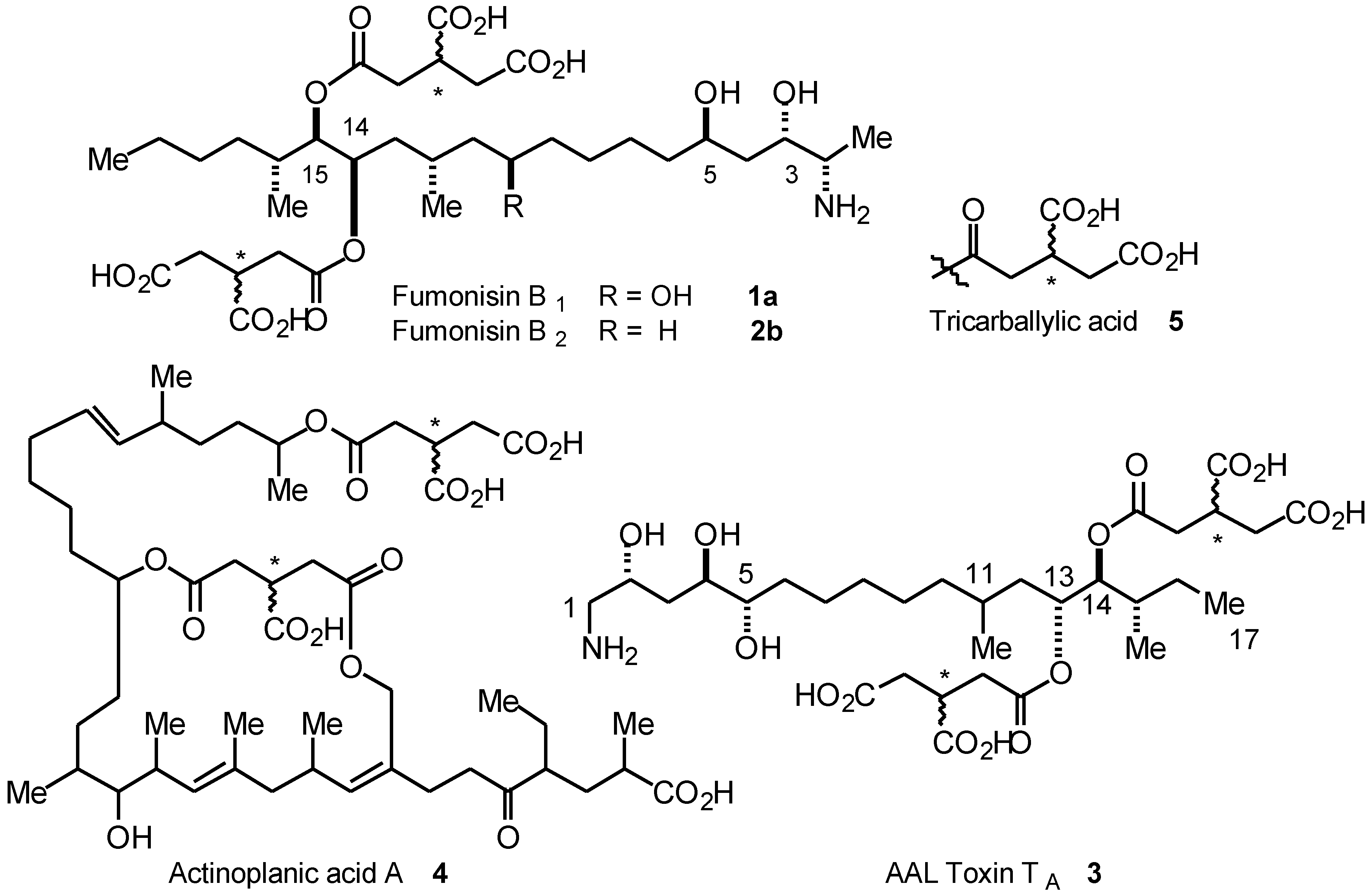
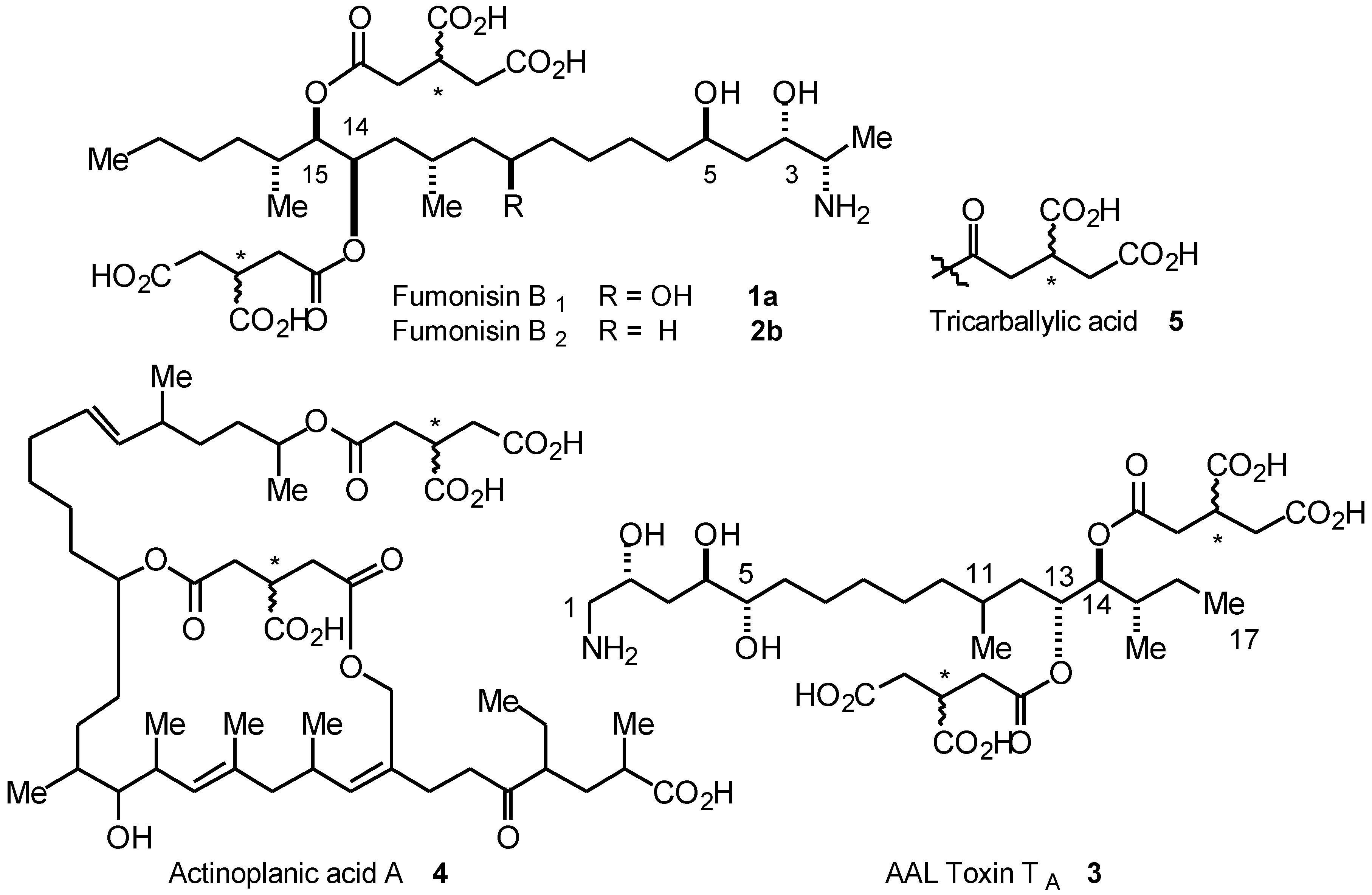
Results and Discussion
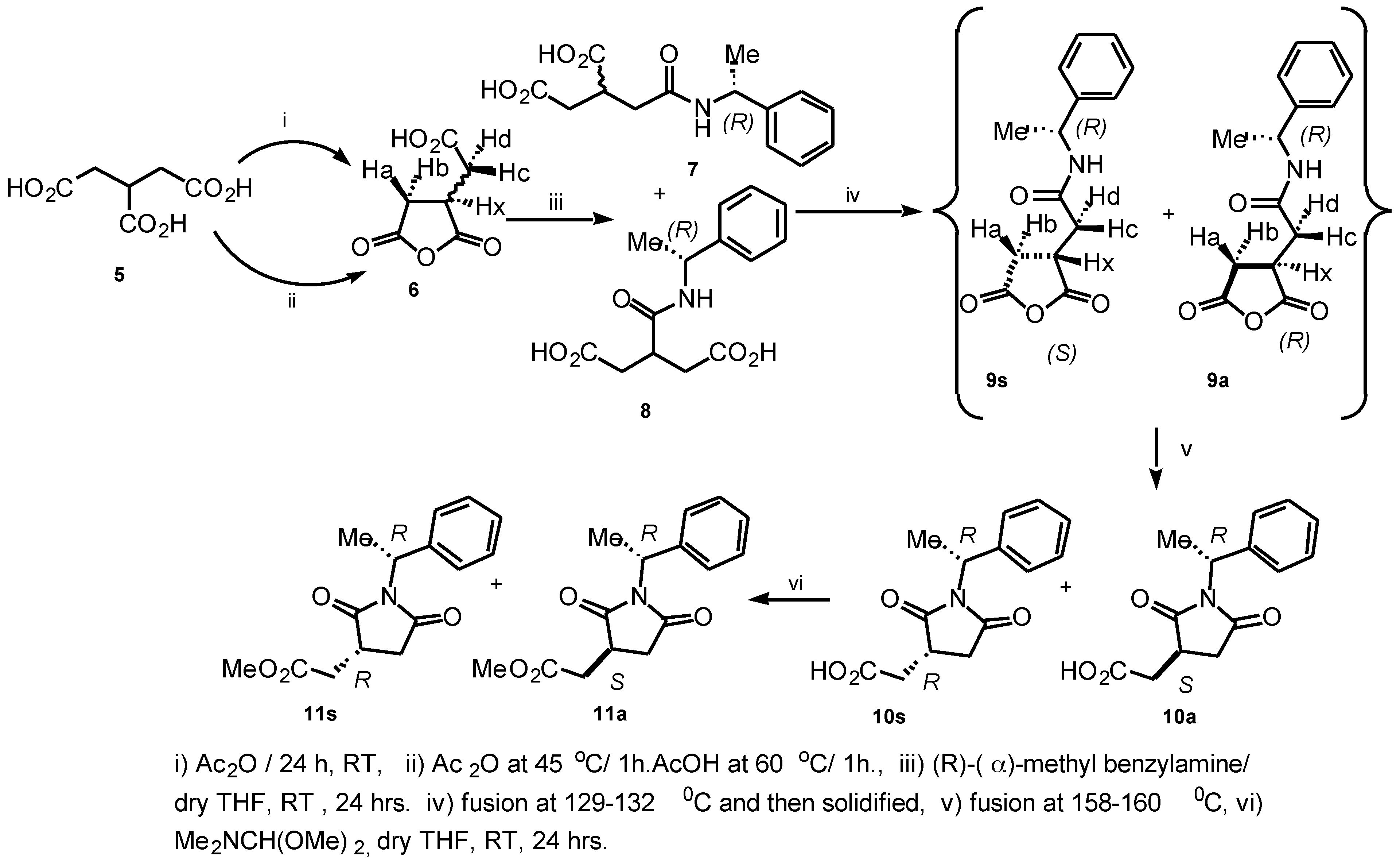
