Synthesis of Liquid-Phase Phosphine Reagent (2)
A concern with the original reagent
1 was the carbamate ester moiety incorporated as a linker between the triarylphosphine and the PEG support. The known base and acid sensitivity of the urethane linkage, coupled with worries about its lability in the presence of Lewis acids and metallating reagents, linked any further exploitation of PEG-supported triarylphosphines to an immediate replacement of this carbamate ester group. An aryl-alkyl ether moiety was chosen as the replacement linker, the chemical stability of which is comparable to that of the poly(ethylene oxide) backbone, therefore reagent
2 became the functionalized polymer of choice [
19].
The synthetic strategy towards
2 involves an initial concise preparation of the key hydroxyphosphine
3 [20], followed by its attachment to PEG
via the dimesylate
4 [21]. 4-Bromo-phenol
5 was protected as the TBDMS ether
6, that was then phosphinylated under standard conditions [
22] to give the triarylphosphine
7. The silyl ether was removed and the resultant hydroxyphosphine
3 was obtained in 87% yield for the three steps. Mesylate
4 was obtained by heating (50°C) a neat solution of PEG
3400 8 in methanesulfonyl chloride. No base was required under these conditions, which had the benefit that the purification of
6 was simplified from its original preparation [
21]. Etherification between
4 and
3 was performed in rigorously degassed DMF. Phosphine
2 was then isolated by addition of the reaction mixture into degassed diethyl ether, followed by filtration and sequential washing of the polymeric precipitate with
i-Pr alcohol (to remove salts) followed by diethyl ether to give reagent
2 in 92% yield, based on the weight of polymer isolated.
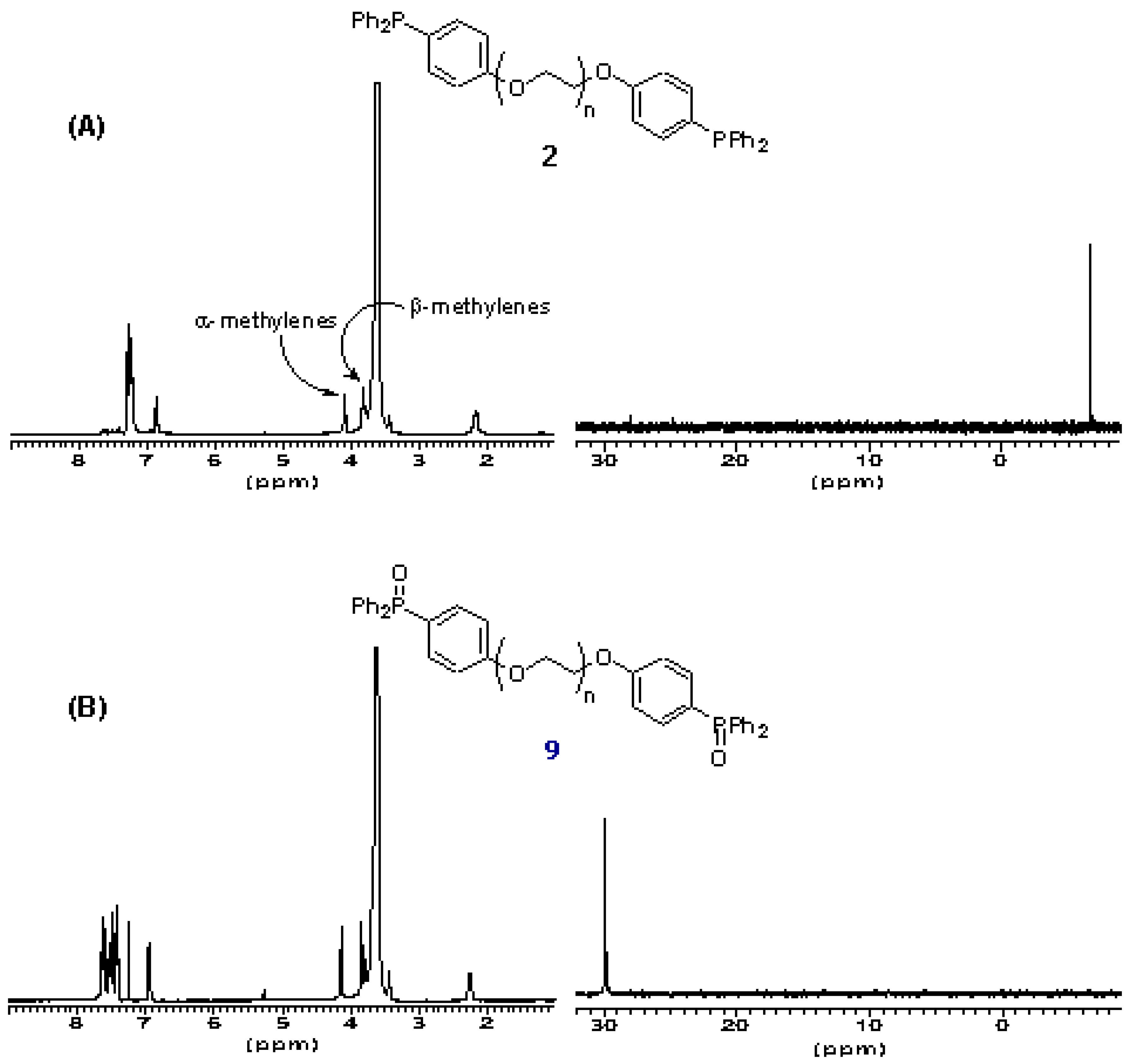
The oxidation state of the phosphine termini of 2 was routinely quantified by 31P NMR after certain periods of storage. This revealed that even after prolonged exposure to air (2-3 weeks) little oxidation to the phosphine oxide (< 5%) occurs.
