

Sulfide Oxidation to Sulfone Using Sodium Chlorite and Hydrochloric Acid in Organic Solvents


Abstract
:1. Introduction
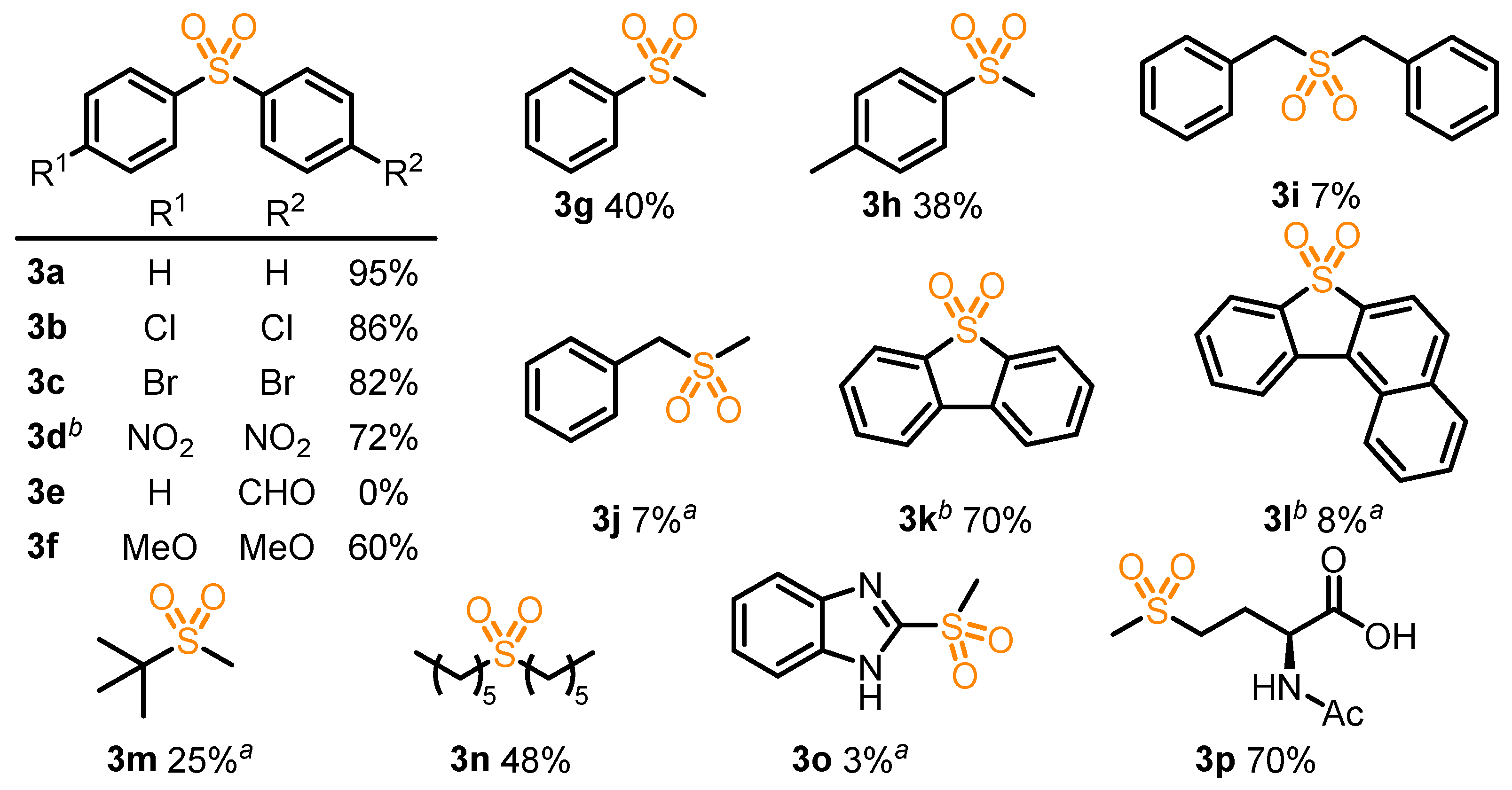
2. Results and Discussion
3. Materials and Methods
3.1. Materials
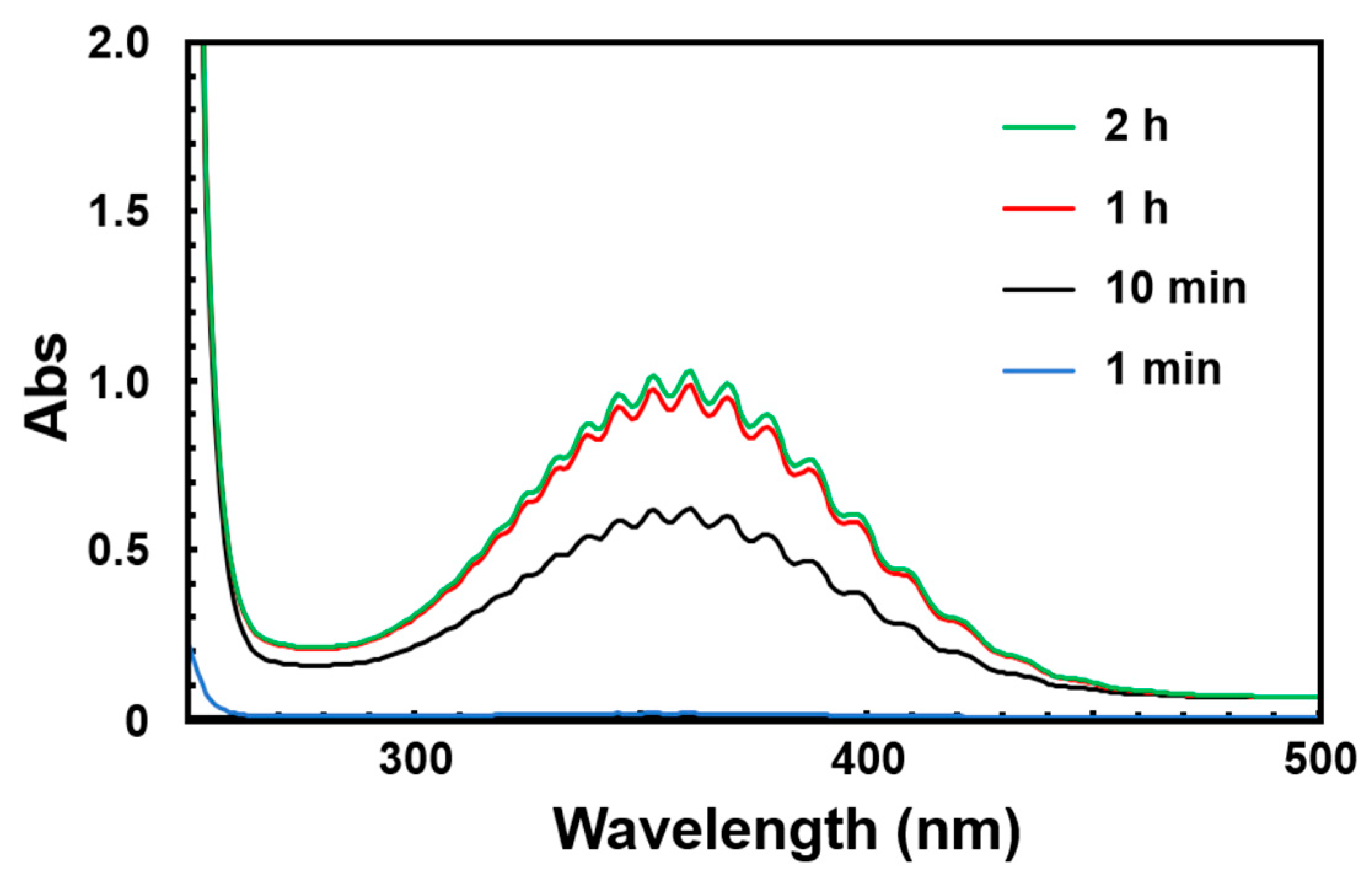
3.2. UV–Vis Absorption Spectral Measurements
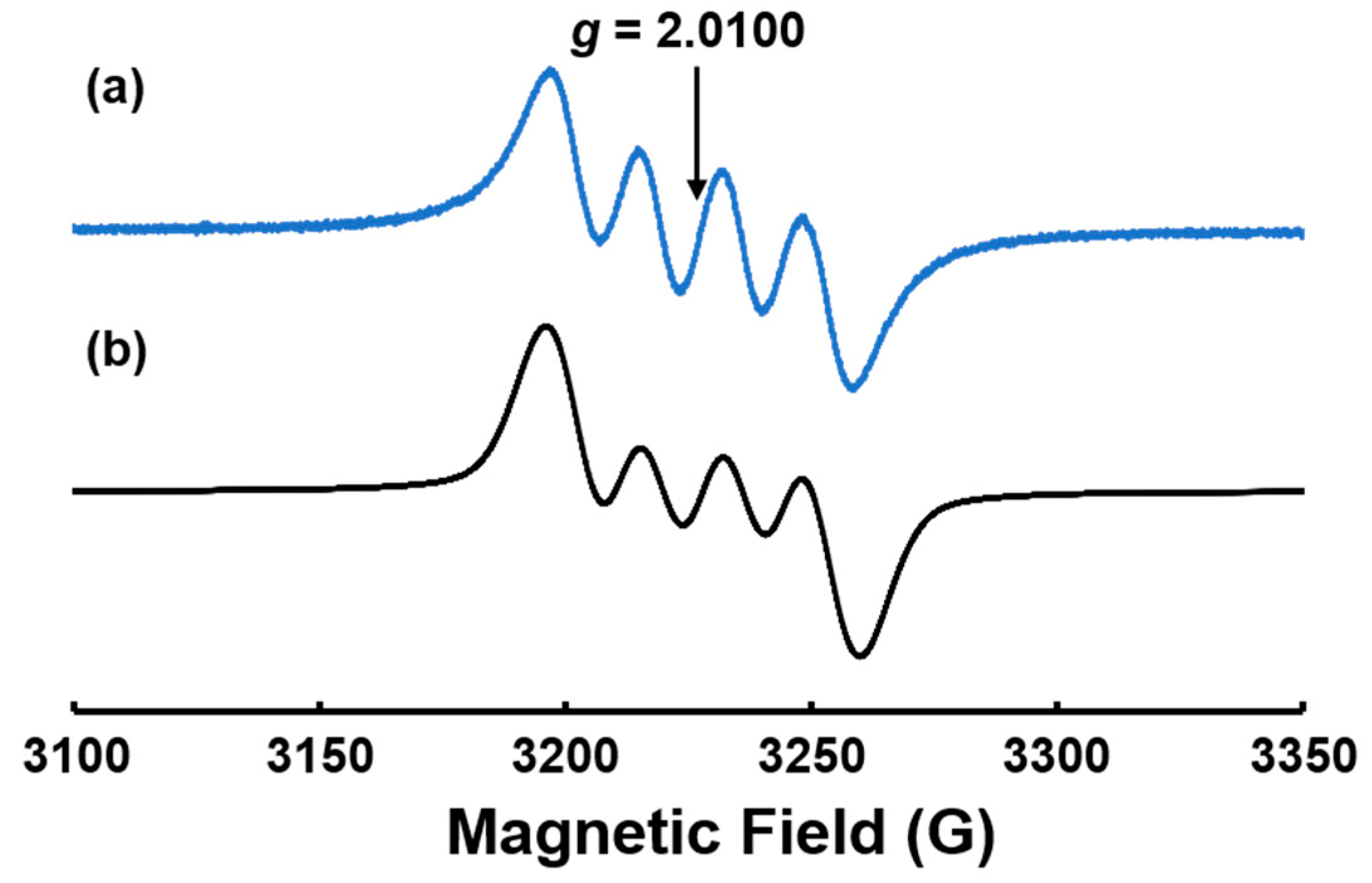
3.3. ESR Measurements
3.4. General Procedure for the Synthesis of 3a
3.5. Spectroscopic Data of Products
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feng, M.; Tang, B.; Liang, S.H.; Jiang, X. Sulfur Containing Scaffolds in Drugs: Synthesis and Application in Medicinal Chemistry. Curr. Top. Med. Chem. 2016, 16, 1200–1216. [Google Scholar] [CrossRef] [PubMed]
- Tayu, M.; Rahmanudin, A.; Perry, G.J.P.; Khan, R.U.; Tate, D.J.; Marcial-Hernandez, R.; Shen, Y.; Dierking, I.; Janpatompong, Y.; Aphichatpanichakul, S.; et al. Modular Synthesis of Unsymmetrical [1]Benzothieno [3,2-b][1]benzothiophene Molecular Semiconductors for Organic Transistors. Chem. Sci. 2022, 13, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.-Y.; Li, M.; Zhou, T.-P.; Huang, J.-R. Benzo [b]naphtho[1, 2-d]thiophene Sulfoxides: Biomimetic Synthesis, Photophysical Properties, and Applications. Angew. Chem. Int. Ed. 2022, 61, e202203908. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.-W.; Liang, S.; Manolikakes, G. Recent Advances in the Synthesis of Sulfones. Synthesis 2016, 48, 1939–1973. [Google Scholar]
