
Implications of Mucin-Type O-Glycosylation in Alzheimer’s Disease


Abstract
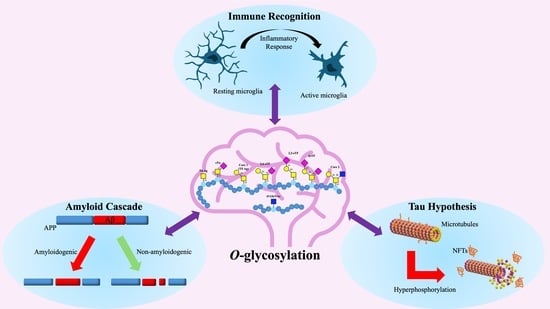
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Alzheimer’s Disease
1.2. Hypotheses Surrounding Alzheimer’s Disease Onset
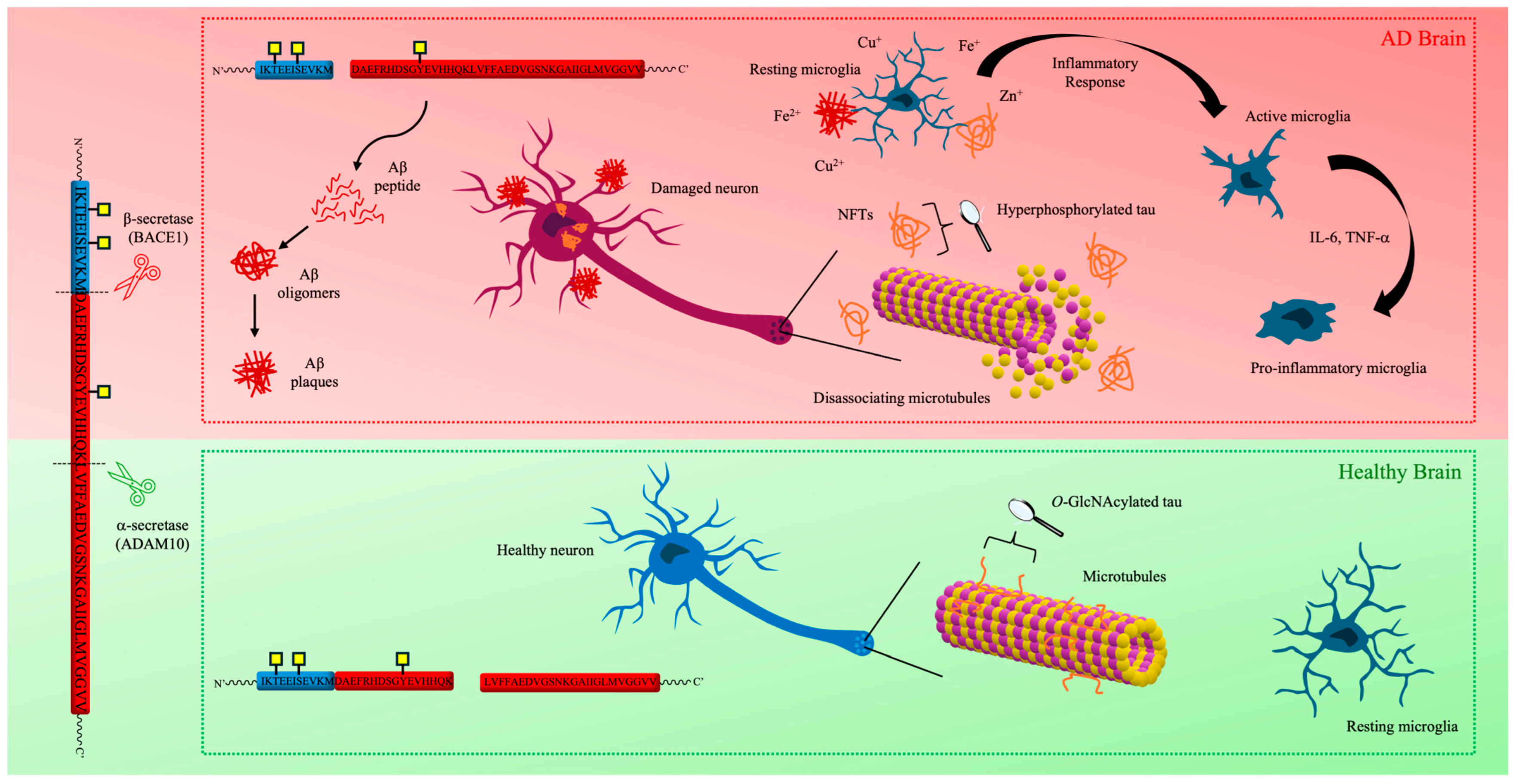
1.2.1. Tau Hypothesis
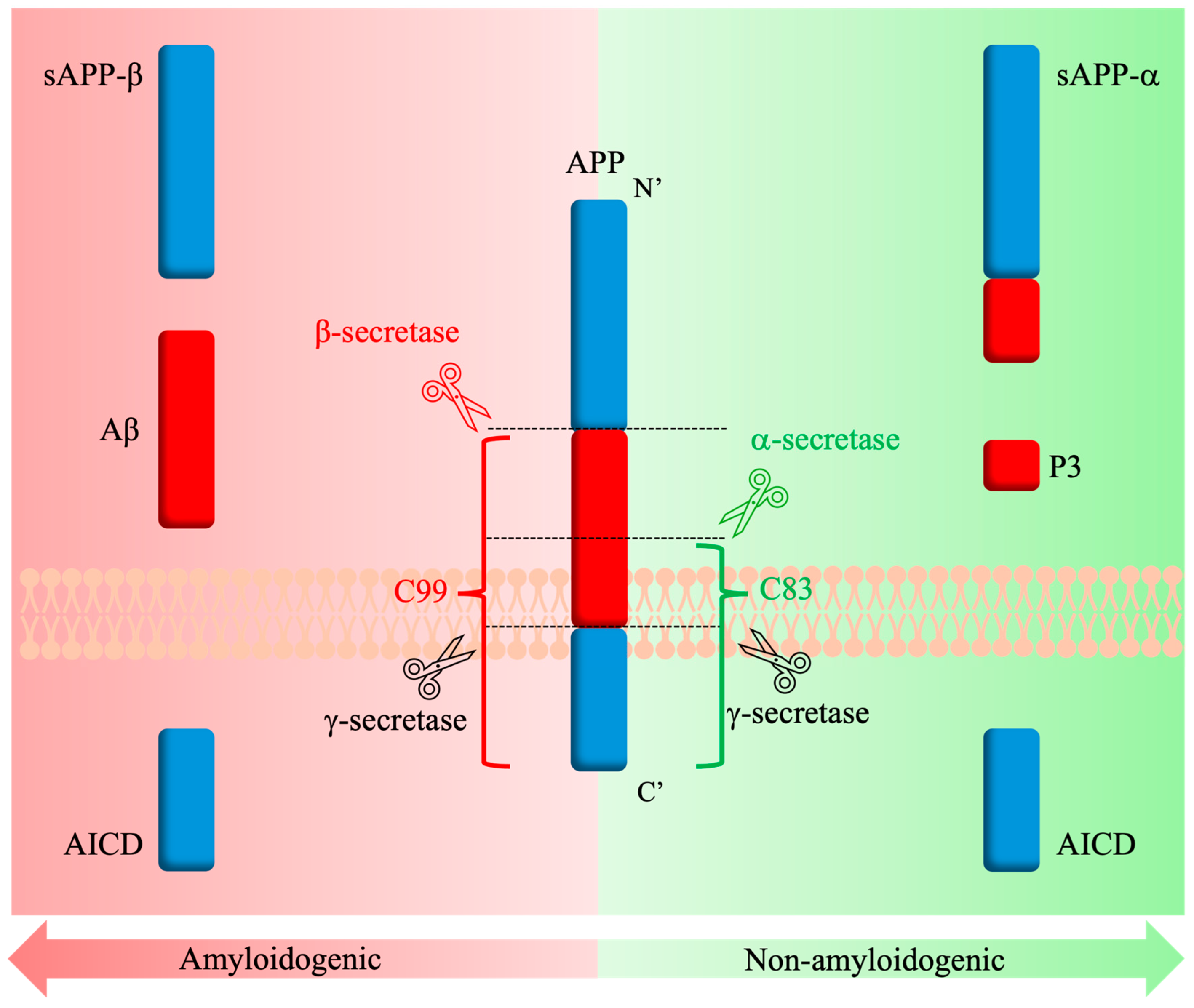
1.2.2. The Amyloid Cascade Hypothesis
1.2.3. Neuroinflammation Cascade
1.2.4. Metal Hypothesis
2. Post-Translational Modification in Alzheimer’s Disease
2.1. Methylation
2.2. Ubiquitylation
2.3. Acetylation
2.4. Phosphorylation
2.5. N-Glycosylation
2.6. O-Glycosylation
3. Implications of APP Mucin-Type O-Glycosylation in APP Processing
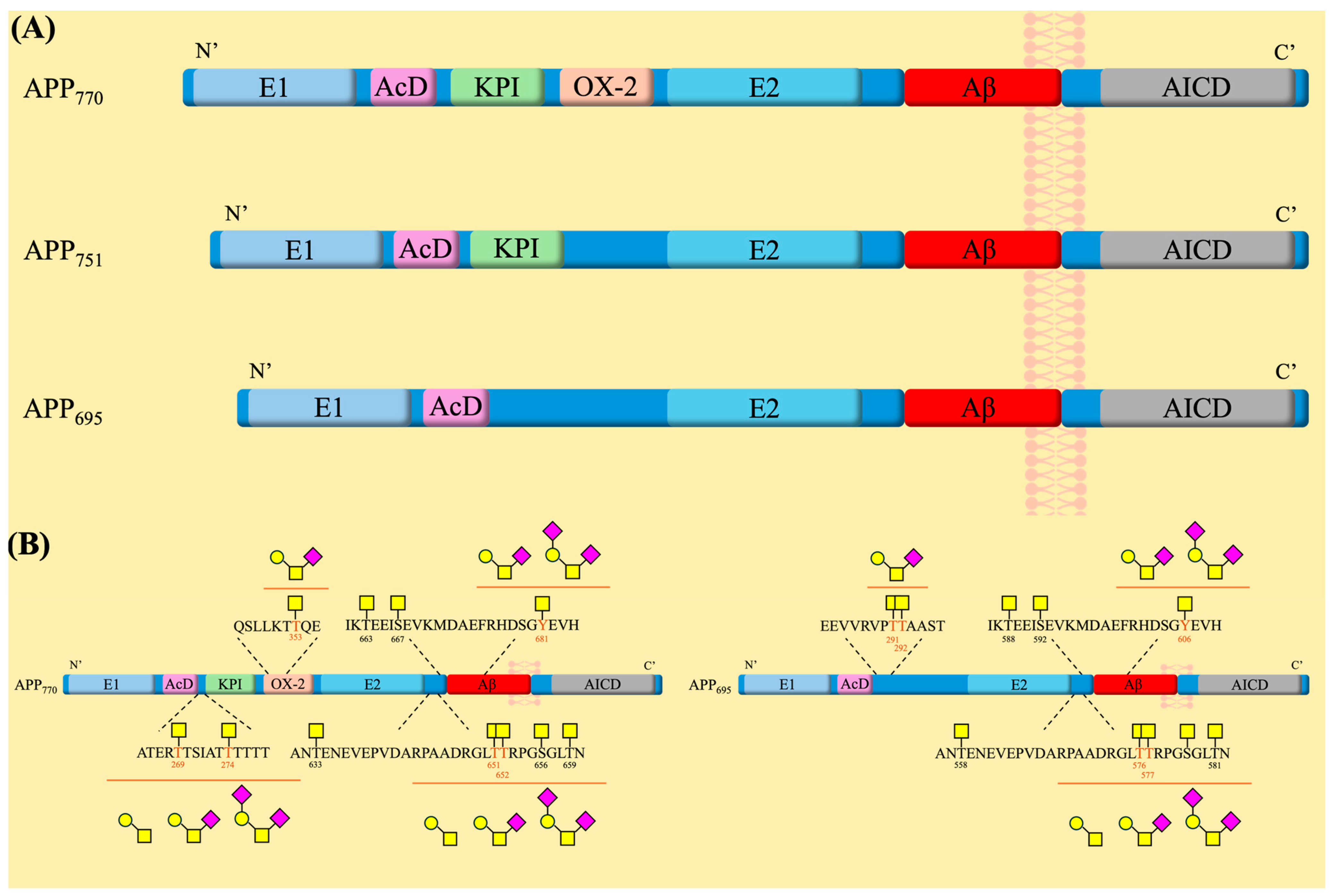
3.1. APP Structure and Physiological Functions
3.2. Role of Mucin-Type O-Glycosylation in APP Processing and AD Pathology
4. Regulation of Immune Responses and Neuroinflammation by O-Glycan Binding Proteins
4.1. Immune Homeostasis
4.2. The CNS and the Neuroimmune Signaling Interplay
4.3. Glycan-Binding Proteins and Immune Response
4.3.1. Galectins and O-Glycans
4.3.2. Siglecs and Sialylated O-Glycans
4.3.3. C-Type Lectins and O-Glycans
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Silva, M.V.F.; de Loures, C.M.G.; Alves, L.C.V.; de Souza, L.C.; Borges, K.B.G.; Carvalho, M.d.G. Alzheimer’s Disease: Risk Factors and Potentially Protective Measures. J. Biomed. Sci. 2019, 26, 33. [Google Scholar] [CrossRef] [PubMed]
- 2024 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2024, 20, 3708–3821. [CrossRef] [PubMed]
- Gustavsson, A.; Norton, N.; Fast, T.; Frölich, L.; Georges, J.; Holzapfel, D.; Kirabali, T.; Krolak-Salmon, P.; Rossini, P.M.; Ferretti, M.T.; et al. Global Estimates on the Number of Persons across the Alzheimer’s Disease Continuum. Alzheimer’s Dement. 2023, 19, 658–670. [Google Scholar] [CrossRef]
- Kingston, A.; Comas-Herrera, A.; Jagger, C. Forecasting the Care Needs of the Older Population in England over the next 20 Years: Estimates from the Population Ageing and Care Simulation (PACSim) Modelling Study. Lancet Public Health 2018, 3, e447–e455. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s Disease Drug Development Pipeline: 2019. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 272–293. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Osse, A.M.L.; Cammann, D.; Powell, J.; Chen, J. Anti-Amyloid Monoclonal Antibodies for the Treatment of Alzheimer’s Disease. BioDrugs 2024, 38, 5–22. [Google Scholar] [CrossRef]
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Monkul Nery, E.S.; et al. Donanemab in Early Symptomatic Alzheimer Disease. JAMA 2023, 330, 512. [Google Scholar] [CrossRef]
- Karran, E.; De Strooper, B. The Amyloid Hypothesis in Alzheimer Disease: New Insights from New Therapeutics. Nat. Rev. Drug Discov. 2022, 21, 306–318. [Google Scholar] [CrossRef]
- Stelzmann, R.A.; Norman Schnitzlein, H.; Reed Murtagh, F. An English Translation of Alzheimer’s 1907 Paper, “Über Eine Eigenartige Erkankung Der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef]
- Tagarelli, A.; Piro, A.; Tagarelli, G.; Lagonia, P.; Quattrone, A. Alois Alzheimer: A Hundred Years after the Discovery of the Eponymous Disorder. Int. J. Biomed. Sci. 2006, 2, 196–204. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s Disease: Initial Report of the Purification and Characterization of a Novel Cerebrovascular Amyloid Protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaupt, G.; Mcdonald, B.L.; Beyreuthert, K. Amyloid Plaque Core Protein in Alzheimer Disease and Down Syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Kidd, M. Alzheimer’s Disease-an Electron Microscopical Study. Brain 1964, 87, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Kidd, M. Paired Helical Filaments in Electron Microscopy of Alzheimer’s Disease. Nature 1963, 197, 192–193. [Google Scholar] [CrossRef]
- Terry, R.D. The FIne Structure of Neurofibrillary Tangles in Alzheimer’s Disease. J. Neuropathol. Exp. Neurol. 1963, 22, 629–642. [Google Scholar] [CrossRef]
- Terry, R.; Gonatas, N.; Weiss, M. Ultrastructural Studies in Alzheimer’s Presenile Dementia. Am. J. Pathol. 1964, 44, 269–297. [Google Scholar]
- Iqbal, K.; Grundke-Iqbal, I. Discoveries of Tau, Abnormally Hyperphosphorylated Tau and Others of Neurofibrillary Degeneration: A Personal Historical Perspective. J. Alzheimer’s Dis. 2006, 9, 219–242. [Google Scholar] [CrossRef]
- Johnson, G.V.; Jenkins, S.M. Tau Protein in Normal and Alzheimer’s Disease Brain. J. Alzheimer’s Dis. 1999, 1, 307–328. [Google Scholar] [CrossRef]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging-Alzheimer’s Association Guidelines for the Neuropathologic Assessment of Alzheimer’s Disease: A Practical Approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef]
- Deture, M.A.; Dickson, D.W. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Mandelkow, E.-M.; Mandelkow, E. Biochemistry and Cell Biology of Tau Protein in Neurofibrillary Degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef]
- Kirschner, M. The Discovery of Tau Protein. Cytoskeleton 2024, 81, 78–82. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A Protein Factor Essential for Microtubule Assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Neve, R.L.; Kosik, K.S. The Microtubule Binding Domain of Tau Protein. Neuron 1989, 2, 1615–1624. [Google Scholar] [CrossRef] [PubMed]
- Brandt, R.; Léger, J.; Lee, G. Interaction of Tau with the Neural Plasma Membrane Mediated by Tau’s Amino-Terminal Projection Domain. J. Cell Biol. 1995, 131, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Alquezar, C.; Arya, S.; Kao, A.W. Tau Post-Translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation. Front. Neurol. 2021, 11, 595532. [Google Scholar] [CrossRef]
- Neddens, J.; Temmel, M.; Flunkert, S.; Kerschbaumer, B.; Hoeller, C.; Loeffler, T.; Niederkofler, V.; Daum, G.; Attems, J.; Hutter-Paier, B. Phosphorylation of Different Tau Sites during Progression of Alzheimer’s Disease. Acta Neuropathol. Commun. 2018, 6, 52. [Google Scholar] [CrossRef]