The derivatization level of the polymer is also determined by inspection of the
1H NMR of
2. The chemical shift of α-methylene protons of PEG and its derivatives is a function of the moiety attached to the termini hydroxyl groups. For underivatized PEG
3400, the chemical shift of these α-protons is δ 3.8 ppm, for PEG-mesylate
4, this changes to δ 4.35 ppm (also detectable are the β-methylene protons δ 3.74 ppm) and for the phosphine terminated polymer
2 this changes to δ 4.16 ppm (β-methylene protons δ 3.64 ppm). Inspection of the
1H NMR of
2 shows that no mesylate
4 remains. Therefore there is high confidence that the polymeric phosphine
2 is quantitatively substituted with terminal triarylphosphine residues (
ca. 0.5 mmol g
-1). The ease of NMR-characterization of soluble polymer reagent
2 offers a considerable advantage over its solid-phase homologue which, in order to determine the oxidation state of the phosphine center, must involve either gel- or solid-phase NMR, or single-bead FTIR techniques [
23].
Liquid-Phase Ozonide Reduction
Figure 2.
A) 1H and 31P NMR spectra of PEG-supported triarylphosphine 2. B) 1H and 31P NMR spectra of PEG-supported triarylphosphine 9.
Figure 2.
A) 1H and 31P NMR spectra of PEG-supported triarylphosphine 2. B) 1H and 31P NMR spectra of PEG-supported triarylphosphine 9.
As an entry level into the utility of
2 in organic chemistry, the mild chemical decomposition of ozonides was selected. Ozonolysis of alkenes has a broad scope of application in organic chemistry, and the range of methods for destruction of the intermediary ozonides is equally broad. However, the two major methods of choice: Zn/acetic acid or dimethyl sulfide have limitations, a result of potential chemical substituent sensitivity in the former case, or noxious reagents and high-boiling point byproducts in the latter. For these reasons PPh
3 is an excellent alternative [
24]. It has received only limited use in solution-phase chemistry however, because of the problems of removing both the excess PPh
3 and reaction byproduct, triphenylphosphine oxide, from the reaction mixture. The use of polystyrene-supported PPh
3 as a stoichiometric reagent for this reaction has been reported as an alternative and gives good to excellent yields of aldehydes [
25]. Therefore an important part of this study was a direct comparison between this new liquid-phase approach with
2, and a solid-phase approach with commercially available PPh
3.
A range of alkenes
10a-
e were treated with ozone. The incipient ozonides were then decomposed into the product aldehydes (
11a-
e) by addition of one of three reagents: PPh
3, polystyrene-supported PPh
3, or PEG-supported PPh
3 (
2). The results are shown below (
Table 1).
The isolation procedure for the PEG-supported reagent involved a simple precipitation of the spent reagent into ether, and concentration. This consistently removed > 99% of the polymer byproduct. Passage of the ether concentrate through a short silica pad completely removed the remaining polymer traces, giving the product aldehydes in analytically pure form. The solid-phase reagent was separated from the reaction mixture by filtration. The product was isolated by then washing the resin with volumes of CH2Cl2. The solution-phase reactions were purified by silica gel chromatography to remove the excess PPh3 and the byproduct PPh3O.