- Voutyritsa, E.; Triandafillidi, I.; Kokotos, C.G. Green Organocatalytic Oxidation of Sulfides to Sulfoxides and Sulfones. Synthesis 2017, 49, 917–924. [Google Scholar]
- Mangaonkar, S.R.; Singh, F.V. Selective Oxidation of Organosulphides using m-CPBA as oxidant. Pharma Chem. 2016, 8, 419–423. [Google Scholar]
- Tong, Q.-L.; Fan, Z.-F.; Yang, J.-W.; Li, Q.; Chen, Y.-X.; Cheng, M.-S.; Liu, Y. The Selective Oxidation of Sulfides to Sulfoxides or Sulfones with Hydrogen Peroxide Catalyzed by a Dendritic Phosphomolybdate Hybrid. Catalysts 2019, 9, 791. [Google Scholar] [CrossRef]
- Khodaei, M.M.; Bahrami, K.; Khedri, M. The efficient and chemoselective MoO3-catalyzed oxidation of sulfides to sulfoxides and sulfones with H2O2. Can. J. Chem. 2007, 85, 7–11. [Google Scholar] [CrossRef]
- Maleki, B.; Hemmati, S.; Sedrpoushan, A.; Ashrafi, S.S.; Veisi, H. Selective synthesis of sulfoxides and sulfones from sulfides using silica bromide as the heterogeneous promoter and hydrogen peroxide as the terminal oxidant. RSC Adv. 2014, 4, 40505–40510. [Google Scholar] [CrossRef]
- Khurana, J.M.; Panda, A.K.; Ray, A.; Gogia, A. Rapid Oxidation of Sulfides and Sulfoxides with Sodium Hypochlorite. Org. Prep. Proced. Int. 1996, 28, 234. [Google Scholar] [CrossRef]
- Khurana, J.M.; Nand, B. Aqueous sodium hypochlorite mediated chemoselective oxidation of chalcogenides to monoxides and dioxides by microwave exposure. Can. J. Chem. 2010, 88, 906–909. [Google Scholar] [CrossRef]