- Köpke, E.; Tung, Y.C.; Shaikh, S.; Alonso, A.C.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-Associated Protein Tau. Abnormal Phosphorylation of a Non-Paired Helical Filament Pool in Alzheimer Disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Hwo, S.-Y.; Kirschner, M.W. Physical and Chemical Properties of Purified Tau Factor and the Role of Tau in Microtubule Assembly. J. Mol. Biol. 1977, 116, 227–247. [Google Scholar] [CrossRef]
- Lindwall, G.; Cole, R.D. Phosphorylation Affects the Ability of Tau Protein to Promote Microtubule Assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar] [CrossRef]
- Tenreiro, S.; Eckermann, K.; Outeiro, T.F. Protein Phosphorylation in Neurodegeneration: Friend or Foe? Front. Mol. Neurosci. 2014, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Selkoe, D.J. Aβ Oligomers—A Decade of Discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Guo, Z. Alzheimer’s Aβ42 and Aβ40 Peptides Form Interlaced Amyloid Fibrils. J. Neurochem. 2013, 126, 305–311. [Google Scholar] [CrossRef]
- Wang, L.; Eom, K.; Kwon, T. Different Aggregation Pathways and Structures for Aβ40 and Aβ42 Peptides. Biomolecules 2021, 11, 198. [Google Scholar] [CrossRef]
- Tolar, M.; Hey, J.; Power, A.; Abushakra, S. Neurotoxic Soluble Amyloid Oligomers Drive Alzheimer’s Pathogenesis and Represent a Clinically Validated Target for Slowing Disease Progression. Int. J. Mol. Sci. 2021, 22, 6355. [Google Scholar] [CrossRef]
- Chen, G.; Xu, T.; Yan, Y.; Zhou, Y.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Herrup, K. The Case for Rejecting the Amyloid Cascade Hypothesis. Nat. Neurosci. 2015, 18, 794–799. [Google Scholar] [CrossRef]
- Dickson, D.W. The Pathogenesis of Senile Plaques. J. Neuropathol. Exp. Neurol. 1997, 56, 321–339. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Translating Cell Biology into Therapeutic Advances in Alzheimer’s Disease. Nature 1999, 399, A23–A31. [Google Scholar] [CrossRef]
- Barage, S.H.; Sonawane, K.D. Amyloid Cascade Hypothesis: Pathogenesis and Therapeutic Strategies in Alzheimer’s Disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Kepp, K.P.; Robakis, N.K.; Høilund-Carlsen, P.F.; Sensi, S.L.; Vissel, B. The Amyloid Cascade Hypothesis: An Updated Critical Review. Brain 2023, 146, 3969–3990. [Google Scholar] [CrossRef]
- Shen, X.-N.; Niu, L.-D.; Wang, Y.-J.; Cao, X.-P.; Liu, Q.; Tan, L.; Zhang, C.; Yu, J.-T. Inflammatory Markers in Alzheimer’s Disease and Mild Cognitive Impairment: A Meta-Analysis and Systematic Review of 170 Studies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 590–598. [Google Scholar] [CrossRef]
- Garland, E.F.; Hartnell, I.J.; Boche, D. Microglia and Astrocyte Function and Communication: What Do We Know in Humans? Front. Neurosci. 2022, 16, 824888. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Tago, H.; McGeer, E.G. Reactive Microglia in Patients with Senile Dementia of the Alzheimer Type Are Positive for the Histocompatibility Glycoprotein HLA-DR. Neurosci. Lett. 1987, 79, 195–200. [Google Scholar] [CrossRef]
- Tooyama, I.; Kimura, H.; Akiyama, H.; McGeer, P.L. Reactive Microglia Express Class I and Class II Major Histocompatibility Complex Antigens in Alzheimer’s Disease. Brain Res. 1990, 523, 273–280. [Google Scholar] [CrossRef]
- Dani, M.; Wood, M.; Mizoguchi, R.; Fan, Z.; Walker, Z.; Morgan, R.; Hinz, R.; Biju, M.; Kuruvilla, T.; Brooks, D.J.; et al. Microglial Activation Correlates in Vivo with Both Tau and Amyloid in Alzheimer’s Disease. Brain 2018, 141, 2740–2754. [Google Scholar] [CrossRef]
- Kabba, J.A.; Xu, Y.; Christian, H.; Ruan, W.; Chenai, K.; Xiang, Y.; Zhang, L.; Saavedra, J.M.; Pang, T. Microglia: Housekeeper of the Central Nervous System. Cell. Mol. Neurobiol. 2018, 38, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Al-Qahtani, A.A.; Alhamlan, F.S.; Al-Qahtani, A.A. Pro-Inflammatory and Anti-Inflammatory Interleukins in Infectious Diseases: A Comprehensive Review. Trop. Med. Infect. Dis. 2024, 9, 13. [Google Scholar] [CrossRef]
- van Kooyk, Y.; Ilarregui, J.M.; van Vliet, S.J. Novel Insights into the Immunomodulatory Role of the Dendritic Cell and Macrophage-Expressed C-Type Lectin MGL. Immunobiology 2015, 220, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Ilarregui, J.M.; Kooij, G.; Rodríguez, E.; van der Pol, S.M.A.; Koning, N.; Kalay, H.; van der Horst, J.C.; van Vliet, S.J.; García-Vallejo, J.J.; de Vries, H.E.; et al. Macrophage Galactose-Type Lectin (MGL) Is Induced on M2 Microglia and Participates in the Resolution Phase of Autoimmune Neuroinflammation. J. Neuroinflamm. 2019, 16, 130. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, S.J.; Saeland, E.; van Kooyk, Y. Sweet Preferences of MGL: Carbohydrate Specificity and Function. Trends Immunol. 2008, 29, 83–90. [Google Scholar] [CrossRef]
- Hanisch, F.-G. O-Glycosylation of the Mucin Type. Biol. Chem. 2001, 382, 143–149. [Google Scholar] [CrossRef]
- Guevara, J.; Espinosa, B.; Zenteno, E.; Vázquez, L.; Luna, J.; Perry, G.; Mena, R. Altered Glycosylation Pattern of Proteins in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 1998, 57, 905–914. [Google Scholar] [CrossRef]
- Bush, A.I.; Tanzi, R.E. Therapeutics for Alzheimer’s Disease Based on the Metal Hypothesis. Neurotherapeutics 2008, 5, 421–432. [Google Scholar] [CrossRef]
- Babić Leko, M.; Langer Horvat, L.; Španić Popovački, E.; Zubčić, K.; Hof, P.R.; Šimić, G. Metals in Alzheimer’s Disease. Biomedicines 2023, 11, 1161. [Google Scholar] [CrossRef]
- Chen, L.-L.; Fan, Y.-G.; Zhao, L.-X.; Zhang, Q.; Wang, Z.-Y. The Metal Ion Hypothesis of Alzheimer’s Disease and the Anti-Neuroinflammatory Effect of Metal Chelators. Bioorg. Chem. 2023, 131, 106301. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, D.; Yu, Q.; Johnson, J.; Shipman, R.; Zhong, X.; Huang, J.; Yu, Q.; Zetterberg, H.; Asthana, S.; et al. In-Depth Site-Specific O-Glycosylation Analysis of Glycoproteins and Endogenous Peptides in Cerebrospinal Fluid (CSF) from Healthy Individuals, Mild Cognitive Impairment (MCI), and Alzheimer’s Disease (AD) Patients. ACS Chem. Biol. 2022, 17, 3059–3068. [Google Scholar] [CrossRef]