In all cases studied, the yields of aldehyde were highest with the soluble polymer-supported reagent 2 and with the exception of 11d the soluble reagent gave higher yields than the solid-phase reagent. This result may be a composite of three factors; increased reducing power of the soluble polymer-supported phosphine 2 when compared to both the solid- and solution-phase reagents, a result of the pethoxyether functionality which links the reagent to the soluble polymer support, the homogeneous nature of the ensuing chemical process when compared to the hetereogeneous resin reagent and the ease of isolation and purification of the products when compared with solution-phase strategy.
Liquid-Phase Wittig Reactions in aqua
Reagent
2 was subsequently transformed into the polymer-supported phosphonium salt
12 suitable for Wittig olefination in aqueous solution. Organic chemistry in water has been the focus of considerable efforts in recent years [
26,
27,
28]. The Wittig olefination reaction has also been much studied throughout the evolution of crosslinked polymer-supported reagents and considerable success has been achieved [
29,
30,
31,
32,
33,
34]. Bernard and Ford [
34] noted an inverse correlation between the yield of alkene obtained from polymer-supported Wittig reagents with aldehydes and ketones in solution and the degree of divinylbenzene (DVB) cross-linking in the resin. Progressing from 0.5-20% DVB crosslinking,
i.e. from gel to macroporous resins, can result in a fall of 20% in the yield of alkenes. These results suggested that a soluble polymer-supported phosphonium salt may give excellent yields in the Wittig reaction.
The physical properties of the PEG backbone are such that the PEG-supported phosphonium salt
12 is eminently water soluble and offer the tantalizing prospect of facilitating the first soluble polymersupported Wittig olefination reactions
in aqua. Treatment of PEG-supported phosphine
2 with benzylbromide, followed by precipitation into diethyl ether yielded the soluble polymer-supported benzyl triarylphosphonium salt
12 in 81% yield (based on the weight of polymer obtained). This polymeric phosphonium salt
12 is completely soluble in water at pH 7. The reaction of this novel Wittig salt (
12) was then studied with a range of aldehydes (
11a and
13a-
d) in aqueous sodium hydroxide solution (
Scheme 2,
Table 2).
The isolated yields of the alkenes 14a-e by this method were good to excellent, proving that the rate of reaction of the ylide with the substrate aldehydes is indeed much faster than decomposition to 9. Isolation of the stilbenes (14a-e) was achieved by partitioning the reaction mixture with CH2Cl2, followed by removal of the water by addition of anhydrous magnesium sulfate, and then precipitation of the polymer supported triphenylphosphine oxide byproduct 9 into diethyl ether and concentration of the etheric solution to dryness.
To explore the scope of this reaction two components were varied: base strength and temperature. Increasing the base-strength (from 1 N to 2 N NaOH) neither affected the yield of stilbenes (14a-e) nor the E:Z isomer distribution. However, elevating the temperature (up to 90°C) resulted in improved yields of 14d and as to be expected an increase in the E:Z ratio for the stilbene products.
Poly(ethylene glycol) as a Soluble Polymer Matrix for the Stille Cross Coupling Reaction
The Stille [
39,
40] cross-coupling reaction possesses wide scope and functional group tolerance. In addition, the requisite tin reagents are easily prepared and relatively stable when compared to most organometallic reagents. These properties make this particular process very attractive as a route to many carbon-carbon bond forming reactions under relatively ambient conditions. The importance of this reaction within the sphere of polymer-supported chemistry has been demonstrated with the recent emergence of solid- [
41,
42,
43] and fluorous-phase [
44,
45] approaches.
