Introduction
An important goal of computational chemistry research is the design of molecules for specific applications. Factors that must be taken into account include shape, size, electronic properties and reactivity. For any physical and chemical properties, the molecular shape largely determines the final function, and this is the rationale for conformational studies. The search for materials with large values of polarizabilities and other optical properties has attracted much interest from the experimental and the theoretical points of view [
1,
2,
3]. An important consequence of various chemical processes involving charge redistribution is the changes in intermolecular interactions of the molecules involved [
4,
5,
6,
7].
Molecular polarization is essential for the description of both optical properties and intermolecular interactions [
8,
9,
10,
11,
12,
13]. In addition, the intramolecular force fields used in molecular mechanics are usually based on the assumption that the interaction between non-bonded atoms can be analyzed in the same way as intermolecular interactions [
14,
15,
16]. A possible improvement of conventional force fields could come from the introduction of an induction term [
17]. Modern theoretical chemistry, with the help of powerful computers, can attempt to become a substitute for difficult experimental determinations [
18]. Classical models such as the dipole interaction model and its improvements, which were investigated among others by Applequist [
19,
20] are able to predict molecular polarizabilities with surprising accuracy. Because of their simplicity they can be conveniently implemented in molecular force fields to account for the non-additivity effects in the intermolecular interactions [
14,
15,
16,
21,
22,
23].
In the theory of higher-order polarizabilities, when the external electric field is non-uniform, the hamiltonian)
(
Ea ,
Eab, ... ) and the energy
W(
Ea ,
Eab, ... ) may be expressed as a power series in the electric field,
Ea, its gradient,
Eab, its hessian,
Eabc,
etc.:[
24].
where
a,
b,
c,… denote Cartesian components, and the Einstein convention of summation over repeated indices is implied here and in all subsequent equations. The electric multipole expansion is formed by the dipole (
) and quadrupole (
) moments; the polarizability expansion is formed by the dipole-dipole (
), dipole-quadrupole (
) and quadrupole-quadrupole (
) polarizabilities. In the following equations, a partial derivative with respect to
Eabc… is the mathematical operation of taking the derivative with respect to
Eabc… while keeping all other components of the field and its derivatives constant. The total electric multipole moments are
In previous articles [
25,
26,
27,
28,
29,
30,
31] we proposed a method for the calculation of
,
,
and
that was applied for the study of benzothiazole (A)-benzobisthiazole (B) linear oligomers 26-28]. In Ref.26 we calculated fully-planar linear oligomers in the series A-B1-A to A-B
13-A and extrapolated the results to give predictions for polybenzobisthiazole A-B
•-A. This choice was prompted by the importance of this heteroaromatic structure in rodlike polymeric chains that possess interesting mechanical and optical properties.
It should be noted that polymers do not usually have well-defined molecular structures [
18]. A sample of a polymer will contain molecules having possibly a variety of structures. Rational polymer design technologies rely on a detailed structural knowledge of oligomers at the atomic level. Although the strong C-C, C-N and C-S bonds maintain the integrity of the polymer, A-B
•-A has a high degree of flexibility because rotation can occur about the intercycle bonds between monomeric units in the polymer chain. Thus the polymer chain can adopt a large number of configurations in three-dimensional space. The torsion angles of intercycle bonds play a crucial part in determining properties, for these determine the overall polarity of the macromolecule. In Ref.27 we examined the torsional effects on
,
and
for A-B
13-A. We characterized three model rotational isomers: (1) the fully-planar (000) conformation; (2) a rotational isomer where each unit is rotated 10°, rotations being performed in the alternate directions (+-+); and (3) a rotational isomer where each unit is rotated 10°, all rotations being performed in the same direction (+++). We dealt with small torsion angles because the molecular polarizabilities were due to
p-p overlap between successive monomeric units in the one-dimensional chain and this overlap fell sharply for big rotations. The main conclusion of this previous work was that small torsional changes in A-B
13-A affect
and
, and, specially,
. In particular,
was changed by rotation and
was revealed to be highly sensitive to
. This previous work allowed us to ask ourselves whether changes in
,
,
and
holds for smaller angle rotations. So, in this article we have studied the polarizabilities of the fully-planar 000 and the +++ oligomers where each unit is rotated from 1° to 10°.