- Boix, C.P.; Lopez-Font, I.; Cuchillo-Ibañez, I.; Sáez-Valero, J. Amyloid Precursor Protein Glycosylation Is Altered in the Brain of Patients with Alzheimer’s Disease. Alzheimer’s Res. Ther. 2020, 12, 96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ma, C.; Chin, L.-S.; Li, L. Integrative Glycoproteomics Reveals Protein N-Glycosylation Aberrations and Glycoproteomic Network Alterations in Alzheimer’s Disease. Sci. Adv. 2020, 6, eabc5802. [Google Scholar] [CrossRef]
- Suttapitugsakul, S.; Stavenhagen, K.; Donskaya, S.; Bennett, D.A.; Mealer, R.G.; Seyfried, N.T.; Cummings, R.D. Glycoproteomics Landscape of Asymptomatic and Symptomatic Human Alzheimer’s Disease Brain. Mol. Cell. Proteom. 2022, 21, 100433. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, D.M.; Montagna, D.R.; Gu, Y.; Selkoe, D.J.; Wolfe, M.S. Nicastrin Functions to Sterically Hinder γ-Secretase–Substrate Interactions Driven by Substrate Transmembrane Domain. Proc. Natl. Acad. Sci. USA 2016, 113, E509–E518. [Google Scholar] [CrossRef]
- Vanoni, O.; Paganetti, P.; Molinari, M. Consequences of Individual N-Glycan Deletions and of Proteasomal Inhibition on Secretion of Active BACE. Mol. Biol. Cell 2008, 19, 4086–4098. [Google Scholar] [CrossRef]
- Frenkel-Pinter, M.; Shmueli, M.D.; Raz, C.; Yanku, M.; Zilberzwige, S.; Gazit, E.; Segal, D. Interplay between Protein Glycosylation Pathways in Alzheimer’s Disease. Sci. Adv. 2017, 3, e1601576. [Google Scholar] [CrossRef]
- Müller, M.M. Post-Translational Modifications of Protein Backbones: Unique Functions, Mechanisms, and Challenges. Biochemistry 2018, 57, 177–185. [Google Scholar] [CrossRef]
- Keenan, E.K.; Zachman, D.K.; Hirschey, M.D. Discovering the Landscape of Protein Modifications. Mol. Cell 2021, 81, 1868–1878. [Google Scholar] [CrossRef]
- Fuchs, S.M. Chemically Modified Tandem Repeats in Proteins: Natural Combinatorial Peptide Libraries. ACS Chem. Biol. 2013, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.-J.; Dammer, E.B.; Wang, G.; Seyfried, N.T.; Levey, A.I. Proteomics of Protein Post-Translational Modifications Implicated in Neurodegeneration. Transl. Neurodegener. 2014, 3, 23. [Google Scholar] [CrossRef]
- Gupta, R.; Sahu, M.; Srivastava, D.; Tiwari, S.; Ambasta, R.K.; Kumar, P. Post-Translational Modifications: Regulators of Neurodegenerative Proteinopathies. Ageing Res. Rev. 2021, 68, 101336. [Google Scholar] [CrossRef]
- Schaffert, L.-N.; Carter, W.G. Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review. Brain Sci. 2020, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, H.-L.; Wang, J.-Z.; Liu, R.; Wang, X. Abnormal Protein Post-Translational Modifications Induces Aggregation and Abnormal Deposition of Protein, Mediating Neurodegenerative Diseases. Cell Biosci. 2024, 14, 22. [Google Scholar] [CrossRef]
- Lee, J.M.; Hammarén, H.M.; Savitski, M.M.; Baek, S.H. Control of Protein Stability by Post-Translational Modifications. Nat. Commun. 2023, 14, 201. [Google Scholar] [CrossRef] [PubMed]
- Rowe, E.M.; Xing, V.; Biggar, K.K. Lysine Methylation: Implications in Neurodegenerative Disease. Brain Res. 2019, 1707, 164–171. [Google Scholar] [CrossRef]
- Thomas, S.N.; Funk, K.E.; Wan, Y.; Liao, Z.; Davies, P.; Kuret, J.; Yang, A.J. Dual Modification of Alzheimer’s Disease PHF-Tau Protein by Lysine Methylation and Ubiquitylation: A Mass Spectrometry Approach. Acta Neuropathol. 2012, 123, 105–117. [Google Scholar] [CrossRef]
- Xiao, X.; Liu, X.; Jiao, B. Epigenetics: Recent Advances and Its Role in the Treatment of Alzheimer’s Disease. Front. Neurol. 2020, 11, 538301. [Google Scholar] [CrossRef]
- Guan, P.-P.; Wang, P. The Involvement of Post-Translational Modifications in Regulating the Development and Progression of Alzheimer’s Disease. Mol. Neurobiol. 2023, 60, 3617–3632. [Google Scholar] [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef]
- Bai, N.; Li, N.; Cheng, R.; Guan, Y.; Zhao, X.; Song, Z.; Xu, H.; Yi, F.; Jiang, B.; Li, X.; et al. Inhibition of SIRT2 Promotes APP Acetylation and Ameliorates Cognitive Impairment in APP/PS1 Transgenic Mice. Cell Rep. 2022, 40, 111062. [Google Scholar] [CrossRef] [PubMed]
- Tracy, T.; Claiborn, K.C.; Gan, L. Regulation of Tau Homeostasis and Toxicity by Acetylation. In Tau Biology; Springer: Singapore, 2019; pp. 47–55. [Google Scholar]
- Rubin, C.S.; Rosen, O.M. Protein Phosphorylation. Annu. Rev. Biochem. 1975, 44, 831–887. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Ram, M.; Kumar, R.; Prasad, R.; Roy, B.K.; Singh, K.K. Phosphorylation: Implications in Cancer. Protein J. 2017, 36, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pang, K.; Wang, W.; Qin, J.; Shi, Z.; Hao, L.; Ma, Y.; Xu, H.; Wu, Z.; Pan, D.; Chen, Z.; et al. Role of Protein Phosphorylation in Cell Signaling, Disease, and the Intervention Therapy. MedComm 2022, 3, e175. [Google Scholar] [CrossRef]
- Zhang, T.; Chen, D.; Lee, T.H. Phosphorylation Signaling in APP Processing in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 21, 209. [Google Scholar] [CrossRef]
- Schneider, A.; Biernat, J.; von Bergen, M.; Mandelkow, E.; Mandelkow, E.-M. Phosphorylation That Detaches Tau Protein from Microtubules (Ser262, Ser214) Also Protects It against Aggregation into Alzheimer Paired Helical Filaments. Biochemistry 1999, 38, 3549–3558. [Google Scholar] [CrossRef]
- Schwarz, F.; Aebi, M. Mechanisms and Principles of N-Linked Protein Glycosylation. Curr. Opin. Struct. Biol. 2011, 21, 576–582. [Google Scholar] [CrossRef]
- Stanley, P.; Moremen, K.W.; Lewis, N.E.; Taniguchi, N.; Aebi, M. N-Glycans. In Essentials of Glycobiology, 4th ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2022; ISBN 9781621824213. [Google Scholar]
- Esmail, S.; Manolson, M.F. Advances in Understanding N-Glycosylation Structure, Function, and Regulation in Health and Disease. Eur. J. Cell Biol. 2021, 100, 151186. [Google Scholar] [CrossRef]
- Lin, T.; van Husen, L.S.; Yu, Y.; Tjernberg, L.O.; Schedin-Weiss, S. Lack of N-Glycosylation Increases Amyloidogenic Processing of the Amyloid Precursor Protein. Glycobiology 2022, 32, 506–517. [Google Scholar] [CrossRef]