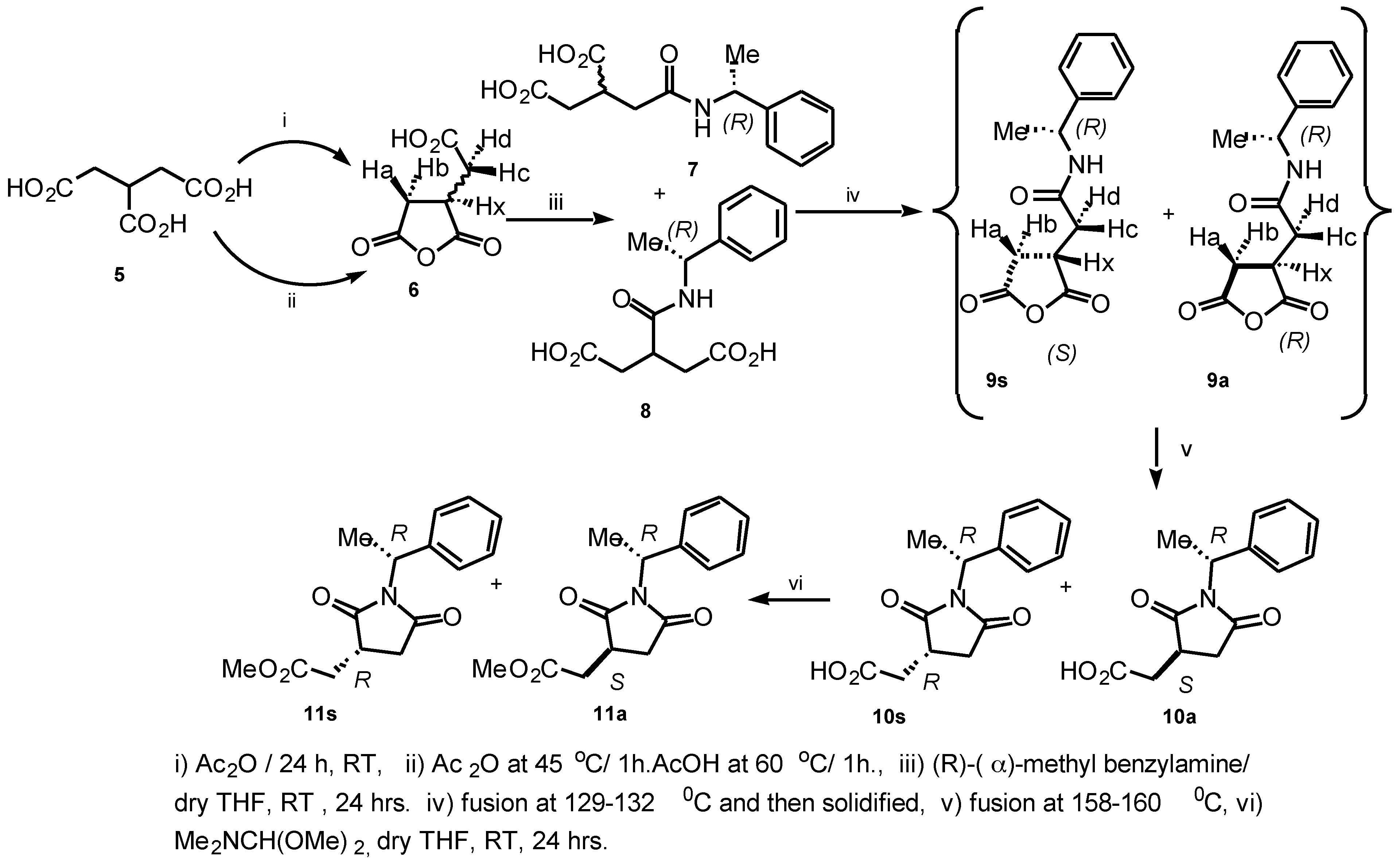
Ring-opening of 2-(carboxymethyl) succinic anhydride
6 with (
R)-(
α)-methylbenzylamine in dry THF (RT, 24 h) gave a white sticky material of diastereomeric and regioisomeric mixture of amides upon concentration (
Scheme 1).