In the next section we present the description of the electrostatic properties used in this study. Following that, the interacting induced dipoles polarization model for the calculation of molecular polarizabilities is presented. Next, the geometric descriptors and topological indices of molecules are described as calculated by the TOPO program. Following that, results are presented and discussed. The last section summarizes our conclusions.
Electrostatic properties
Atomic net charges and polarizabilities are calculated from their
s and
p contributions. The
s net charges and polarizabilities are estimated by the principle of electronegativity equalization [
32,
33,
34,
35] but applied bond-by-bond in the molecule [
25,
26,
27,
28,
29,
30,
31]. The
p net charges and polarizabilities are evaluated with the method of Hückel. Dipole (
) and tensor quadrupole (
) moments have been calculated from the point (atomic) distribution of net charges. The molecule is brought into its principal inertial coordinate system.
It is well known that intercycle
p conjugation vanishes for perpendicular cycles. So, the Hückel
b parameter should decrease for out-of-plane conformations, tending to zero for perpendicular polycyclic structures. Therefore, if the functional relation of
p conjugation parameters with respect to the dihedral angles is known, improvements can be made in the simple Hückel calculations [
36]. Mulliken [
37,
38] has proposed that the Hückel
b parameter is proportional to the overlap integral
S and has shown that in diatomic systems the proportionality holds rather well at bond distances of interest. For Slater overlap integrals [
39]
b as a function of distance has been calculated [
40].
Although there is no convincing evidence as to which
b-distance curve to use, Mulliken’s success with diatomic molecules [
37,
38,
41] is suggestive, as is a successful application to calculation of ionization potentials [
42]. Thus, in first approximation,
b is proportional to the overlap integral
S between two fragments. This is certainly a crude approximation but often used to deduce the tendency in the variation of
b. It should be noted that
S between two parallel orbitals with no spherical symmetry varies as the cosine of their relative torsion angle
q. Acceptance of the assumption further implies that
b can be evaluated, in first approximation, between
pz orbitals twisted from coplanarity by an angle
q as [
43,
44,
45,
46]
Thus, it is convenient to define a standard b0 (being equal to the b parameter for benzene) for the planar q dihedral angles and to modify other b parameters (in units of the standard b0) with the use of this dimensionless function.
Of course, this method is not exact. It should be remarked that the rotation of a torsion angle can generate conformational changes. For example, the bond distance can vary and the overlap between two atomic orbitals decreases exponentially when the distance between them increases. So, the overlap integral
S not only depends on the relative orientation of the
pz orbitals directly involved in the fragment-fragment bonding, but also on second-neighbour overlaps between
pz orbitals each belonging to a different fragment. The proper
b function in this case is considerably different from the cosine function being used. An improved estimate of the
b function can be expected from some kind of empirical information. Joachim
et al. [
47] have evaluated the electronic coupling,
Vab, of the binuclear mixed valence M
II-L-M
III complex [(NH
3)
5Ru-bipyridyl-Ru(NH
3)
5]
5+ in the valence bond electronic states M
II-L-M
III (a) and M
III-L-M
II (b). When a pyridine ring rotates around the ligand axis,
p-p Vab (θ) can be best fitted by a cos
1.15 θ function. From this observation we assume that the
b function is universal and has the same form as the electronic coupling function,
Vab, for complex [(NH
3)
5Ru-bipyridyl-Ru(NH
3)
5]
5+:
Interacting induced dipoles polarization model for molecular polarizabilities
The computation of molecular polarizabilities has been carried out by the interacting induced dipoles polarization model presented elsewhere [
14,
15,
16,
25,
26,
27,
28]. It calculates tensor effective anisotropic point polarizabilities by the method of Applequist
et al. [
19,
20,
48,
49,
50]. One considers the molecule as being made up of