- Sato, Y.; Naito, Y.; Grundke-Iqbal, I.; Iqbal, K.; Endo, T. Analysis of N-glycans of Pathological Tau: Possible Occurrence of Aberrant Processing of Tau in Alzheimer’s Disease. FEBS Lett. 2001, 496, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zaidi, T.; Iqbal, K.; Grundke-Iqbal, I.; Merkle, R.K.; Gong, C.-X. Role of Glycosylation in Hyperphosphorylation of Tau in Alzheimer’s Disease. FEBS Lett. 2002, 512, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Kizuka, Y.; Kitazume, S.; Taniguchi, N. N-Glycan and Alzheimer’s Disease. Biochim. Biophys. Acta—Gen. Subj. 2017, 1861, 2447–2454. [Google Scholar] [CrossRef]
- Haltiwanger, R.S.; Wells, L.; Freeze, H.H.; Jafar-Nejad, H.; Okajima, T.; Stanley, P. Other Classes of Eukaryotic Glycans. In Essentials of Glycobiology, 4th ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2022; ISBN 9781621824213. [Google Scholar]
- Brockhausen, I.; Schachter, H.; Stanley, P. O-GalNAc Glycans. In Essentials of Glycobiology, 2nd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2009; ISBN 9780879697709. [Google Scholar]
- Taherzadeh, G.; Dehzangi, A.; Golchin, M.; Zhou, Y.; Campbell, M.P. SPRINT-Gly: Predicting N- and O-Linked Glycosylation Sites of Human and Mouse Proteins by Using Sequence and Predicted Structural Properties. Bioinformatics 2019, 35, 4140–4146. [Google Scholar] [CrossRef]
- Pakhrin, S.C.; Chauhan, N.; Khan, S.; Upadhyaya, J.; Beck, M.R.; Blanco, E. Prediction of Human O-Linked Glycosylation Sites Using Stacked Generalization and Embeddings from Pre-Trained Protein Language Model. Bioinformatics 2024, 40, btae643. [Google Scholar] [CrossRef]
- Torres, C.R.; Hart, G.W. Topography and Polypeptide Distribution of Terminal N-Acetylglucosamine Residues on the Surfaces of Intact Lymphocytes. Evidence for O-Linked GlcNAc. J. Biol. Chem. 1984, 259, 3308–3317. [Google Scholar] [CrossRef]
- Lefebvre, T.; Ferreira, S.; Dupont-Wallois, L.; Bussière, T.; Dupire, M.-J.; Delacourte, A.; Michalski, J.-C.; Caillet-Boudin, M.-L. Evidence of a Balance between Phosphorylation and O-GlcNAc Glycosylation of Tau Proteins—A Role in Nuclear Localization. Biochim. Biophys. Acta—Gen. Subj. 2003, 1619, 167–176. [Google Scholar] [CrossRef]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.W.; Gong, C.-X. O-GlcNAcylation Regulates Phosphorylation of Tau: A Mechanism Involved in Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804–10809. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Liu, F.; Iqbal, K. O-GlcNAcylation: A Regulator of Tau Pathology and Neurodegeneration. Alzheimer’s Dement. 2016, 12, 1078–1089. [Google Scholar] [CrossRef]
- Griffith, L.S.; Mathes, M.; Schmitz, B. Β-Amyloid Precursor Protein Is Modified with O-linked N-acetylglucosamine. J. Neurosci. Res. 1995, 41, 270–278. [Google Scholar] [CrossRef]
- Jacobsen, K.T.; Iverfeldt, K. O-GlcNAcylation Increases Non-Amyloidogenic Processing of the Amyloid-β Precursor Protein (APP). Biochem. Biophys. Res. Commun. 2011, 404, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.S.; Kwon, O.-H.; Chung, S. O-GlcNAcylation of Amyloid-β Precursor Protein at Threonine 576 Residue Regulates Trafficking and Processing. Biochem. Biophys. Res. Commun. 2017, 490, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.T.; Ten Hagen, K.G. Mucin-Type O-Glycosylation during Development. J. Biol. Chem. 2013, 288, 6921–6929. [Google Scholar] [CrossRef]
- Bagdonaite, I.; Pallesen, E.M.H.; Nielsen, M.I.; Bennett, E.P.; Wandall, H.H. Mucin-Type O-GalNAc Glycosylation in Health and Disease. In The Role of Glycosylation in Health and Disease; Springer: Berlin/Heidelberg, Germany, 2021; pp. 25–60. [Google Scholar]
- Bennett, E.P.; Mandel, U.; Clausen, H.; Gerken, T.A.; Fritz, T.A.; Tabak, L.A. Control of Mucin-Type O-Glycosylation: A Classification of the Polypeptide GalNAc-Transferase Gene Family. Glycobiology 2012, 22, 736–756. [Google Scholar] [CrossRef]
- Beckwith, D.M.; Cudic, M. Tumor-Associated O-Glycans of MUC1: Carriers of the Glyco-Code and Targets for Cancer Vaccine Design. Semin. Immunol. 2020, 47, 101389. [Google Scholar] [CrossRef]
- Lohmueller, J.J.; Sato, S.; Popova, L.; Chu, I.M.; Tucker, M.A.; Barberena, R.; Innocenti, G.M.; Cudic, M.; Ham, J.D.; Cheung, W.C.; et al. Antibodies Elicited by the First Non-Viral Prophylactic Cancer Vaccine Show Tumor-Specificity and Immunotherapeutic Potential. Sci. Rep. 2016, 6, 31740. [Google Scholar] [CrossRef]
- Perdivara, I.; Petrovich, R.; Allinquant, B.; Deterding, L.J.; Tomer, K.B.; Przybylski, M. Elucidation of O-Glycosylation Structures of the β-Amyloid Precursor Protein by Liquid Chromatography−Mass Spectrometry Using Electron Transfer Dissociation and Collision Induced Dissociation. J. Proteome Res. 2009, 8, 631–642. [Google Scholar] [CrossRef]
- Halim, A.; Brinkmalm, G.; Rüetschi, U.; Westman-Brinkmalm, A.; Portelius, E.; Zetterberg, H.; Blennow, K.; Larson, G.; Nilsson, J. Site-Specific Characterization of Threonine, Serine, and Tyrosine Glycosylations of Amyloid Precursor Protein/Amyloid β-Peptides in Human Cerebrospinal Fluid. Proc. Natl. Acad. Sci. USA 2011, 108, 11848–11853. [Google Scholar] [CrossRef] [PubMed]
- Akasaka-Manya, K.; Manya, H. The Role of APP O-Glycosylation in Alzheimer’s Disease. Biomolecules 2020, 10, 1569. [Google Scholar] [CrossRef]
- Shi, J.; Ku, X.; Zou, X.; Hou, J.; Yan, W.; Zhang, Y. Comprehensive Analysis of O-Glycosylation of Amyloid Precursor Protein (APP) Using Targeted and Multi-Fragmentation MS Strategy. Biochim. Biophys. Acta—Gen. Subj. 2021, 1865, 129954. [Google Scholar] [CrossRef]
- Tachida, Y.; Iijima, J.; Takahashi, K.; Suzuki, H.; Kizuka, Y.; Yamaguchi, Y.; Tanaka, K.; Nakano, M.; Takakura, D.; Kawasaki, N.; et al. O-GalNAc Glycosylation Determines Intracellular Trafficking of APP and Aβ Production. J. Biol. Chem. 2023, 299, 104905. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.; Regmi, D.; Ormaza, D.; Ayyalasomayajula, R.; Vela, N.; Mundim, G.; Du, D.; Minond, D.; Cudic, M. Mucin-Type O-Glycosylation Proximal to β-Secretase Cleavage Site Affects APP Processing and Aggregation Fate. Front. Chem. 2022, 10, 859822. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wei, Q.; Xia, W.; He, C.; Zhang, Q.; Huang, L.; Wang, X.; Sun, Y.; Ma, Y.; Zhang, X.; et al. O-Glycosylation Induces Amyloid-β To Form New Fibril Polymorphs Vulnerable for Degradation. J. Am. Chem. Soc. 2021, 143, 20216–20223. [Google Scholar] [CrossRef]