- Kirihara, M.; Okada, T.; Sugiyama, Y.; Akiyoshi, M.; Matsunaga, T.; Kimura, Y. Sodium Hypochlorite Pentahydrate Crystals (NaOCl·5H2O): A Convenient and Environmentally Benign Oxidant for Organic Synthesis. Org. Process Res. Dev. 2017, 21, 1925–1937. [Google Scholar] [CrossRef]
- Fukuda, N.; Ikemoto, T. Imide-Catalyzed Oxidation System: Sulfides to Sulfoxides and Sulfones. J. Org. Chem. 2010, 75, 4629–4631. [Google Scholar] [CrossRef] [PubMed]
- Vogt, H.; Balej, J.; Bennett, J.E.; Wintzer, P.; Sheikh, S.A.; Gallone, P.; Vasudevan, S.; Pelin, K. Chlorine Oxides and Chlorine Oxygen Acids. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Itabashi, Y.; Ogata, S.; Shimada, Y.; Kondo, M.; Nishiwaki, N.; Inoue, T.; Asahara, H.; Ohkubo, K. Tetrabutylammonium Chlorite as an Efficient Oxidant for Controlled Oxidation of Sulfides to Sulfoxides. Chem. Eur. J. 2025, 31, e202404279. [Google Scholar] [CrossRef]
- Itabashi, Y.; Asahara, H.; Ohkubo, K. Chlorine-radical-mediated C–H Oxygenation Reaction Under Light Irradiation. Chem. Commun. 2023, 59, 7506–7517. [Google Scholar] [CrossRef]
- Ohkubo, K.; Hirose, K. Light-Driven C–H Oxygenation of Methane into Methanol and Formic Acid by Molecular Oxygen Using a Perfluorinated Solvent. Angew. Chem. Int. Ed. 2018, 57, 2126–2129. [Google Scholar] [CrossRef]
- Deshwal, B.R.; Jo, H.-D.; Lee, H.-K. Reaction Kinetics of Decomposition of Acidic Sodium Chlorite. Can. J. Chem. Eng. 2004, 82, 619–632. [Google Scholar] [CrossRef]
