Abstract
Several new pyrido[2,3,4-kl]acridines were synthesized by reacting naphthoquinone, juglone or cyclohexan-1,3-dione with β,β’-diaminoketones in a biomimetic reaction. The structure of all new compounds was elucidated by NMR and MS spectroscopy. Electrophilic substitution, mainly nitration, of the various compounds was undertaken and the substitution positions determined. A series of derivatives was prepared and their cytotoxicity towards P-388 mouse lymphoma cells analysed. The most cytotoxic derivatives were found to have IC50’s of 0.05 and 0.1 ug/ml.
Keywords:
Pyrido[2; 3; 4-kl]acridines; Biomimetic synthesis; NMR; Electrophilic nitration; Cytotoxicity Introduction
Over the last 15 years more than 50 pyridoacridine alkaloids, based on the 4H-pyrido[2,3,4-kl]acridone (1) skeleton (Figure 1), have been isolated from marine organisms [1]. Almost all natural pyridoacridines have been reported to possess significant cytotoxicity against cultured tumor cells [1]. This motivated us to synthesize and study some of the compounds of this group. In 1993 we reported a biomimetic synthesis of the pyrido[2,3,4-kl]acridine ring system by the reaction of β,β’-diamino-ketones with a variety of quinones and diketones [2][3]. Using this method we synthesized the marine alkaloid ascididemin (2)[4], eilatin (3)[3] (Figure 1) and also new pyridoacridine skeletons such as benzoascididemin and isoeilatin [5].
Here we report a biomimetic synthesis of additional new pyridoacridines and a study of their reactions with electrophiles or amines (in the case of the quinoneimines). Most of the new pyridoacridines were tested for in-vitro activity against tumor cells and some of them were found to be highly cytotoxic.
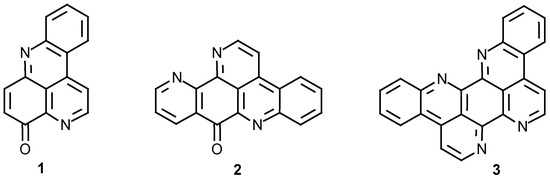
Figure 1.
Figure 1.
Results and Discussion
Several new pyridoacridines were synthesized in a two step reaction of β,β’-diaminoketones with quinones. Thus, addition of 2,2’-diaminobenzophenone 4a or 4b to 1,4-naphthoquinone afforded in the first step the arylaminonaphthoquinones 5a and 5b respectively, in approximately 50% yield (Scheme 1).
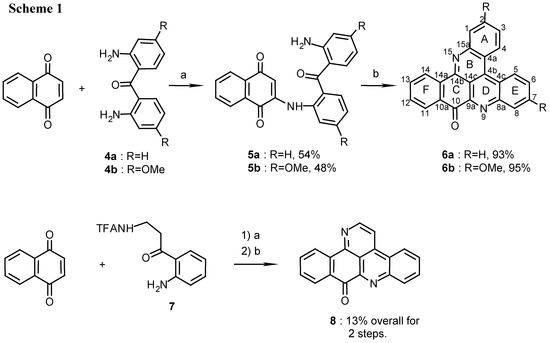
Scheme 1.
Scheme 1.
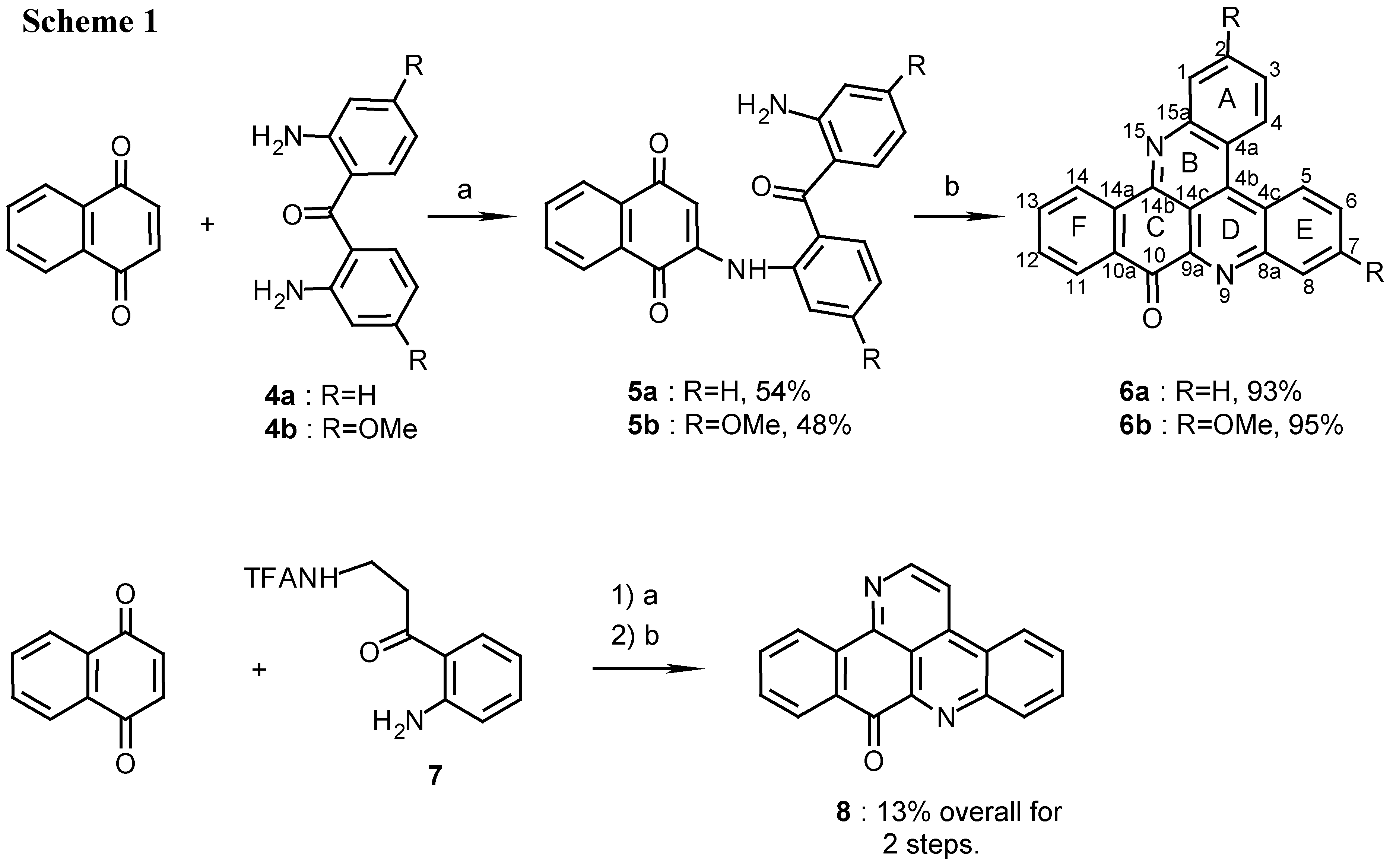
