Introduction
(
3R,4S)-4-Methyl-3-hexanol (
I) is the pheromone of the ant
Tetramorium impurum. In 1981, Pasteels et al. isolated this compound together with 4-methyl-3-hexanone, from the heads of the all adult castes of this ant [
1]. The absolute configuration of
I was determined to be
3R,4S by GLC analysis of its (S)-α-acetoxypropionate. On the other hand, the ant
Manica mutica uses (S)-4-Methyl-3-hexanone (
II) as an alarm pheromone [
2]. So far a few syntheses of both
I [
3] and
II [
4,
5,
6] have been reported in literature. However, it is observed that both the compounds possess a chiral methyl branching with an α-oxygenated carbon centre that is found in many other natural products. Hence, we have chosen these two compounds as representative synthetic targets in order to demonstrate the usefulness of a simple synthetic strategy developed by us to generate such functional moieties
via a chiron approach. We felt that (
R)-2,3-cyclohexylideneglyceraldehyde (
1), which had earlier been prepared by us on a practical scale [
7] should be an ideal chiron for this endeavour.
Results and Discussion
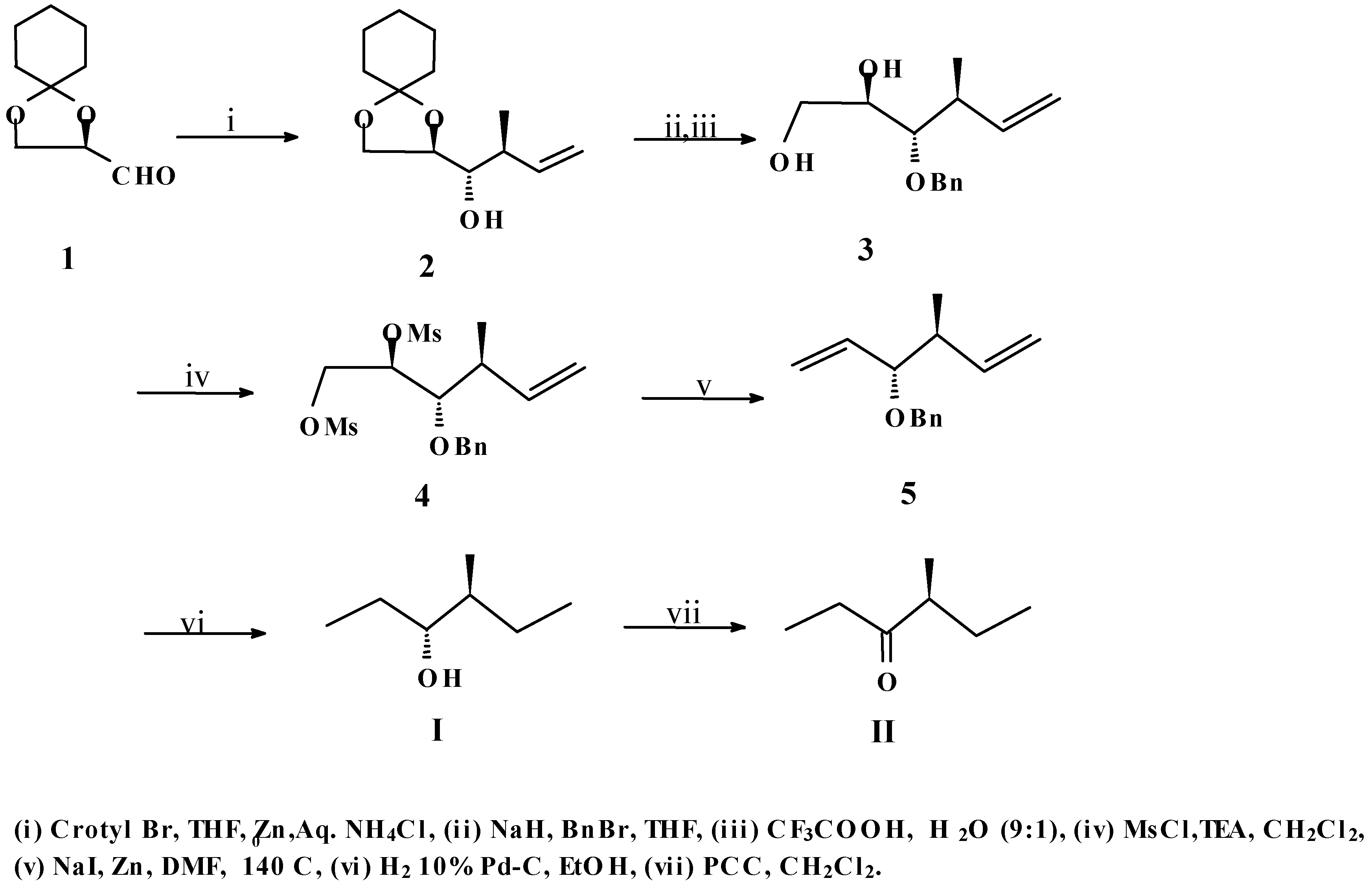
In an earlier communication,
1 has been crotylated by us to afford reasonably good amounts of compound
2 [
8]. We felt that
2, having the required carbon skeleton along with the correct stereochemistry at the appropriate positions as found in both
I and
II, had good prospects to be a useful precursor for the synthesis of both target molecules. To prove this point, compound
2 was first benzylated and subsequently deketalised to produce the diol
3. Dimesylation of both the free hydroxyl groups of
3 afforded compound
4. Next, the elimination of the vicinal disulfonyloxy groups of
4 was carried out by treatment with excess Zn dust and NaI following a reported procedure [
9] to give the diene
5. This step was followed by catalytic hydrogenation of
