.K.; Karimov, A.M.; Skripkin, M.Y.; Burkov, K.A.; Tveryanovich, Y.S.; Tarnovsky, A.N. Photochemistry of Copper(II) Chlorocomplexes in Acetonitrile: Trapping the Ligand-to-Metal Charge Transfer Excited Staterelaxations Pathways. Chem. Phys. Lett. 2014, 615, 105–110. [Google Scholar] [CrossRef]
- Ding, H.W.; Wang, S.; Qin, X.C.; Wang, J.; Song, H.R.; Zhao, Q.C.; Song, S.J. Design, Synthesis, and Biological Evaluation of Some Novel 4-Aminoquinazolines as Pan-PI3K Inhibitors. Bioorg. Med. Chem. 2019, 27, 2729–2740. [Google Scholar] [CrossRef]
- Qiu, X.P.; Korchagina, E.V.; Rolland, J.; Winnik, F.M. Synthesis of a Poly(N-Isopropylacrylamide) Charm Bracelet Decorated with a Photomobile α-Cyclodextrin Charm. Polym. Chem. 2014, 5, 3656–3665. [Google Scholar] [CrossRef]
- Mazaki, Y.; Mutai, K. Charge-Transfer Interaction and Molecular Arrangement in the Crystals of 2-(p-Nitrophenoxy)Ethyl Ethers and Amines. Bull. Chem. Soc. Jpn. 1995, 68, 3247–3254. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burcevs, A.; Jonusauskas, G.; Novosjolova, I.; Turks, M. Synthesis of Purine-1,4,7,10-Tetraazacyclododecane Conjugate and Its Complexation Modes with Copper(II). Molecules 2025, 30, 1612. https://doi.org/10.3390/molecules30071612
Burcevs A, Jonusauskas G, Novosjolova I, Turks M. Synthesis of Purine-1,4,7,10-Tetraazacyclododecane Conjugate and Its Complexation Modes with Copper(II). Molecules. 2025; 30(7):1612. https://doi.org/10.3390/molecules30071612
Chicago/Turabian StyleBurcevs, Aleksejs, Gediminas Jonusauskas, Irina Novosjolova, and Māris Turks. 2025. "Synthesis of Purine-1,4,7,10-Tetraazacyclododecane Conjugate and Its Complexation Modes with Copper(II)" Molecules 30, no. 7: 1612. https://doi.org/10.3390/molecules30071612
APA StyleBurcevs, A., Jonusauskas, G., Novosjolova, I., & Turks, M. (2025). Synthesis of Purine-1,4,7,10-Tetraazacyclododecane Conjugate and Its Complexation Modes with Copper(II). Molecules, 30(7), 1612. https://doi.org/10.3390/molecules30071612