Reagents and conditions: (a) CeCl 3·7H2O, Air, E tOH, reflux, 9 h; (b) 25% NH 4OH, MeOH, R.T., 7 days.
The reaction took place in the presence of catalytic amounts of cerous chloride while air was bubbled through the solution to oxidize the intermediate hydroquinone [6,7]. In the second step, treatment of compounds 5a and 5b in methanol with NH4OH at room temperature for 7 days gave the corresponding compounds 6a and 6b in over 90% yield (Scheme 1).
The structures of 6a and 6b, possessing the required molecular ions (m/z 332 and 392, respectively) were confirmed by 1D and mainly COSY, HMQC and HMBC 2D-NMR spectra (See Table 1 for the HMBC correlations and the Experimental section for the proton and carbon chemical shift assignments).
Characteristic were the resonances of C-10 and C-14b of the quinoneimine system (ring C) and the down-field proton resonances of the spatially close protons H-4 and H-5 (δH 9.09 and 9.18 ppm, respectively, for 6a and δH 8.90 and 8.98 ppm, respectively, for 6b)[5].
Three four-spin systems were observed in the 1H-NMR spectrum of 6a belonging to rings A, E and F. Rings A and E, bearing the spatially close H-4 and H-5 protons, were distinguished from ring F by NOE measurements. The differentiation between rings A and E was achieved from an NOE between H-1 and H-14 (about 3.7Å apart). This NOE was also the key for determining the structure of the nitration products 21, 23a and 23b, as described below. A second reaction that was performed with naphthoquinone was its reaction with TFA-kynuramine (7) [4] (Scheme 1). This reaction afforded 9H-benzo[i]pyrido[2,3,4-kl]acridin-9-one (deaza-ascididemin, 8), earlier synthesized by Zjawiony by a four step reaction [8]. The structure of compound 8 (m/z 282) was confirmed by its NMR data (see Experimental) and comparison with the data in the literature [8].
A second naphthoquinone that was tested was juglone. Reacting juglone (5-hydroxy-1,4-naphthoquinone) with diaminobenzophenone 4a afforded, in a regioselective reaction, a single addition product 9 in 80% yield (Scheme 2).
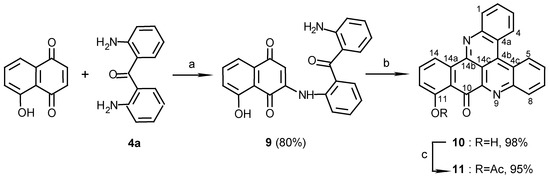
Scheme 2.
Scheme 2.
Reagents and conditions: (a) CeCl 3·7H2O, Air, E tOH, R.T., 3 days; (b) E t 3N, MeOH, R.T., 10 days; (c) Ac 2O, pyridine, R.T., 24 h.