The
1H NMR spectrum allowed us to suggest that the mixture of diastereomeric and regioisomeric amides were the
α-amide and
β-amide derivatives
7 and
8, respectively. NMR data for these structures
7 and
8 were consistent with the results described by Hoye et al.[
8] for the determination of absolute configuration of 3-substituted carboxylic acids (
β-chiral acids). The resulting products
7 and
8 were melted at 129-132°C, re-solidified by cooling, transferred into a NMR tube and finally analyzed. The spectrum shows doublet peaks downfield for (RC
Hc-R') and (RC
Hd-R') at δ 2.86 (dd,
J = 6.86, 16.90 Hz) and δ 3.30 (dd,
J = 9.75, 18.54 Hz) of the compound
9s, respectively. The
1H NMR spectrum of
9a was virtually the same as for
9s with the following differences: δ (RC
Hc-R') and (RC
Hd-R') at δ 2.75 (dd, J = 9.76, 17.90 Hz) and δ 3.22 (dd, J = 9.93, 18.42 Hz) ppm. This was considered to be due to the anisotropic shielding effect of the aromatic ring of the phenyl group attached to the succinimide. The major product of the diastereomeric mixture of anhydride was assigned to be
9s (1.3:1). The intermediate compounds
9s/
9a were re-melted at 158-160°C, and gave the expected assignment of the diastereomeric mixture of succinimide derivatives
10s/
10a in a 1:1 ratio. The product was quite pure after spectroscopic analysis and no attempt was made to purify this diastereomeric mixture further. Esterification of the diastereomeric mixture
10s/
10a was achieved with
N,N'-dimethylformamide dimethylacetal in dry THF (RT, 24h). Concentration gave a white sticky material of diastereomeric mixture
11s/
11a in a 1.2:1 ratio. The diastereomeric mixture was purified by MPLC (1:9 Hexane:EtOAc) to provide compounds (
R)-2-(methoxycarbonylmethyl)-
N-(
R)-1-(1-phenylethyl)-succin-imide
11s and (
S)-2-(methoxycarbonylmethyl)-
N-(
R)-1-(1-phenyl-ethyl) succinimide