- Dunn, R.C.; Simon, J.D. Excited-State Photoreactions of Chlorine Dioxide in Water. J. Am. Chem. Soc. 1992, 114, 4856–4860. [Google Scholar] [CrossRef]
- Ohkubo, K.; Hirose, K.; Shibata, T.; Takamori, K.; Fukuzumi, S. Dihydroxylation of Styrene by Sodium Chlorite with Scandium Triflate. J. Phys. Org. Chem. 2017, 30, e3619. [Google Scholar] [CrossRef]
- Ozawa, T.; Miura, Y.; Ueda, J. Oxidation of Spin-Traps by Chlorine Dioxide (ClO2) Radical in Aqueous Solutions: First ESR Evidence of Formation of New Nitroxide Radicals. Free Radic. Biol. Med. 1996, 20, 837–841. [Google Scholar] [CrossRef]
- Giraldo Isaza, L.; Mortha, G.; Marlin, N.; Molton, F.; Duboc, C. ClO2-Mediated Oxidation of the TEMPO Radical: Fundamental Considerations of the Catalytic System for the Oxidation of Cellulose Fibers. Molecules 2023, 28, 6631. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Du, J.; Zhang, Y.; Chang, H.; Gao, W.; Wei, W. Synthesis and nano-Pd catalyzed chemoselective oxidation of symmetrical and unsymmetrical sulfides. Org. Biomol. Chem. 2019, 17, 3048–3055. [Google Scholar] [CrossRef] [PubMed]
- Kirihara, M.; Yamamoto, J.; Noguchi, T.; Itou, A.; Naito, S.; Hirai, Y. Tantalum(V) or Niobium(V) Catalyzed Oxidation of Sulfides with 30% Hydrogen Peroxide. Tetrahedron 2009, 65, 10477–10484. [Google Scholar] [CrossRef]
- Gogoi, S.R.; Boruah, J.J.; Sengupta, G.; Saikia, G.; Ahmed, K.; Bania, K.K.; Islam, N.S. Peroxoniobium(V)-catalyzed Selective Oxidation of Sulfides with Hydrogen Peroxide in Water: A Sustainable Approach. Catal. Sci. Technol. 2015, 5, 595–610. [Google Scholar] [CrossRef]