- Rebelo, A.L.; Chevalier, M.T.; Russo, L.; Pandit, A. Role and Therapeutic Implications of Protein Glycosylation in Neuroinflammation. Trends Mol. Med. 2022, 28, 270–289. [Google Scholar] [CrossRef]
- Gabriele, R.M.C.; Abel, E.; Fox, N.C.; Wray, S.; Arber, C. Knockdown of Amyloid Precursor Protein: Biological Consequences and Clinical Opportunities. Front. Neurosci. 2022, 16, 835645. [Google Scholar] [CrossRef]
- Zhang, Y.; Thompson, R.; Zhang, H.; Xu, H. APP Processing in Alzheimer’s Disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ma, Q.; Zhang, Y.; Xu, H. Proteolytic Processing of Alzheimer’s Β-amyloid Precursor Protein. J. Neurochem. 2012, 120, 9–21. [Google Scholar] [CrossRef]
- Pfundstein, G.; Nikonenko, A.G.; Sytnyk, V. Amyloid Precursor Protein (APP) and Amyloid β (Aβ) Interact with Cell Adhesion Molecules: Implications in Alzheimer’s Disease and Normal Physiology. Front. Cell Dev. Biol. 2022, 10, 969547. [Google Scholar] [CrossRef]
- Coburger, I.; Dahms, S.O.; Roeser, D.; Gührs, K.-H.; Hortschansky, P.; Than, M.E. Analysis of the Overall Structure of the Multi-Domain Amyloid Precursor Protein (APP). PLoS ONE 2013, 8, e81926. [Google Scholar] [CrossRef]
- Buoso, E.; Lanni, C.; Schettini, G.; Govoni, S.; Racchi, M. β-Amyloid Precursor Protein Metabolism: Focus on the Functions and Degradation of Its Intracellular Domain. Pharmacol. Res. 2010, 62, 308–317. [Google Scholar] [CrossRef]
- Ring, S.; Weyer, S.W.; Kilian, S.B.; Waldron, E.; Pietrzik, C.U.; Filippov, M.A.; Herms, J.; Buchholz, C.; Eckman, C.B.; Korte, M.; et al. The Secreted β-Amyloid Precursor Protein Ectodomain APPsα Is Sufficient to Rescue the Anatomical, Behavioral, and Electrophysiological Abnormalities of APP-Deficient Mice. J. Neurosci. 2007, 27, 7817–7826. [Google Scholar] [CrossRef]
- Orobets, K.S.; Karamyshev, A.L. Amyloid Precursor Protein and Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 14794. [Google Scholar] [CrossRef] [PubMed]
- Li, N.-M.; Liu, K.-F.; Qiu, Y.-J.; Zhang, H.-H.; Nakanishi, H.; Qing, H. Mutations of Beta-Amyloid Precursor Protein Alter the Consequence of Alzheimer’s Disease Pathogenesis. Neural Regen. Res. 2019, 14, 658. [Google Scholar] [CrossRef] [PubMed]
- Holsinger, R.M.D.; McLean, C.A.; Beyreuther, K.; Masters, C.L.; Evin, G. Increased Expression of the Amyloid Precursor Β-secretase in Alzheimer’s Disease. Ann. Neurol. 2002, 51, 783–786. [Google Scholar] [CrossRef]
- TCW, J.; Goate, A.M. Genetics of β-Amyloid Precursor Protein in Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2017, 7, a024539. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Koo, E.H. Biology and Pathophysiology of the Amyloid Precursor Protein. Mol. Neurodegener. 2011, 6, 27. [Google Scholar] [CrossRef]
- Kuhn, P.-H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Roßner, S.; Lichtenthaler, S.F. ADAM10 Is the Physiologically Relevant, Constitutive α-Secretase of the Amyloid Precursor Protein in Primary Neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. β-Secretase Cleavage of Alzheimer’s Amyloid Precursor Protein by the Transmembrane Aspartic Protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef]
- Ling, Y.; Morgan, K.; Kalsheker, N. Amyloid Precursor Protein (APP) and the Biology of Proteolytic Processing: Relevance to Alzheimer’s Disease. Int. J. Biochem. Cell Biol. 2003, 35, 1505–1535. [Google Scholar] [CrossRef]
- Olsen, O.; Kallop, D.Y.; McLaughlin, T.; Huntwork-Rodriguez, S.; Wu, Z.; Duggan, C.D.; Simon, D.J.; Lu, Y.; Easley-Neal, C.; Takeda, K.; et al. Genetic Analysis Reveals That Amyloid Precursor Protein and Death Receptor 6 Function in the Same Pathway to Control Axonal Pruning Independent of β-Secretase. J. Neurosci. 2014, 34, 6438–6447. [Google Scholar] [CrossRef]
- Tiwari, M.K.; Kepp, K.P. Modeling the Aggregation Propensity and Toxicity of Amyloid-β Variants. J. Alzheimer’s Dis. 2015, 47, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Jan, A.; Gokce, O.; Luthi-Carter, R.; Lashuel, H.A. The Ratio of Monomeric to Aggregated Forms of Aβ40 and Aβ42 Is an Important Determinant of Amyloid-β Aggregation, Fibrillogenesis, and Toxicity. J. Biol. Chem. 2008, 283, 28176–28189. [Google Scholar] [CrossRef] [PubMed]
- García-González, L.; Pilat, D.; Baranger, K.; Rivera, S. Emerging Alternative Proteinases in APP Metabolism and Alzheimer’s Disease Pathogenesis: A Focus on MT1-MMP and MT5-MMP. Front. Aging Neurosci. 2019, 11, 244. [Google Scholar] [CrossRef] [PubMed]
- Rivera, S.; García-González, L.; Khrestchatisky, M.; Baranger, K. Metalloproteinases and Their Tissue Inhibitors in Alzheimer’s Disease and Other Neurodegenerative Disorders. Cell. Mol. Life Sci. 2019, 76, 3167–3191. [Google Scholar] [CrossRef]
- Baranger, K.; Marchalant, Y.; Bonnet, A.E.; Crouzin, N.; Carrete, A.; Paumier, J.-M.; Py, N.A.; Bernard, A.; Bauer, C.; Charrat, E.; et al. MT5-MMP Is a New pro-Amyloidogenic Proteinase That Promotes Amyloid Pathology and Cognitive Decline in a Transgenic Mouse Model of Alzheimer’s Disease. Cell. Mol. Life Sci. 2016, 73, 217–236. [Google Scholar] [CrossRef]
- Paumier, J.-M.; Py, N.A.; García-González, L.; Bernard, A.; Stephan, D.; Louis, L.; Checler, F.; Khrestchatisky, M.; Baranger, K.; Rivera, S. Proamyloidogenic Effects of Membrane Type 1 Matrix Metalloproteinase Involve MMP-2 and BACE-1 Activities, and the Modulation of APP Trafficking. FASEB J. 2019, 33, 2910–2927. [Google Scholar] [CrossRef]
- Pilat, D.; Paumier, J.-M.; García-González, L.; Louis, L.; Stephan, D.; Manrique, C.; Khrestchatisky, M.; Di Pasquale, E.; Baranger, K.; Rivera, S. MT5-MMP Promotes Neuroinflammation, Neuronal Excitability and Aβ Production in Primary Neuron/Astrocyte Cultures from the 5xFAD Mouse Model of Alzheimer’s Disease. J. Neuroinflamm. 2022, 19, 65. [Google Scholar] [CrossRef]
- Becker-Pauly, C.; Pietrzik, C.U. The Metalloprotease Meprin β Is an Alternative β-Secretase of APP. Front. Mol. Neurosci. 2017, 9, 159. [Google Scholar] [CrossRef]
- Bien, J.; Jefferson, T.; Čaušević, M.; Jumpertz, T.; Munter, L.; Multhaup, G.; Weggen, S.; Becker-Pauly, C.; Pietrzik, C.U. The Metalloprotease Meprin β Generates Amino Terminal-Truncated Amyloid β Peptide Species. J. Biol. Chem. 2012, 287, 33304–33313. [Google Scholar] [CrossRef]