11a in 82% yield. We have developed a reliable method for determining the absolute configuration of carboxylic acids containing a stereogenic center at C(3) of the tricarballylic acid derivatives.[
8,
9].
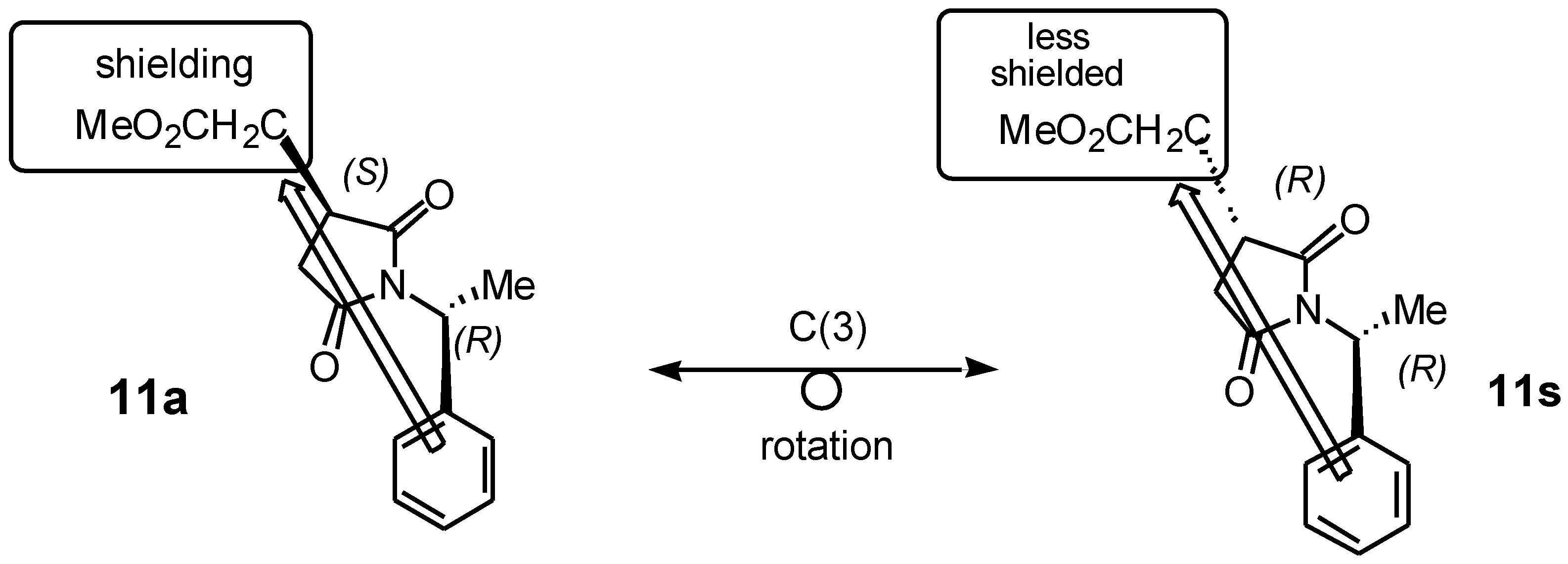
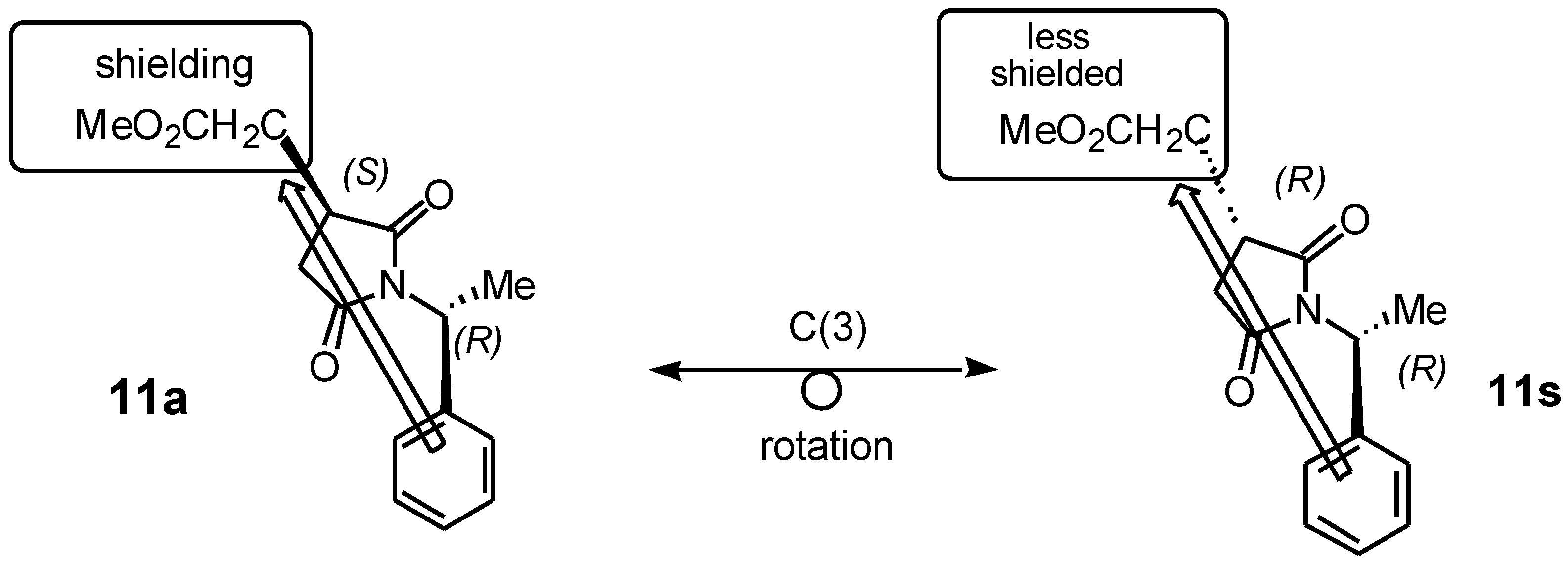
Figure 2 shows the application of this method to identify a heterotopic group attached to C(3) that contains readily distinguishable
1H NMR resonances in the diastereomeric pair of 1-arylethylsuccinimides (e.g. the methoxycarbonyl methyl group in
11). The C(3)-configuration has been deduced by comparing the relative shifts of these resonances. For example, in the
anti isomer
11 of the CH
2CO
2Me-succinimide of generic
anti-(3S) diastereomer, the methoxyl resonance will be observed at higher field than in the
syn -(3
R) diastereomer.
Similarly, the 1H NMR spectrum of compounds 11s/11a shows the (CO2Me) of 11s as a singlet downfield (high chemical shift) at δ (3.66) and the 11a was a singlet upfield (low chemical shift) at δ (3.62), due to the anisotropic shielding effect of the aromatic ring of the phenyl group attached to the succinimide derivatives. We attempted to further separate these diastereomers and confirmed the present of two diastereomers by using gas chromatography at different temperatures (250, 270°C) and also with different retention times, but we obtained a broad peak which included a shoulder of the other diastereomer. HPLC was used for the separation of those diastereomeric mixture of 11s/11a, unfortunately those diastereomeric mixture was enriched to each other.
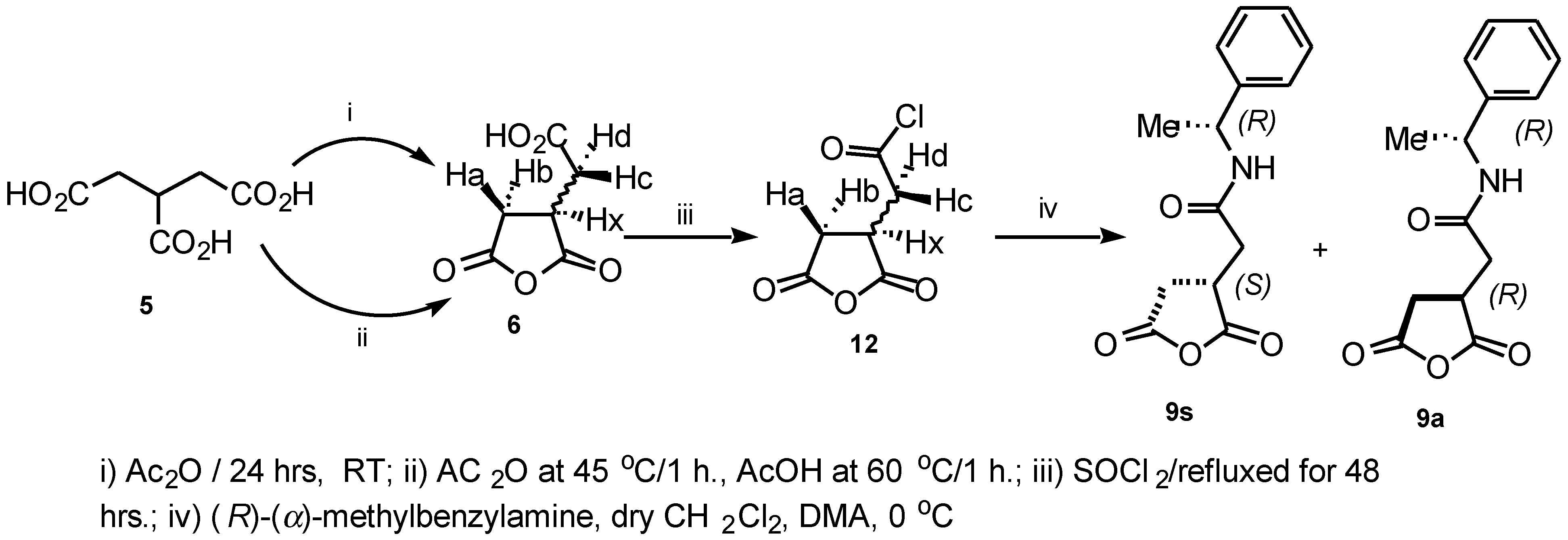
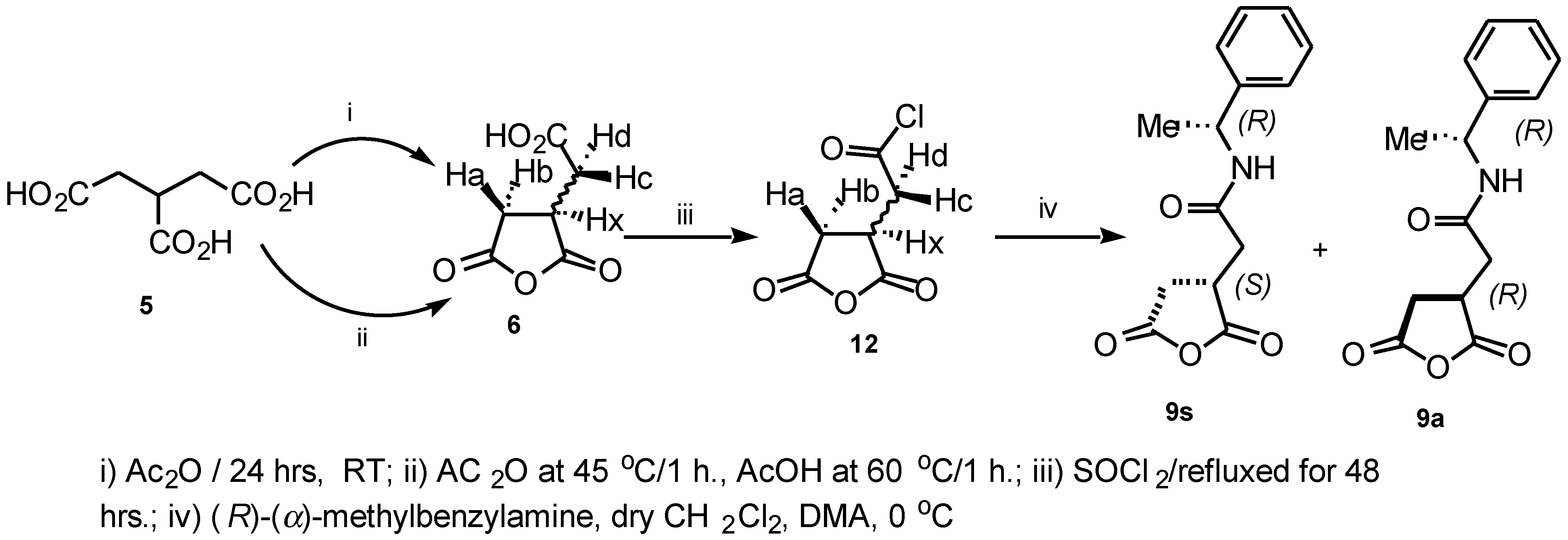
The intermediate compounds
9s and
9a were also prepared via a different route as shown in (
Scheme 2), by treatment of the 1-(chloroformyl) propane-2,3-dicarboxylic acid anhydride
12 with (
R)-
α-methylbenzylamine in the presence of DMA and CH
2Cl