- Lutz, M.; Wenzler, M.; Likhotvorik, I. An Efficient Oxidation of Sulfides to Sulfones with Urea–Hydrogen Peroxide in the Presence of Phthalic Anhydride in Ethyl Acetate. Synthesis 2018, 50, 2231–2234. [Google Scholar]
- Bhanuchandra, M.; Murakami, K.; Vasu, D.; Yorimitsu, H.; Osuka, A. Transition-Metal-Free Synthesis of Carbazoles and Indoles by an SNAr-Based “Aromatic Metamorphosis” of Thiaarenes. Angew. Chem. Int. Ed. 2015, 54, 10234–10238. [Google Scholar] [CrossRef]
- Toda, N.; Asano, S.; Barbas, C.F. Rapid, Stable, Chemoselective Labeling of Thiols with Julia–Kocieński-like Reagents: A Serum-Stable Alternative to Maleimide-Based Protein Conjugation. Angew. Chem. Int. Ed. 2013, 52, 12592–12596. [Google Scholar] [CrossRef]
- Doherty, S.; Knight, J.G.; Carroll, M.A.; Ellison, J.R.; Hobson, S.J.; Stevens, S.; Hardacre, C.; Goodrich, P. Efficient and Selective Hydrogen Peroxide-mediated Oxidation of Sulfides in Batch and Segmented and Continuous Flow Using a Peroxometalate-based Polymer Immobilised Ionic Liquid Phase Catalyst. Green Chem. 2015, 17, 1559–1571. [Google Scholar] [CrossRef]
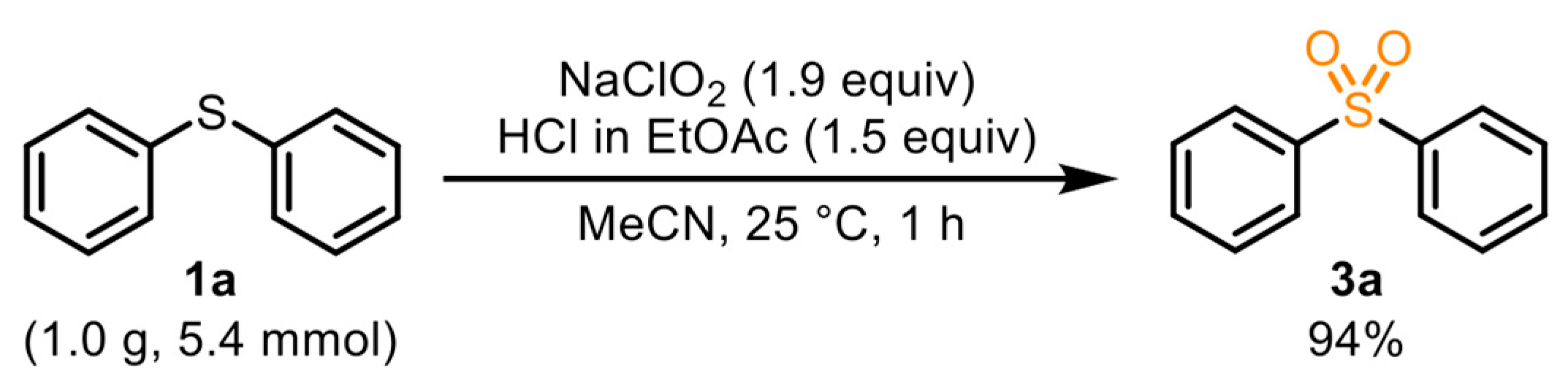

{kind=link}
{kind=link}
{kind=link}
{kind=link}
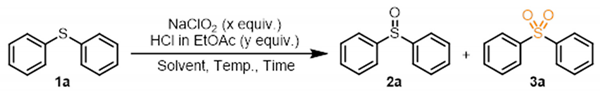 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | x | y | Solvent | Conc. of 1a (M) | Temp. (°C) | Time | NMR Yield a (%) | ||
| 1a | 2a | 3a | |||||||
| 1 | 5.0 | 4.0 | MeCN | 0.040 | 25 | 1 h | 0 | 0 | 96 |