- Menon, P.K.; Koistinen, N.A.; Iverfeldt, K.; Ström, A.-L. Phosphorylation of the Amyloid Precursor Protein (APP) at Ser-675 Promotes APP Processing Involving Meprin β. J. Biol. Chem. 2019, 294, 17768–17776. [Google Scholar] [CrossRef]
- Werny, L.; Grogro, A.; Bickenbach, K.; Bülck, C.; Armbrust, F.; Koudelka, T.; Pathak, K.; Scharfenberg, F.; Sammel, M.; Sheikhouny, F.; et al. MT1-MMPand ADAM10 /17 Exhibit a Remarkable Overlap of Shedding Properties. FEBS J. 2023, 290, 93–111. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, T.; Čaušević, M.; auf dem Keller, U.; Schilling, O.; Isbert, S.; Geyer, R.; Maier, W.; Tschickardt, S.; Jumpertz, T.; Weggen, S.; et al. Metalloprotease Meprin β Generates Nontoxic N-Terminal Amyloid Precursor Protein Fragments in Vivo. J. Biol. Chem. 2011, 286, 27741–27750. [Google Scholar] [CrossRef] [PubMed]
- Marengo, L.; Armbrust, F.; Schoenherr, C.; Storck, S.E.; Schmitt, U.; Zampar, S.; Wirths, O.; Altmeppen, H.; Glatzel, M.; Kaether, C.; et al. Meprin β Knockout Reduces Brain Aβ Levels and Rescues Learning and Memory Impairments in the APP/Lon Mouse Model for Alzheimer’s Disease. Cell. Mol. Life Sci. 2022, 79, 168. [Google Scholar] [CrossRef]
- Liu, X.-H.; Liu, X.-T.; Wu, Y.; Li, S.-A.; Ren, K.-D.; Cheng, M.; Huang, B.; Yang, Y.; Liu, P.-P. Broadening Horizons: Exploring the Cathepsin Family as Therapeutic Targets for Alzheimer’s Disease. Aging Dis. 2024, 16, 2. [Google Scholar] [CrossRef] [PubMed]
- Embury, C.M.; Dyavarshetty, B.; Lu, Y.; Wiederin, J.L.; Ciborowski, P.; Gendelman, H.E.; Kiyota, T. Cathepsin B Improves SS-Amyloidosis and Learning and Memory in Models of Alzheimer’s Disease. J. Neuroimmune Pharmacol. 2017, 12, 340–352. [Google Scholar] [CrossRef]
- Oberstein, T.J.; Utz, J.; Spitzer, P.; Klafki, H.W.; Wiltfang, J.; Lewczuk, P.; Kornhuber, J.; Maler, J.M. The Role of Cathepsin B in the Degradation of Aβ and in the Production of Aβ Peptides Starting With Ala2 in Cultured Astrocytes. Front. Mol. Neurosci. 2021, 13, 615740. [Google Scholar] [CrossRef]
- Zhao, J.; Lang, M. New Insight into Protein Glycosylation in the Development of Alzheimer’s Disease. Cell Death Discov. 2023, 9, 314. [Google Scholar] [CrossRef]
- Liu, F.; Xu, K.; Xu, Z.; de las Rivas, M.; Wang, C.; Li, X.; Lu, J.; Zhou, Y.; Delso, I.; Merino, P.; et al. The Small Molecule Luteolin Inhibits N-Acetyl-α-Galactosaminyltransferases and Reduces Mucin-Type O-Glycosylation of Amyloid Precursor Protein. J. Biol. Chem. 2017, 292, 21304–21319. [Google Scholar] [CrossRef]
- Haukedal, H.; Freude, K.K. Implications of Glycosylation in Alzheimer’s Disease. Front. Neurosci. 2021, 14, 625348. [Google Scholar] [CrossRef]
- Goettig, P. Effects of Glycosylation on the Enzymatic Activity and Mechanisms of Proteases. Int. J. Mol. Sci. 2016, 17, 1969. [Google Scholar] [CrossRef]
- Goth, C.K.; Vakhrushev, S.Y.; Joshi, H.J.; Clausen, H.; Schjoldager, K.T. Fine-Tuning Limited Proteolysis: A Major Role for Regulated Site-Specific O-Glycosylation. Trends Biochem. Sci. 2018, 43, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhang, Q.; Xu, S.; Yu, Y. The Alteration and Role of Glycoconjugates in Alzheimer’s Disease. Front. Aging Neurosci. 2024, 16, 1398641. [Google Scholar] [CrossRef] [PubMed]
- Tomita, S.; Kirino, Y.; Suzuki, T. Cleavage of Alzheimer’s Amyloid Precursor Protein (APP) by Secretases Occurs after O-Glycosylation of APP in the Protein Secretory Pathway. J. Biol. Chem. 1998, 273, 6277–6284. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Kurosaka, A. Mucin-Type Glycosylation as a Regulatory Factor of Amyloid Precursor Protein Processing. J. Biochem. 2019, 165, 205–208. [Google Scholar] [CrossRef]
- Akasaka-Manya, K.; Kawamura, M.; Tsumoto, H.; Saito, Y.; Tachida, Y.; Kitazume, S.; Hatsuta, H.; Miura, Y.; Hisanaga, S.; Murayama, S.; et al. Excess APP O. -Glycosylation by GalNAc-T6 Decreases Aβ Production. J. Biochem. 2017, 161, 99–111. [Google Scholar] [CrossRef]
- Yang, K.; Yang, Z.; Chen, X.; Li, W. The Significance of Sialylation on the Pathogenesis of Alzheimer’s Disease. Brain Res. Bull. 2021, 173, 116–123. [Google Scholar] [CrossRef]
- Fastenau, C.; Bunce, M.; Keating, M.; Wickline, J.; Hopp, S.C.; Bieniek, K.F. Distinct Patterns of Plaque and Microglia Glycosylation in Alzheimer’s Disease. Brain Pathol. 2024, 34, e13267. [Google Scholar] [CrossRef]
- Cai, Y.; Liu, J.; Wang, B.; Sun, M.; Yang, H. Microglia in the Neuroinflammatory Pathogenesis of Alzheimer’s Disease and Related Therapeutic Targets. Front. Immunol. 2022, 13, 856376. [Google Scholar] [CrossRef]
- Wang, R.; Lan, C.; Benlagha, K.; Camara, N.O.S.; Miller, H.; Kubo, M.; Heegaard, S.; Lee, P.; Yang, L.; Forsman, H.; et al. The Interaction of Innate Immune and Adaptive Immune System. MedComm 2024, 5, e714. [Google Scholar] [CrossRef]
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An Introduction to Immunology and Immunopathology. Allergy Asthma Clin. Immunol. 2018, 14, 49. [Google Scholar] [CrossRef]
- Ousman, S.S.; Kubes, P. Immune Surveillance in the Central Nervous System. Nat. Neurosci. 2012, 15, 1096–1101. [Google Scholar] [CrossRef]
- Morimoto, K.; Nakajima, K. Role of the Immune System in the Development of the Central Nervous System. Front. Neurosci. 2019, 13, e714. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Ma, L.; Kaarela, T.; Li, Z. Neuroimmune Crosstalk in the Central Nervous System and Its Significance for Neurological Diseases. J. Neuroinflamm. 2012, 9, 594. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A Polarizing Question: Do M1 and M2 Microglia Exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Gabriela Sánchez, M.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gómez-Nicola, D.; Luheshi, G.; Vallières, L.; et al. Dark Microglia: A New Phenotype Predominantly Associated with Pathological States. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zong, S.; Cui, X.; Wang, X.; Wu, S.; Wang, L.; Liu, Y.; Lu, Z. The Effects of Microglia-Associated Neuroinflammation on Alzheimer’s Disease. Front. Immunol. 2023, 14, 1117172. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, G.; Luo, Y.; Jiang, L.; Chi, H.; Tian, G. Neuroinflammation in Alzheimer’s Disease: Insights from Peripheral Immune Cells. Immun. Ageing 2024, 21, 38. [Google Scholar] [CrossRef]