2 at 0°C. The reaction mixture was vigorously stirred at room temperature overnight to provide a good yield of the crude product which was purified further by flash chromatography (2:1 Hexane:EtOAc) to afford the diastereoisomeric mixture in 60% yield.
Experimental
General
1H and
13C NMR spectra were taken on an IBM NR-200 or IBM NR-300-AF spectrometer. Chemical shifts are given in ppm (δ-scale), coupling constants (J) in hertz.
1H-chemical shifts (300MHz) were referenced to the residual CHCl
3 signal (7.24).
13C-chemical shifts (75 MHz) were referenced to CDCl
3 (77.0). Optical rotations were measured on a Perkin Elmer 1420 ratio recording spectrometer, 1-2% solution in CHCl
3. MPLC refers to medium pressure liquid chromatography (25-60 psi) using hand packed columns of E. Merck silica gel (230-400 mesh) and a Fluid Metering Inc. pump, an ISCO Model 2361 gradient programmer, and a differential refractive index detector (Waters R401). Flash column chromatography was performed as described by Still [
10] using E. Merck silica gel (230-400 mesh). The R
f value reported for TLC analysis was determined on Macherey-Nagel 0.25 mm layer fluorescent UV254 plates with the indicated solvent system. Infrared IR spectra listed as recorded ‘neat’ refer to a thin film of material on NaCl disks. Infrared spectra were recorded on a MIDAC Prospect spectrometer. IR spectrum peaks are reported in cm
-1. Elemental analyses were performed by M-H-W Laboratories (Phoenix, AZ) and the Central Lab.of Ain Shams University, Cairo, Egypt. Tandem gas chromatography/ low resolution mass spectrometry (GC/LRMS) using electron impact (EI) ionization was performed on a Hewlett-Packard 5890 series II gas chromatography and 5971A mass selective detector at 70 eV. Gas chromatography retention time is reported along with the capillary column configuration using the following convention: Column C = DB-5.6 m x 0.1 mm ID x 0.1 m film + 1 guard column with a 1 mL/in flow rate.