5 along with concomitant debenzylation to yield the pheromone
I. This compound was subsequently oxidized with PCC [
10] to afford target
II (
Scheme 1).
Experimental
General
All boiling points are uncorrected. The IR spectra were scanned with a Perkin-Elmer model 837 spectrophotometer. The 1H-NMR spectra were recorded in CDCl3 on a Bruker-200 MHz instrument. All the anhydrous reactions were carried out under an argon atmosphere using freshly distilled solvents. In all the cases solvents were later removed under reduced pressure. Unless otherwise mentioned, the organic extracts were dried over anhydrous Na2SO4.
(2R, 3S, 4S)-3-Benzyloxy-4-methyl-5-hexene-1,2-diol (3)
Compound 2 (3.39 g, 0.015 mol) in THF ( 50 mL) ) was added dropwise over a period of 1.5 h to a stirred suspension of NaH (720 mg of a 50% suspension in oil, 0.015 mole, washed with hexane) in THF (100 mL). The mixture was gently heated to 50°C for 1 h more. Benzyl bromide (2.6 g, 0.015 mol) in THF (50 mL) was then added dropwise to the stirred mixture at 50°C over a period of 3 h. After stirring for 30 min more, the mixture was treated with water. Usual extraction and solvent removal afforded the crude residue in quantitative yield. This was dissolved in 90% aqueous trifluroacetic acid (20 mL) and the mixture was stirred for 6 hr at 0°C and then diluted with water. The mixture was extracted with CHCl3. The combined organic layer was washed with water to remove residual acid, then with brine and finally dried. Solvent removal and chromatography of the residue (silica gel, 0-5% MeOH in CHCl3 ) afforded pure 3 (2.68 g, 75.5%). [α]23 + 27.8 (c 1.05, CHCl3); IR (film) 3500, 3075, 3005, 1365, 1178, 1095, 910 cm-1; 1H-NMR (CDCl3 ) δ 1.12 (d, J = 6.6 Hz, 3H), 2.4-2.6 (m, 1H), 3.16 (bs, D2O exchangeable, 2H), 3.4-3.6 (m, 1H), 3.86-4.2 (m, 3H), 4.4-4.6 (m, 2H), 5.0-5.2 (m, 2H), 5.5-5.7 (m, 1H), 7.2-7.5 (m, 5H). Anal. Calcd. For C14H20O3 : %C 71.16, %H 8.53; Found: %C 70.88, %H 8.75.