| 2 | 5.0 | 4.0 | EtOAc | 0.040 | 25 | 1 h | 0 | 0 | 96 |
| 3 | 5.0 | 4.0 | Toluene | 0.040 | 25 | 1 h | 0 | 45 | 30 |
| 4 b | 5.0 | 4.0 | H2O | 0.040 | 25 | 1 h | 0 | 58 | 35 |
| 5 | 5.0 | 0 | EtOAc | 0.040 | 60 | 24 h | 100 | 0 | 0 |
| 6 | 5.0 | 4.0 | EtOAc | 0.20 | 25 | 1 h | 0 | 14 | 80 |
| 7 | 5.0 | 4.0 | MeCN | 0.20 | 25 | 1 h | 0 | 0 | 95 |
| 8 | 5.0 | 4.0 | MeCN | 0.20 | 25 | 10 min | 0 | 9 | 85 |
| 9 | 5.0 | 4.0 | MeCN | 0.20 | 25 | 1 h | 0 | 43 | 50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Itabashi, Y.; Ogata, S.; Inoue, T.; Asahara, H.; Ohkubo, K. Sulfide Oxidation to Sulfone Using Sodium Chlorite and Hydrochloric Acid in Organic Solvents. Molecules 2025, 30, 1912. https://doi.org/10.3390/molecules30091912
Itabashi Y, Ogata S, Inoue T, Asahara H, Ohkubo K. Sulfide Oxidation to Sulfone Using Sodium Chlorite and Hydrochloric Acid in Organic Solvents. Molecules. 2025; 30(9):1912. https://doi.org/10.3390/molecules30091912
Chicago/Turabian StyleItabashi, Yuki, Shuto Ogata, Tsuyoshi Inoue, Haruyasu Asahara, and Kei Ohkubo. 2025. "Sulfide Oxidation to Sulfone Using Sodium Chlorite and Hydrochloric Acid in Organic Solvents" Molecules 30, no. 9: 1912. https://doi.org/10.3390/molecules30091912
APA StyleItabashi, Y., Ogata, S., Inoue, T., Asahara, H., & Ohkubo, K. (2025). Sulfide Oxidation to Sulfone Using Sodium Chlorite and Hydrochloric Acid in Organic Solvents. Molecules, 30(9), 1912. https://doi.org/10.3390/molecules30091912