N atoms
(i, j, k,…), each of which acts as a point particle located at the nucleus and responds to an electric field only by the induction of a dipole moment which is a linear function of the local field. If a Cartesian component of the field owing to the permanent multipole moments is
, then the induced moment
in atom
i is
where
ai is the polarizability of atom
i and
is the symmetrical field gradient tensor,
, where
e is the charge of the proton and the subscripts
a,
b,
c,… stand for the Cartesian components
x,
y,
z. The set of coupled linear Equations (1) for the induced dipole moments can conveniently be expressed in compact matrix equation form, if one introduces the 3
N × 3
N matrices
and
, with elements
and
(
being the Kronecker
d), respectively. To suppress the restriction in the sum, the diagonal elements
are defined as zero. Similarly,
and
are 3
N × 1 column vectors with elements
and
. Equation (1) is thus written,
where
is the 3
N × 3
N -dimensional unit matrix. This matrix equation can be solved for the induced dipoles as
Here the symmetrical
many-body polarizability matrix,
, has been introduced:
The compact matrix equation
=
is equivalent to the
N matrix equations:
Let the molecule be in a uniform applied field, so that
=
for all
j. Then
The coefficient of
in this equation is seen to be an effective (hence, additive) polarizability of unit
i,
. The total moment induced in the molecule
is
from which it is seen that the molecular polarizability tensor
is
The most noticeable feature of molecular polarizability surfaces is a curve of discontinuity along which the polarizability approaches ±• under certain conditions [
51,
52]. The significance of a polarizability of ±• is that the molecule is in a state of resonance and absorbs energy from the applied field. This behaviour of the model can be understood from its close relation to the classical system of
N coupled oscillators, which likewise shows resonance under conditions other than the resonance conditions of the isolated oscillators [
53,
54,
55,
56].
Voisin [
57] wrote a computer program called PAPID for the computation of molecular polarizabilities. A database of atomic polarizabilities has been built up from coupled Hartree-Fock calculations of a series of molecules containing the main functional groups which are presented in organic molecules [
17], and has been implemented in the Block Data of the program [
58,
59]. In this work, we have implemented the following improvements in PAPID.
1. We have used a damping function in the calculation of the symmetrical field gradient tensor in order to prevent the polarizability from going to infinity [
17]. Thus, if the distance
r between atoms
i and
j is less than the cut-off value,
s = 1.662(α
i , the field gradient tensor is now evaluated as
where
v = r/s.
2. We have neglected the interaction between bonded atoms and atoms with a distance lying out of an interval defined by [
rinf,
r sup] [
17]. The starting values for this interval are [0,10
30] and we increment
rinf if resonance conditions are detected. In addition, we have improved this feature using the following algorithm [
25,
26,
27,
28]:
Step 1: Set rinf to zero.
Step 2: Neglect interaction for atoms with distance lying in the interval [rinf, rsup] .
Step 3: If a test indicates resonance conditions, then increment rinf by 0.529Å and continue from Step 2.
Step 4: Stop.
After the application the algorithm, the typical value of rinf is 6 a.u. Since the results depend on the choice of these parameters, which in fact control the interaction between atoms, we have minimized this effect using a value for rsup which is never reached, and a value for rinf which is the minimum allowed for each model and molecule, compatible with the condition of no resonance.
3. We have also implemented the following tests indicating a resonance condition [
25,
26,
27,
28]: (a) test whether the
matrix is singular [
17]: det
= 0; (b) test whether the
matrix is not defined positive
(i.e., some
eigenvalue is less than or equal to zero) [
17]; (c) test whether the
matrix is not defined positive; and (d) test whether some effective
matrix is not defined positive.
The values of
are calculated with this model. We give particular attention to the average molecular polarizability
and the anisotropy
g [
17].