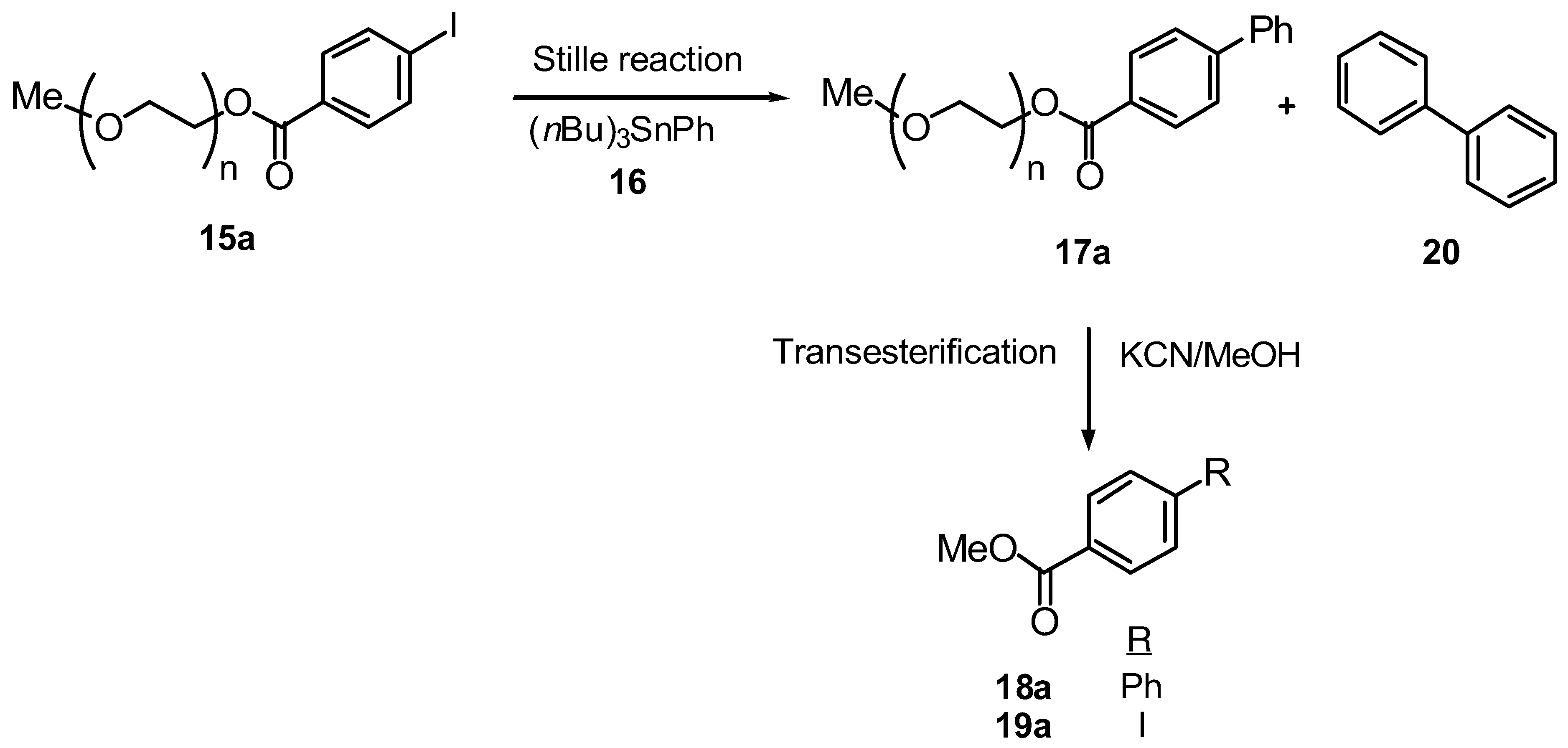
The defining feature of the Stille coupling is the use of a trialkyltin reagent in a palladium-catalyzed coupling with either a halide or triflate. As with all tin chemistry, the benefits associated with these reagents are to some extent outweighed by the problems associated with their removal after reaction completion. The tri-n-butyl derivatives are more difficult to remove from a reaction mixture than the methyl congeners due to their low volatility and poor water solubility. Therefore there is an increasing need for methodologies which facilitate the separation of the side-product tributyltin derivatives from the required adducts following the Stille reaction. Our aim was to study the potential of poly(ethylene glycol) as a soluble matrix for the electrophile component of the reaction. The tributyltin derivative and ‘other’ components of the reaction being in solution could, theoretically, be separated from the polymersupported products by a simple precipitation of the support into a suitable solvent and recovery by filtration.
MeO-PEG
5000 was initially esterified with either
para- or
ortho-iodobenzoic acid using DCC and DMAP to give
para- and
ortho-polymer-supported iodides,
15a-
b respectively, with polymer recoveries being > 95% [
46]. The conversion was quantitative based upon
1H NMR spectroscopic analysis of
15a-
b.