- Pan, X.; Zhu, Y.; Lin, N.; Zhang, J.; Ye, Q.; Huang, H.; Chen, X. Microglial Phagocytosis Induced by Fibrillar β-Amyloid Is Attenuated by Oligomeric β-Amyloid: Implications for Alzheimer’s Disease. Mol. Neurodegener. 2011, 6, 45. [Google Scholar] [CrossRef]
- Yang, T.; Li, S.; Xu, H.; Walsh, D.M.; Selkoe, D.J. Large Soluble Oligomers of Amyloid β-Protein from Alzheimer Brain Are Far Less Neuroactive Than the Smaller Oligomers to Which They Dissociate. J. Neurosci. 2017, 37, 152–163. [Google Scholar] [CrossRef]
- d’Errico, P.; Ziegler-Waldkirch, S.; Aires, V.; Hoffmann, P.; Mezö, C.; Erny, D.; Monasor, L.S.; Liebscher, S.; Ravi, V.M.; Joseph, K.; et al. Microglia Contribute to the Propagation of Aβ into Unaffected Brain Tissue. Nat. Neurosci. 2022, 25, 20–25. [Google Scholar] [CrossRef]
- Walker, L.C.; Schelle, J.; Jucker, M. The Prion-Like Properties of Amyloid-β Assemblies: Implications for Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2016, 6, a024398. [Google Scholar] [CrossRef] [PubMed]
- Morales, I.; Jiménez, J.M.; Mancilla, M.; Maccioni, R.B. Tau Oligomers and Fibrils Induce Activation of Microglial Cells. J. Alzheimer’s Dis. 2013, 37, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Long, H.-Z.; Zhou, Z.-W.; Cheng, Y.; Luo, H.-Y.; Li, F.-J.; Xu, S.-G.; Gao, L.-C. The Role of Microglia in Alzheimer’s Disease From the Perspective of Immune Inflammation and Iron Metabolism. Front. Aging Neurosci. 2022, 14, 888989. [Google Scholar] [CrossRef]
- Lee, S.-H.; Meilandt, W.J.; Xie, L.; Gandham, V.D.; Ngu, H.; Barck, K.H.; Rezzonico, M.G.; Imperio, J.; Lalehzadeh, G.; Huntley, M.A.; et al. Trem2 Restrains the Enhancement of Tau Accumulation and Neurodegeneration by β-Amyloid Pathology. Neuron 2021, 109, 1283–1301.e6. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Salih, D. TREM2-Mediated Activation of Microglia Breaks Link between Amyloid and Tau. Lancet Neurol. 2021, 20, 416–417. [Google Scholar] [CrossRef]
- Garner, O.B.; Baum, L.G. Galectin–Glycan Lattices Regulate Cell-Surface Glycoprotein Organization and Signalling. Biochem. Soc. Trans. 2008, 36, 1472–1477. [Google Scholar] [CrossRef]
- Pinho, S.S.; Alves, I.; Gaifem, J.; Rabinovich, G.A. Immune Regulatory Networks Coordinated by Glycans and Glycan-Binding Proteins in Autoimmunity and Infection. Cell. Mol. Immunol. 2023, 20, 1101–1113. [Google Scholar] [CrossRef]
- Teichberg, V.I.; Silman, I.; Beitsch, D.D.; Resheff, G. A Beta-D-Galactoside Binding Protein from Electric Organ Tissue of Electrophorus Electricus. Proc. Natl. Acad. Sci. USA 1975, 72, 1383–1387. [Google Scholar] [CrossRef]
- Liu, F.-T.; Stowell, S.R. The Role of Galectins in Immunity and Infection. Nat. Rev. Immunol. 2023, 23, 479–494. [Google Scholar] [CrossRef]
- Liu, F.-T.; Rabinovich, G.A. Galectins as Modulators of Tumour Progression. Nat. Rev. Cancer 2005, 5, 29–41. [Google Scholar] [CrossRef]
- Di Lella, S.; Sundblad, V.; Cerliani, J.P.; Guardia, C.M.; Estrin, D.A.; Vasta, G.R.; Rabinovich, G.A. When Galectins Recognize Glycans: From Biochemistry to Physiology and Back Again. Biochemistry 2011, 50, 7842–7857. [Google Scholar] [CrossRef] [PubMed]
- Tazhitdinova, R.; Timoshenko, A.V. The Emerging Role of Galectins and O-GlcNAc Homeostasis in Processes of Cellular Differentiation. Cells 2020, 9, 1792. [Google Scholar] [CrossRef]
- Troncoso, M.F.; Elola, M.T.; Blidner, A.G.; Sarrias, L.; Espelt, M.V.; Rabinovich, G.A. The Universe of Galectin-Binding Partners and Their Functions in Health and Disease. J. Biol. Chem. 2023, 299, 105400. [Google Scholar] [CrossRef]
- Nielsen, M.I.; Stegmayr, J.; Grant, O.C.; Yang, Z.; Nilsson, U.J.; Boos, I.; Carlsson, M.C.; Woods, R.J.; Unverzagt, C.; Leffler, H.; et al. Galectin Binding to Cells and Glycoproteins with Genetically Modified Glycosylation Reveals Galectin–Glycan Specificities in a Natural Context. J. Biol. Chem. 2018, 293, 20249–20262. [Google Scholar] [CrossRef] [PubMed]
- Dimitroff, C.J. Galectin-Binding O-Glycosylations as Regulators of Malignancy. Cancer Res. 2015, 75, 3195–3202. [Google Scholar] [CrossRef]
- Nguyen, J.T.; Evans, D.P.; Galvan, M.; Pace, K.E.; Leitenberg, D.; Bui, T.N.; Baum, L.G. CD45 Modulates Galectin-1-Induced T Cell Death: Regulation by Expression of Core 2 O-Glycans. J. Immunol. 2001, 167, 5697–5707. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.C.; Pang, M.; Hsu, D.K.; Liu, F.-T.; de Vos, S.; Gascoyne, R.D.; Said, J.; Baum, L.G. Galectin-3 Binds to CD45 on Diffuse Large B-Cell Lymphoma Cells to Regulate Susceptibility to Cell Death. Blood 2012, 120, 4635–4644. [Google Scholar] [CrossRef]
- Ilarregui, J.M.; Croci, D.O.; Bianco, G.A.; Toscano, M.A.; Salatino, M.; Vermeulen, M.E.; Geffner, J.R.; Rabinovich, G.A. Tolerogenic Signals Delivered by Dendritic Cells to T Cells through a Galectin-1-Driven Immunoregulatory Circuit Involving Interleukin 27 and Interleukin 10. Nat. Immunol. 2009, 10, 981–991. [Google Scholar] [CrossRef]
- Siew, J.J.; Chern, Y. Microglial Lectins in Health and Neurological Diseases. Front. Mol. Neurosci. 2018, 11, 158. [Google Scholar] [CrossRef]
- Boza-Serrano, A.; Vrillon, A.; Minta, K.; Paulus, A.; Camprubí-Ferrer, L.; Garcia, M.; Andreasson, U.; Antonell, A.; Wennström, M.; Gouras, G.; et al. Galectin-3 Is Elevated in CSF and Is Associated with Aβ Deposits and Tau Aggregates in Brain Tissue in Alzheimer’s Disease. Acta Neuropathol. 2022, 144, 843–859. [Google Scholar] [CrossRef]
- Tao, C.-C.; Cheng, K.-M.; Ma, Y.-L.; Hsu, W.-L.; Chen, Y.-C.; Fuh, J.-L.; Lee, W.-J.; Chao, C.-C.; Lee, E.H.Y. Galectin-3 Promotes Aβ Oligomerization and Aβ Toxicity in a Mouse Model of Alzheimer’s Disease. Cell Death Differ. 2020, 27, 192–209. [Google Scholar] [CrossRef] [PubMed]
- Siew, J.J.; Chen, H.-M.; Chiu, F.-L.; Lee, C.-W.; Chang, Y.-M.; Chen, H.-L.; Nguyen, T.N.A.; Liao, H.-T.; Liu, M.; Hagar, H.-T.; et al. Galectin-3 Aggravates Microglial Activation and Tau Transmission in Tauopathy. J. Clin. Investig. 2024, 134, e165523. [Google Scholar] [CrossRef]
- Angata, T.; Varki, A. Discovery, Classification, Evolution and Diversity of Siglecs. Mol. Aspects Med. 2023, 90, 101117. [Google Scholar] [CrossRef] [PubMed]