A fully operative version of the PAPID program [
57] including the whole interacting induced dipoles polarization model has been implemented [
30,
31] into the MM2 program for molecular mechanics [
60,
61]. We have called the new version MMID [
14,
15,
16]. A description of the implementation has been reported elsewhere [
25].
Geometric descriptors and topological indices
The TOPO program for the theoretical simulation of molecular shape has been described elsewhere [
25,
26,
27,
30,
31,
62]. The surface of molecules can be represented by the external surface of a set of overlapping spheres with appropriate radii, centred on the nuclei of the atoms [
63,
64,
65,
66,
67]. If the radii are those of van der Waals [
68], the
bare (solvent free) molecular surface is obtained [
63,
64]; if the radii are those of van der Waals plus the effective radius of a solvent molecule, the solvent-accessible molecular surface is now obtained [
65].
The characterization of the molecular shape is helped by the calculation of geometric descriptors and topological indices of the molecules [
63,
64]. The molecule is treated as a solid in space defined by tracing spheres about the atomic nuclei. It is computationally enclosed in a graduated rectangular box and the geometric descriptors evaluated by counting points within the solid or close to chosen surfaces. The molecular volume
V is concurrently approximated as
V =
P ·
GRID3, where
P is the number of points within the molecular volume (within distance
RX of any atomic nucleus
X) and
GRID is the size of the mesh grid.
As a first approximation, the bare molecular surface area could be calculated as
S ≈
Q ·
GRID2, where
Q is the number of points close to the bare surface area (within a distance between
RX and
RX +
GRID of any atomic nucleus
X). However, the estimate has been improved [
64]: if the point falls exactly on the surface of one of the atomic spheres, it accounts indeed for
GRID2 units of area on the bare molecular surface. This is because the ed at the nucleus and respondsommodate 4π
RX2/
GRID2 points. When a point falls beyond the surface, it represents
GRID2 units of area on the surface of a sphere of radius
R >
RX, not on the surface of atom
X. On the surface of
X it accounts only for a fraction of this quantity, namely,
GRID2(
RX/
R)
2. The total bare surface area is, therefore, calculated as
S =
F ·
GRID2, where
F is the sum of elements
AF defined as
AF =
R2X/
R2(
I) for those points close enough to the surface of any atom
X.
R2X is the squared radius of atom
X and
R2(
I) is the squared distance of point
I from the atomic nucleus
X. Meyer [
64] has written a program including the OEPP (one element per point) sub-program that allows the calculation of the molecular volume and bare surface area. We have written a version of this sub-program (we have called it ATOEPP) that allows the atom-to-atom partition analysis of these geometric descriptors into their atomic component parts [
25,
26,
27,
28,
29,
30,
31].
Two topological indices of molecular shape can be now calculated:
G and
G´ [
64]. The ratio
G =
Se/
S has been interpreted as a descriptor of molecular globularity;
Se is the surface area of a sphere whose volume is equal to the molecular volume
V. The ratio
G´ =
S/V descriptor of molecular rugosity. has been interpreted as a descriptor of molecular rugosity.
The importance of the solvent validates the assumption that the properties of the systems solvated in water are strongly related to the contact surface between solute and water molecules [
65,
67]. We can propose a new molecular geometric descriptor that is the
solvent-accessible surface AS [
65]. This surface is denoted when a spherical probe is allowed to roll on the outside while maintaining contact with the bare molecular surface. The continuous sheet defined by the locus of the centre of the probe is the accessible surface. Alternatively, the accessible surface is calculated as the bare molecular surface by using pseudo-atoms, whose van der Waals radii were increased by the radius
R of the probe. The
accessibility is a dimensionless quantity varying between 0 and 1 and represents the ratio of the accessible surface area in a particular structure to the accessible surface area of the same atom when isolated from the molecule [
65].
Fractal surfaces [
69] provide a means for characterizing the irregularity of molecular surfaces [
70]. The area of the solvent-accessible surface AS depends on the value of the probe radius
R. The
fractal dimension D of the molecules may be obtained according to Lewis and Rees [
70] as
The fractal dimension, D, provides a quantitative indication of the degree of surface accessibility toward different solvents. The larger the D value the faster the accessible surface area drops with an increase in solvent molecular size.