Propane-1,2,3-tricarboxylic Acid (1,2-Anhydride) 6
Tricarballylic acid
5 (1.76 g, 0.01 mol) was dissolved in acetic anhydride (1.78 mL, 2.04 g, 0.02 mol). The reaction mixture was stirred overnight at room temperature, then excess acetic anhydride was removed under reduced pressure and the product was recrystallized twice from ethyl acetate to give of the title compound as a white micro-crystalline solid (1.37 g, 0.0087 mol, 77.8% yield) that had mp. 132-133°C, lit.[
11]: 132-134°C.
1H NMR
: (200 MHz, acetone-d
6): δ 2.89 [dd, J = 2.6 and 18.2 Hz, 1H, RC
Ha-R'(ax)], 2.91 [dd, J = 6.6 and 18.6 Hz, 1H, RC
Hc-R'(ax)], 3.002 [dd, J = 5.4 and 18.2 Hz, 1H, RC
Hd-R'(eq)], 3.27 [dd, J = 10.4 and 18.2 Hz, 1H, RC
Hb-R'(eq)], 3.62 [dddd, J = 10.4, 6.6, 5.4, and 2.6 Hz, R-CH
2C
HxCH
2-R'(eq)], 11.3 (s, R-CO
2H) ppm.
13C NMR
: (50 MHz, acetone-d
6): δ 176.7, 174.2, 173.4 , 38.2, 35.3 and 35.2 ppm. IR
: (KBr pellet): 1727 (vs), 1790 (s), and 1833 (m) cm
-1 Anal. Calcd for C
6H
6O
5: C, 45.58; H, 3.84; O, 50.59% Found: C, 45.55; H, 4.08.
3-Carboxypentane-1,5-dioic acid, mono-[1-(R)-(1-phenylethyl)]amide 7, and 3-mono-[(R)-(1-phenylethyl)]amide pentane-1,5-dioic acid 8
Tricarballylic acid anhydride (TCA) 6 (47.4 mg, 0.3 mmol) was placed into an oven dried culture tube (25 mL r.b.), dissolved in dry THF (1.2 mL, 0.25 M), and then treated with (R)-(+)-α-methylbenzyl amine (46.4 μL, 43.6 mg, 0.36 mmol). The resulting solution was stirred overnight at room temperature. The solvent was removed under reduced pressure to give 84.3 mg (92% mass recovery) of a white sticky of diastereoisomer 7 and regioisomeric amides 8 as determined by 1H NMR analysis and no attempt was made to purify this material further and isolate the diastereoisomers.
Compound 7
1H NMR (200 MHz, acetone-d6, from the mixture) : δ 1.43−1.40 (d, J = 7.0 Hz, 3H, Ar CHCH3), 2.43−2.54 (m, 2H, RCH2-CONH), 2.61-2.68 (m, 2H, RCH2-CO2H), 3.18-3.22 (m, 1H, RCH2CHCH2-R'), 5.03 (dq, J = 6.8 Hz, ArCH-), 5.34 (d, J = 7.23 Hz, -NH-), 7. 16-7.52 (m, 5H, Ph-H) ppm.
Compound 8
1H NMR (200 MHz, acetone-d6, from the mixture) : δ 1.62−1.59 (d, 3H, J = 6.8 Hz, Ar CHCH3), 2.43−2.54 (m, 2H, RCHaHb-CO2H), 2.61−2.68 (m, 2H, RCHa’Hb’-CO2H), 3.2-3.3 (m, 1H, RCHR'), 5.0-5.08 [dq, J = 6.65 Hz, 1H, ArCHNHMe], 5.63 (d, J = 7.23 Hz, 1H, -NH-), 7.19-7.49 (m, Ph-H) ppm.
The mixture of compounds 7 and 8, were placed into an oven dried melting point tube, fused (129-135°C), solidified, and then transferred into the NMR tube. The diastereoisomers were determined by 1H NMR analysis. The structural assignments were made by comparison of the spectral data of compounds 9s/9a which were obtained by the reaction of tricarballylic anhydride acid chloride 12 with (R)-(+)-α-methylbenzyl amine. The ratio of the distereoisomers was ~1:1 according to the integration. No attempt was made to resolve the diastereoisomers further, m.p. 129-135°C. The 1H NMR spectra data was identical consistent with authentic samples 9s/9a in all aspects.
Compond 9s
LRMS (EI): m/z 148 [M+-113, (20)], 106 (20), 94 (20), 77 (25), 44 (100). 1H NMR (200 MHz, acetone-d6): δ 1.62 (d, J = 7.2 Hz, ArCHCH3), 2.65 (dd, J = 6.39 and 16.93 Hz, RCHa-R'), 2.86 (dd, J = 6.86 and 16.90 Hz, RCHc-R'), 3.09 (dd, J = 7.13 and 18.59 Hz, RCHb-R'), 3.30 (dd, J = 9.75 and 18.54 Hz, RCHd-R'), 3.84 (dddd, J = 12.20, 7.31, 5.16, and 2.21 Hz, RCH2CHxCH2-R'), 5.37 (q, J = 7.20 and 14.36 Hz, ArCH-), 7.42 (m, Ar-H) ppm.; Cap gc; tR = 7.21 min.; column: DB-5 5 m+ 1m guard ~1 mL / min.; gum temp prog: To = 50°C / 2 min. / 20°C min.-1 / 250°C / 5 min.