- Ayyalasomayajula, R.; Cudic, M. Targeting Siglec–Sialylated MUC1 Immune Axis in Cancer. Cancers 2024, 16, 1334. [Google Scholar] [CrossRef]
- Crocker, P.R.; Paulson, J.C.; Varki, A. Siglecs and Their Roles in the Immune System. Nat. Rev. Immunol. 2007, 7, 255–266. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, T.K.; Nath, D.; Ziltener, H.J.; Vestweber, D.; Fukuda, M.; van Die, I.; Crocker, P.R. Cutting Edge: CD43 Functions as a T Cell Counterreceptor for the Macrophage Adhesion Receptor Sialoadhesin (Siglec-1). J. Immunol. 2001, 166, 3637–3640. [Google Scholar] [CrossRef]
- Stewart, N.; Daly, J.; Drummond-Guy, O.; Krishnamoorthy, V.; Stark, J.C.; Riley, N.M.; Williams, K.C.; Bertozzi, C.R.; Wisnovsky, S. The Glycoimmune Checkpoint Receptor Siglec-7 Interacts with T-Cell Ligands and Regulates T-Cell Activation. J. Biol. Chem. 2024, 300, 105579. [Google Scholar] [CrossRef]
- Takamiya, R.; Ohtsubo, K.; Takamatsu, S.; Taniguchi, N.; Angata, T. The Interaction between Siglec-15 and Tumor-Associated Sialyl-Tn Antigen Enhances TGF- Secretion from Monocytes/Macrophages through the DAP12-Syk Pathway. Glycobiology 2013, 23, 178–187. [Google Scholar] [CrossRef]
- Büll, C.; Nason, R.; Sun, L.; Van Coillie, J.; Madriz Sørensen, D.; Moons, S.J.; Yang, Z.; Arbitman, S.; Fernandes, S.M.; Furukawa, S.; et al. Probing the Binding Specificities of Human Siglecs by Cell-Based Glycan Arrays. Proc. Natl. Acad. Sci. USA 2021, 118, e2026102118. [Google Scholar] [CrossRef]
- Siew, J.J.; Chern, Y.; Khoo, K.-H.; Angata, T. Roles of Siglecs in Neurodegenerative Diseases. Mol. Aspects Med. 2023, 90, 101141. [Google Scholar] [CrossRef]
- Eskandari-Sedighi, G.; Jung, J.; Macauley, M.S. CD33 Isoforms in Microglia and Alzheimer’s Disease: Friend and Foe. Mol. Aspects Med. 2023, 90, 101111. [Google Scholar] [CrossRef] [PubMed]
- Bu, X.-L.; Sun, P.-Y.; Fan, D.-Y.; Wang, J.; Sun, H.-L.; Cheng, Y.; Zeng, G.-H.; Chen, D.-W.; Li, H.-Y.; Yi, X.; et al. Associations of Plasma Soluble CD22 Levels with Brain Amyloid Burden and Cognitive Decline in Alzheimer’s Disease. Sci. Adv. 2022, 8, eabm5667. [Google Scholar] [CrossRef] [PubMed]
- Nagamine, S.; Yamazaki, T.; Makioka, K.; Fujita, Y.; Ikeda, M.; Takatama, M.; Okamoto, K.; Yokoo, H.; Ikeda, Y. Hypersialylation Is a Common Feature of Neurofibrillary Tangles and Granulovacuolar Degenerations in Alzheimer’s Disease and Tauopathy Brains. Neuropathology 2016, 36, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Zelensky, A.N.; Gready, J.E. The C-type Lectin-like Domain Superfamily. FEBS J. 2005, 272, 6179–6217. [Google Scholar] [CrossRef]
- Fischer, S.; Stegmann, F.; Gnanapragassam, V.S.; Lepenies, B. From Structure to Function—Ligand Recognition by Myeloid C-Type Lectin Receptors. Comput. Struct. Biotechnol. J. 2022, 20, 5790–5812. [Google Scholar] [CrossRef]
- Brown, G.D.; Gordon, S. A New Receptor for β-Glucans. Nature 2001, 413, 36–37. [Google Scholar] [CrossRef]
- Reis e Sousa, C.; Yamasaki, S.; Brown, G.D. Myeloid C-Type Lectin Receptors in Innate Immune Recognition. Immunity 2024, 57, 700–717. [Google Scholar] [CrossRef]
- Rabinovich, G.A.; van Kooyk, Y.; Cobb, B.A. Glycobiology of Immune Responses. Ann. N. Y Acad. Sci. 2012, 1253, 1–15. [Google Scholar] [CrossRef]
- van Vliet, S.J.; Gringhuis, S.I.; Geijtenbeek, T.B.H.; van Kooyk, Y. Regulation of Effector T Cells by Antigen-Presenting Cells via Interaction of the C-Type Lectin MGL with CD45. Nat. Immunol. 2006, 7, 1200–1208. [Google Scholar] [CrossRef]
- Wang, X.; Liu, G.; Gao, Q.; Li, N.; Wang, R. C-type Lectin-like Receptor 2 and Zonulin Are Associated with Mild Cognitive Impairment and Alzheimer’s Disease. Acta Neurol. Scand. 2020, 141, 250–255. [Google Scholar] [CrossRef]
- Meng, D.; Ma, X.; Li, H.; Wu, X.; Cao, Y.; Miao, Z.; Zhang, X. A Role of the Podoplanin-CLEC-2 Axis in Promoting Inflammatory Response After Ischemic Stroke in Mice. Neurotox. Res. 2021, 39, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Szczykutowicz, J. Ligand Recognition by the Macrophage Galactose-Type C-Type Lectin: Self or Non-Self?—A Way to Trick the Host’s Immune System. Int. J. Mol. Sci. 2023, 24, 17078. [Google Scholar] [CrossRef] [PubMed]
- Tumoglu, B.; Keelaghan, A.; Avci, F.Y. Tn Antigen Interactions of Macrophage Galactose-Type Lectin (MGL) in Immune Function and Disease. Glycobiology 2023, 33, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Gibadullin, R.; Farnsworth, D.W.; Barchi, J.J.; Gildersleeve, J.C. GalNAc-Tyrosine Is a Ligand of Plant Lectins, Antibodies, and Human and Murine Macrophage Galactose-Type Lectins. ACS Chem. Biol. 2017, 12, 2172–2182. [Google Scholar] [CrossRef]
- Marcelo, F.; Supekar, N.; Corzana, F.; van der Horst, J.C.; Vuist, I.M.; Live, D.; Boons, G.-J.P.H.; Smith, D.F.; van Vliet, S.J. Identification of a Secondary Binding Site in Human Macrophage Galactose-Type Lectin by Microarray Studies: Implications for the Molecular Recognition of Its Ligands. J. Biol. Chem. 2019, 294, 1300–1311. [Google Scholar] [CrossRef]
- Gabba, A.; Bogucka, A.; Luz, J.G.; Diniz, A.; Coelho, H.; Corzana, F.; Cañada, F.J.; Marcelo, F.; Murphy, P.V.; Birrane, G. Crystal Structure of the Carbohydrate Recognition Domain of the Human Macrophage Galactose C-Type Lectin Bound to GalNAc and the Tumor-Associated Tn Antigen. Biochemistry 2021, 60, 1327–1336. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vela Navarro, N.; De Nadai Mundim, G.; Cudic, M. Implications of Mucin-Type O-Glycosylation in Alzheimer’s Disease. Molecules 2025, 30, 1895. https://doi.org/10.3390/molecules30091895
Vela Navarro N, De Nadai Mundim G, Cudic M. Implications of Mucin-Type O-Glycosylation in Alzheimer’s Disease. Molecules. 2025; 30(9):1895. https://doi.org/10.3390/molecules30091895
Chicago/Turabian StyleVela Navarro, Nancy, Gustavo De Nadai Mundim, and Maré Cudic. 2025. "Implications of Mucin-Type O-Glycosylation in Alzheimer’s Disease" Molecules 30, no. 9: 1895. https://doi.org/10.3390/molecules30091895
APA StyleVela Navarro, N., De Nadai Mundim, G., & Cudic, M. (2025). Implications of Mucin-Type O-Glycosylation in Alzheimer’s Disease. Molecules, 30(9), 1895. https://doi.org/10.3390/molecules30091895