A version of the TOPO program has been implemented into the AMYR program for the theoretical simulation of molecular associations and chemical reactions [
8,
9,
10,
11,
12]. The algorithm allows for the characterization of molecular units and aggregates. All of the geometric descriptors and topological indices but fractal dimension can be now calculated.
Calculation results and discussion
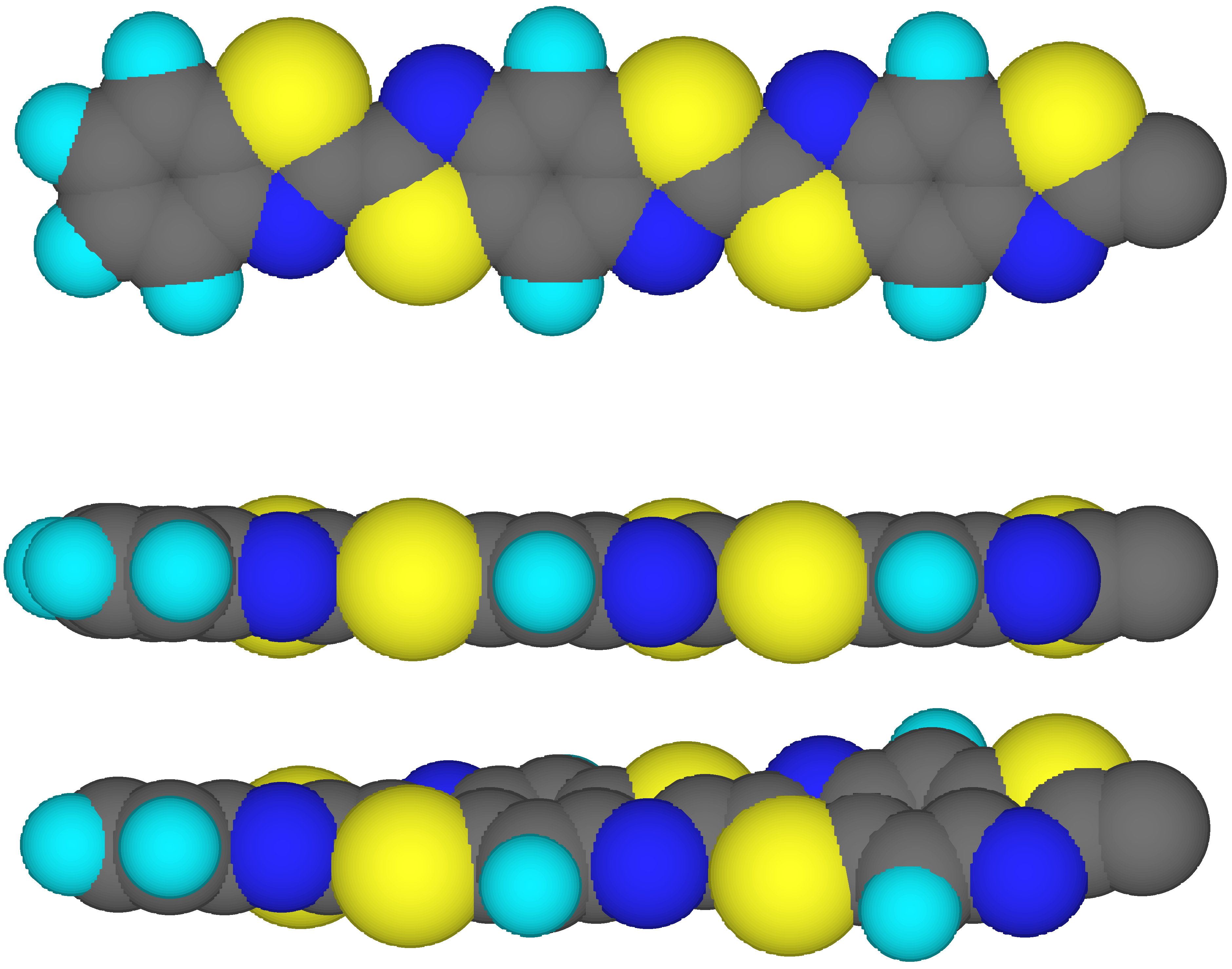
Two model rotational isomers of the benzothiazole (A)-benzobisthiazole (B) linear oligomer A-B
13-A have been characterized: (1) the fully-planar (000) conformation; and (2) a rotational isomer in which each unit is rotated by a fixed angle
f with respect to the previous one. All rotations are performed in the same direction (+++, see
Fig.1). The fixed rotation angle
f has been varied from 1° to 10°. The dipole (
) and quadrupole tensor (
) moments for the rotational isomers are reported in
Table 1. The geometries of the oligomers have been taken from the optimization of the monomeric units with MMID (molecular mechanics with interacting dipoles polarization), a molecular mechanics computer program [
14,
15,
16] based on the MM2 parametrization [
60,
61]. All the linear oligomers are arranged in an all-
trans configuration, in which the series of N=C double bonds, either in the same or in consecutive units, are disposed alternatively. Each rotational isomer is brought into its principal inertial coordinate system. The length
x of the oligomer is defined as the maximal length (corresponding to the translation axis in A-B
•-A), the height
z as its minimal thickness, and its width
y is measured at right angles to the axes indicated by the length and height. The origin is the centre of mass for each conformation.
Three important features in
Table 1 must be remarked. First, the sharp raising of the calculated dipole moments
m even for the smallest distortion from planarity (φ = 1•). Second, the oscillatory behaviour of
m as
f increases. Third, the invariance of the mean quadrupole tensors
(viz., the trace of