A number of efficient reaction conditions for solution-phase Stille couplings have been developed [
39,
40,
47,
48] and initially we utilized a number of these for the reaction between PEG-iodide
15a and tributyl phenyltin (
16) to explore the viability of PEG as a soluble polymer support for this reaction and then to identify the optimal conditions necessary for the PEG-supported Stille variant (
Scheme 3 and
Table 4). Previous work has revealed conflicting evidence regarding the potential for coordination of the PEG polyether backbone to transition metals [
49,
50,
51]. Initially therefore, we had serious concerns that the PEG-backbone may serve to either retard or completely inhibit the Stille reaction
via complexation of either the palladium catalyst, the alkali metal halide or the organostannane reagent.
The efficiency of the initial series of experiments was determined both by
1H NMR analysis of the PEG-biaryl adduct
17a and, following transesterification, by the isolated yields of the biaryl methyl ester
18a and monoaryl ester
19a (resulting from no cross-coupling with
15a). Reactions were conducted in toluene, THF or DMF, with either a Pd(0) or Pd(II) catalyst (0.1 equiv.) in the presence or absence of LiCl (10 equiv.). A three-fold excess of tributyl phenylstannane
16 was used relative to the PEGsupported electrophile
15a. Additional parameters that were modified included temperature, reaction time and reaction concentration. solvent (
Table 1, entries 3 and 6-9) under standard conditions of
15a (20 mM) and 80°C. The results show that DMF is the most superior of the solvents tested for the PEGsupported Stille reaction. Similar to the case with THF, PdCl
2(PPh
3)
2 is the best catalyst (
Table 4, entries 3 and 6) with considerable amounts of the transesterified iodide
19a being recovered after reaction with Pd(0) (58%). In addition, by leaving the reaction to run for 48 h rather than the preliminary 24 h, a considerable increase in the observed biaryl adduct
18a is achieved (89 and 97% respectively) (
Table 4, entries 3 and 8).
In a recent fluorous-phase approach to the Stille reaction, between aryl tin reagents and aryl halides, it was observed that by using LiCl as an additive that the cross-coupling efficiencies were significantly enhanced [
44]. However, the positive effect of LiCl on our PEG-supported Stille variant seems to be only marginal (compare
Table 4, entries 3 and 7). This result is more in line with previous reports which found that LiCl does not usually enhance the reaction between trialkyl tin reagents with aryl halides, but is generally more useful with aryl triflate electrophiles [
52,
53].
The summary of these results is that the use of PdCl2(PPh3)2 in the presence of LiCl in DMF at a reaction concentration of 20 mM for 48 h, or 10 mM for 24 h, gives excellent yields for the monomethoxy-PEG5000-supported variant of the Stille cross-coupling reaction (97 and 98% respectively).
This liquid-phase approach imparts a number of significant advantages over its solution-phase counterpart. The toxic tributyl tin derivatives are easily separated from the PEG-biaryl adduct
17a by precipitation of the polymer-support into either
iso-propyl alcohol or diethyl ether. Polymer recovery was >99% in each case. In addition, the homocoupled biaryl side-product
20, an incontrovertible product in the solution-phase reaction [
39,
40], does not contaminate the soluble-polymer supported biaryl product
17a, because the homocoupling side-reaction takes place in solution and thus the contaminant is removed during the precipitation step. Having optimized the liquid-phase Stille reaction conditions on monomethoxy-PEG
5000, we then sought to examine the scope of this cross coupling process in a parallel format. A range of tributyl stannanes,
16 and
21-
28, were reacted with the PEG-supported iodides
15a-
b in a parallel fashion, furnishing the library of biaryl, heterobiaryl and styryl derivatives
18a-
b and
28a-
b to
34a-
b (
Table 5).
The isolated yields of all library members were good to excellent (69 - 99%) showing that under our pre-optimized conditions vide supra, that the scope of the PEG-supported variant of the Stille reaction is quite broad. Following passage down a short pad of silica gel, the library members (18a-b, 28a-b to 34a-b) were each isolated in > 95% purity.
In each case (
Table 5, entries 1-8) the yield of the
ortho-isomer was lower than its corresponding
para-congener. We rationalize that this phenomenon may be a result, at least in part, of the steric effect imparted by the PEG-backbone polymer chain during ligand transfer. However this effect followed no definitive trend related to the size of the moiety being transferred. suggests that the process involved may be more complex than simple steric retardation. While, in this report, no attempt has been made to re-optimize the reaction conditions to improve the yield of the
ortho-isomers, it is speculated that future studies directed to resolve this issue would involve simply increasing the number of equivalents of the stannane and/or palladium catalyst, whilst perhaps using more dilute reaction conditions.






{kind=link}
{kind=link}
{kind=link}
{kind=link}