The 1H NMR spectrum of 9a was virtually the same as for 9s with the following differences. δ 2.75 (dd, J = 9.76 and 17.90 Hz, RCHc-R'), 3.22 (dd, J = 9.93 and 18.42 Hz, RCHd-R') ppm. Anal. Calcd for C14H15NO4: C, 64.36; H, 5.79; N, 5.36; O, 24.49% Found: C, 64.03; H, 4.99; N, 5.01.
The solidified materials 9s/9a were again melted at 158-160°C, and transferred into a NMR tube. The 1H NMR spectral data was identical with those for compounds 10s and 10a. Tlc: Rf = 0.2 in 2:1:Hex:EtOAc. LRMS (EI): m/z 275 (M+, (100), 260 (<3), 244 (20), 232 (55), 215 (20), 200 (15), 172 (20), 146 (40), 120 (65), 105 (100), 104 (100), 94 (12), 77 (60), 59 (75), and 41 (25). The compounds 10s and 10a were obtained as a diastereoisomeric mixture in 1.2:1 ratio (NMR). (10s) 1H NMR (500 MHz, CDCl3): δ 8.07 (d, 1H, J = 8.5 Hz, Ar-H), 7.87 (d, 1H, J = 8.0 Hz, Ar-H), 7.80 (d, 1H, J = 1 and 7 Hz, Ar-Hp), 5.42 (dq, J = 7.5 Hz, Ar-CH-Me), 3.66 (s, RR’CHCOOCH3)2 3.51 (dddd, J = 2.3, 3.6, 3.8, and 9.3 Hz, R2CHCO2Me), 2.99 (dd, J = 3.73 and 15.31 Hz, RCHaHbCONHCHAr), 2.9-2.75 (m, 2H, RCHaHbCOOMe), 2.47-2.44 (dd, J = 6.0 and 7.5 Hz, 2H, RCHcHdCONHAr), 2.43-2.41 (dd, J = 5.5 and 7.5 Hz, RCHcHdCONHAr), 1.827 -1.812 (d, J = 7.5 Hz, 3H, ArCH-Me) ppm. The 1H NMR spectrum of 10a was virtually the same as that for 10s with the following differences: δ 3.62 (s, 3H, RCH2COOMe) and 1.821-1.807 (d, J = 7.0 Hz, 3H, ArCH-CH3) ppm.; Cap gc: tR = 10.44 min.; column: DB-5 5mL = 1m guard ~ 1mL / min.; gum temp prog: To = 50°C / 2 min. / 20°C min.-1 / 250°C / 5 min.
(R)-2-Carboxymethyl-N-[(R)-1-(phenylethyl)succinimide 11s and (S)-2-Carboxymethyl-N-[(R)-1-(phenylethyl)succinimide 11a
Diastereomeric acid mixture 10s/10a (115 mg, 0.44 mmol) was dissolved or suspended in dry THF (1 mL). N,N’-dimethylformamide dimethylacetal (209 mg, 1.76 mmol, 0.5 mL) was added dropwise to the suspended solution within 20 min. The reaction mixture was allowed to stir overnight at room temperature and then the solvent was removed. The residue was washed with water (15 mL) and then the product was extracted with ethyl acetate, washed with saturated NaHCO3 solution (2x10 mL), and brine (10 mL). The organic layer was dried over MgSO4 anhydrous. The solvent was removed, to afford 110.5 mg of crude semi-oily product. The residue was purified by MPLC (with pure EtOAc) to provide the diastereoisomeric products 11s and 11a (95 mg, 0.35 mmol., 20 mole% and 82% yield). The material proved to be a 1.2:1 mixture of diastereoisomers according to 1H NMR analysis. TLC; Rf = 0.2 in 2:1:Hex:EtOAc.; LRMS (EI): m/z 275 (M+, (100), 260 (<3), 244 (20), 232 (55), 215 (20), 200 (15), 172 (20), 146 (40), 120 (65), 105 (100), 104 (100), 94 (12), 77 (60), 59 (75), and 41 (25).
Compound 11s
1H NMR (500 MHz, CDCl3) : δ 8.07 (d, 1H, J = 8.5 Hz, Ar-H), 7.87 (d, 1H, J = 8.0 Hz, Ar-H), 7.80 (d, 1H, J = 1 and 7 Hz, Ar-Hp), 5.42 (dq, J = 7.5 Hz, Ar-CH-Me), 3.66 (s, RR’CHCOOCH3)2 3.51 (dddd, J = 2.3, 3.6, 3.8, and 9.3,Hz, R2CHCO2Me), 2.99 (dd, J = 3.73 and 15.31 Hz, RCHaHbCONHCHAr), 2.9-2.75 (m, 2H, RCHaHbCOOMe), 2.47-2.44 (dd, J = 6.0 and 7.5 Hz, 2H, RCHcHdCONHAr), 2.43-2.41 (dd, J = 5.5 and 7.5 Hz, RCHcHdCONHAr), 1.827 -1.812 (d, J = 7.5 Hz, 3H, ArCH-Me) ppm.
The 1H NMR spectrum of 11a was virtually the same as for 11s with the following differences: δ 3.62 (s, 3H, RCH2COOMe) and 1.821-1.807 (d, J = 7.0 Hz, 3H, ArCH-CH3) ppm.; Cap gc: tR = 10.44 min.; column: DB-5 5mL = 1m guard ~ 1mL / min; gum temp prog.: To = 50°C / 2 min. / 20°C min.-1°/ 250°C / 5 min. Anal. Calcd for C15H17NO4: C, 66.44; H, 6.22; N, 5.09; O, 23.25% Found: C, 66.55; H, 6.09; N, 4.99.

{kind=link}
{kind=link}
{kind=link}
{kind=link}