). Indeed, this property remains roughly constant about -77 D•Å in spite of a great variation of its three diagonal components. In other words, the anisotropy of
changes as
f increases, but a macroscopic measure of
Q in a fluid phase would not detect such a change. As a measure of the anisotropy of
we propose a Ray’s type asymmetry parameter
k [
71] in the last column of
Table 1. Consider |Θ
1| ≤ |Θ
2| ≤ |Θ
3| which can be relabelled as Θ
m ≤ Θ
int ≤ Θ
M.
k is defined from
The
k parameter becomes -1 for a
prolate symmetric quadrupole tensor (Θ
int = Θ
M) and 1 for an
oblate symmetric quadrupole (Θ
int = Θ
m). Intermediate values arise in asymmetric cases. In most of the cases in
Table 1,
k is negative, indicating a closer similarity of the molecular quadrupoles with a prolate symmetric tensor. We can point out that: (1) the variation of
k as a function of
f shows an oscillatory pattern (with maxim at φ = 1°, 5° and 9°), (2) the positive peaks of
k ( φ = 1°, 5° and 9°) are greater than the negative ones ( φ = 3 ° and 7°), and (3) the origin of the positive
k peaks relies in a strong decrease in Θ
int − Θ
m which makes negative the numerator of
k, while the denominator of
k is always negative.
The molecular
,
and
tensors for the rotational isomers of A-B
13-A have been calculated with the PAPID program [
17] and the results are summarized in
Table 2 Table 3 and
Table 4. In general, the values of
increase with the torsion angle
f. The eigenvector analysis shows that this trend is basically due to the third eigenvalue
a 3 (see the fifth column in
Table 2), which corresponds well to the
axx Cartesian component,
i.e., the component along the chain axis. The anisotropies,
g, of the
tensors (calculated through Equation 3) increase, in general, with the torsion angle
f (from 502 Å
3 to 1513 Å
3). The oligomers are reasonably predicted to be more polarizable along the chain direction than across the bonds (α
xx > α
zz). The values of the higher order tensors
and
, reported in
Table 3 and
Table 4, are also anisotropic, showing more polarizability along the chain direction (|
Ax| > |
Az|, |
Ax,xx| > |
Az,zz| and
Cxx,
xx >
Czz,
zz).