(2R, 3S, 4S)-1,2-Dimesyloxy-3-benzyloxy-4-methyl-hex-5-ene (4)
Methanesulfonyl chloride (1.1 ml, 14.2 mmol) was added at 0°C over a period of 30 min. to a solution of compound 3 (1.9 g, 7 mmol) and triethylamine (5 mL) in CH2Cl2 (50 mL). The mixture was stirred for 4 h and then treated with water. Usual extraction with CH2Cl2 was followed by successive washing of the organic layer with dilute aqueous HCl, water and brine. Solvent removal afforded the residue 4 in sufficiently pure form. This was used in the next step without being purified further. Yield: 2.6 g, 83.6%. [α]23 + 26.45 (c 1.5, CHCl3); IR (film) 3032, 3005, 1654, 1360, 1178, 1003, 910 cm-1; 1H NMR (CDCl3 ) δ 1.12 (d, J = 6.6 Hz, 3H), 2.4-2.6 (m, 1H), 3.03 ( s, 3H), 3.09 (s, 3H), 3.72 (t, J=6.0 Hz, 1H), 4.4-4.6 (m, 5H), 5.0-5.2 (m, 2H), 5.5-5.7 (m, 1H), 7.2-7.5 (m, 5H).
(3S, 4S)-3-Benzyloxy-4-methyl-hex-1,5-diene (5)
A stirred mixture of 4 (2g, 5.1 mmol) in DMF (50 mL), Zn dust (18 g, 0.275 g-atom) and NaI (38 g, 0.255 mol) was heated at 140°C for 5 hr. It was cooled to room temperature, mixed with ether and filtered. After washing the residue thoroughly with ether and water, the aqueous extract was thoroughly extracted with ether. The combined organic extract was washed with water, brine and then dried. Solvent removal and column chromatography (silica gel, 0-15% ether in hexane) of the residue afforded pure 5. Yield: 530mg (51.2%). [α]23 11.25 (c 1.64, CHCl3); IR (film) 3032, 3005, 1654, 995, 925; 1H NMR (CDCl3 ) δ 1.02 (d, J = 6.6 Hz, 3H), 2.4-2.6 (m, 1H), 4.25 (m, 1H), 4.57 (m, 2H), 5.0-5.2 (m, 4H), 5.5-5.7 (m, 2H), 7.2-7.5 (m, 5H). Anal Calcd. For C14H18 O: %C 83.12, %H 8.97; Found: %C 83.39, %H. 9.25.
(3R, 4S)-4-Methyl-3-hexanol (I)
A stirred mixture of 10% Pd-C (100 mg) in a solution of
5 (500 mg) in ethanol (50 mL) (containing a few drops of acetic acid) was subjected to hydrogenation for 40 hr under positive pressure of hydrogen and then filtered. After solvent removal under reduced pressure, the residue was mixed with ether. The organic layer was washed with water and brine and then dried. Solvent removal and column chro-matography (silica gel, 0-20% ethyl acetate in hexane) of the residue afforded pure
I (400 mg, 88%). [α]
23 + 2.9 (c 2.8, CHCl
3); lit. [
3] [α]
22 + 2.5 (c 2.07, CHCl
3); IR (film) 3400, 2968, 2935, 1460, 1380, 1150, 1105, 1040, 970cm
-1;
1H-NMR (CDCl
3 ) δ 0.75-1.10 (9H, m), 1.15-1.8 (m, 5H), (3.0, bs, D
2O exchangeable, 1H), 3.3-3.4 (m, 1H).
(S)-4-Methyl-hexan-3-one (II)
To a stirred suspension of PCC (860 mg, 4 mmol) in CH
2Cl
2 (60 mL) was added a solution of
I (300 mg, 2.6 mmol) in CH
2Cl
2 (50 mL). The mixture was stirred at room temperature for 2.5 hr until the disappearance of
I was complete (TLC). Dry ether (80 mL) was added to the stirred mixture, which was then filtered through a pad of Florisil. Removal of solvent from the filtrate and column chromatography (silica gel, 0-15% ether in hexane) of the residue afforded pure
II (236 mg, 69%). [α]
23 + 30.7 (c 3.4, Et
2O); lit. [
5] [α]
23 + 30.2 (c 3.7, Et
2O, 94% ee); IR (film) 2970, 2944, 2900, 1722, 1470, 1380, 1179, 1105, 1036, 980cm
-1;
1H-NMR (CDCl
3 ) δ 0.75-1.1 (9H, m), 1.2-1.5 (m, 2H), 2.3-2.6 (m, 3H).

{kind=link}