It can be seen from
Table 4 that the dominant components of
increase, in general, with the torsion angle
f. Instead, the behaviour of
is rather complicated as exemplified by the
Ax,xx component. While
and
are sensitive to the length of the oligomer,
is expected to be also sensitive to the molecular dipole moment
m, but no clear correlation can be found, unfortunately, between the behaviour of the calculated
m and the components of
[
25,
26,
27,
28].
The calculation of the geometric descriptors and topological indices for A-B
13-A in
Table 5 and
Table 6 is compared with reference calculations carried out with the GEPOL (GEometry of POLyhedron) program [
72,
73,
74,
75]. This program has been developed in this laboratory by Silla
et al. The GEPOL algorithm calculates three kinds of envelope surfaces: (1) the
van der Waals molecular surface, which is the external surface resulting from a set of spheres centred on the atoms or groups forming the molecule; (2) the
solvent-accessible surface, which is the surface generated by the centre of the solvent, considered as a rigid sphere, when it rolls around the van der Waals surface [
65]; and (3) the
solvent-excluding surface which is composed of two parts: the contact surface and the reentrant surface. The
contact surface is the part of the van der Waals surface of each atom that is accessible to a probe sphere of a given radius. The
reentrant surface is defined as the inward-facing part of the probe sphere when this is simultaneously in contact with more than one atom. The GEPOL program is fast and efficient.
The geometric descriptors for the rotational isomers of A-B
13-A are listed in
Table 5. The value of the molecular volumes
V remain roughly constant with the torsion angle
f. The values compare well with reference calculations carried out with the GEPOL program. However, the molecular surface areas
S slightly increase with
f. The water-accessible surface areas AS, the hydrophilic HLAS contribution, and the side-chain accessible-surface areas AS', clearly increase with
f. However, the hydrophobic HBAS component part of AS decreases with
f. It should be noted that large values of HLAS (and low values of HBAS),
e.g. for φ = 10 °, favour solubility in water, which is a fundamental property for the processibility of the material.
The topological indices for the rotational isomers of A-B
13-A are reported in
Table 6. The globularity index
G is roughly constant with the torsion angle
f. The values compare well with reference calculations carried out with the GEPOL program. The behaviour of the rugosity index
G', the fractal dimension of the solvent-accessible surface
D, and the fractal dimension averaged for
nonburied atoms (those with nonzero accessible surface area)
D′, is the same as has been noticed for
G. The comparison between the TOPO and GEPOL programs is of special interest because the latter does not allow an atom-to-atom partition analysis of the geometric descriptors and topological indices [
25,
26,
27,
28,
29,
30,
31].
The atom-to-atom partition for each thiazolic nitrogen atom in the central unit of the oligomer are reported in
Table 7. The contribution of this nitrogen to the molecular volume
V slightly increases with the torsion angle
f. This torsional effect has a steric cause and is due to the partial opening of the oligomer structure under rotation. The trend of the contribution of the N atom to the molecular surface areas
S, the molecular rugosities
G' and the water-accessible surface areas AS, follows that for the molecular volume. Thus, the
accessibilities of the N atom surfaces increase with
f (until 21.2%). It should be noted that the polar thiazolic nitrogens are the only atoms that contribute to the hydrophilic accessible surface area HLAS, and consequently, the conformer with φ = 10 ° shows the greatest value of HLAS in
Table 5. The contribution of the N atoms to the molecular globularities
G and to the fractal dimensions
D', decrease, in general, with
f. The N atoms contributions to
D' are greater (1.62 − 1.81) than for the molecular values averaged for nonburied atoms (1.56 − 1.57 in the last column of
Table 6). This means that the irregularity of the molecular surface near the N atom is greater than the average on the whole molecule.



















{kind=link}