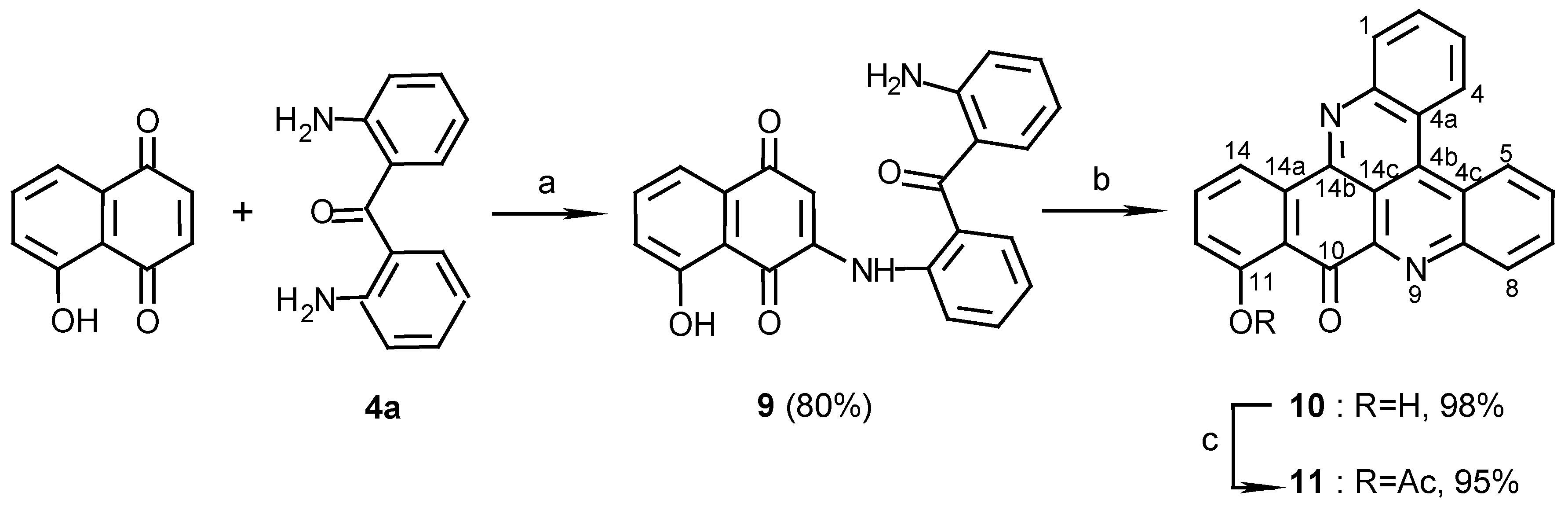
The orientation of this addition was defined by the structure determination of compound 10, obtained in a second step by stirring compound 9 in methanol with Et3N. A key HMBC correlation in the structure elucidation was the one between C-14b (δ 147.5) and H-14 (δ 8.73). For other correlations that assisted with the structure determination see Table 1. The regioselectivity of nucleophilic additions of amines to juglone was observed before by Thomson [9] in the reactions of aniline with the juglone derivatives 5-acetoxy or 5-methoxy-1,4-naphthoquinones. Performing the second step of the latter reaction with ammonia, rather than Et3N, as used for the preparation of compounds 6a and 6b, caused unexpectedly the disappearance of the C-10 carbonyl group. Moreover, acetylation of the obtained pyridoacridine (12) (Scheme 3) gave a mono- (13a) and a diacetate (13b). It is suggested that the carbonyl group of compound 10 is replaced in compound 12 by an imine and indeed, treatment of 10, obtained with the Et3N, with NH3 gave compound 12. The position of the imine group was defined by a HMBC experiment of compound 13a namely from correlations between the 11-hydroxylic proton and carbons C-10a, C-11, and C-12 of ring F (see Table 1).

Table 1.
Long-range CH correlations observed in the HMBC experiments of the benzopyridoacridines
| C# | H# of correlated protons | |||||||
|---|---|---|---|---|---|---|---|---|
| 6a | 6b | 10 | 12 | 13a | 20 | 23a | 23b | |
| 1 | 3 | 3 | 3 | 3 | 3 | |||
| 2 | 4 | 4, OMe | 4 | 4 | 4 | 4 | 4, OMe | 4, OMe |
| 3 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| 4 | 2 | 2 | 2 | 2 | 2 | |||
| 4a | 1, 3 | 1, 3 | 1, 3 | 1, 3 | 1, 3 | 1 | ||
| 4b | 4, 5 | 4, 5 | 4, 5 | 4, 5 | 4, 5 | 4, 5 | 4, 5 | 4, 5 |
| 4c | 6, 8 | 6, 8 | 6, 8 | 6, 8 | 6, 8 | 6, 8 | 6 | 6 |
| 5 | 7 | 7 | 7 | 7 | 7 | |||
| 6 | 8 | 8 | 8 | 8 | 8 | 8 | ||
| 7 | 5 | 5, OMe | 5 | 5 | 5 | 5 | 5, OMe | 5, OMe |
| 8 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 |
| 8a | 5, 7 | 5 | 5, 7 | 5, 7 | 5, 7 | 5, 7 | 5 | 5 |
| 9a | ||||||||
| 10 | 11 | 11 | 11 | 11 | 11 | |||
| 10a | 12, 14 | 12, 14 | 12, 14 | 12, 14 | 12,14,OH | 12, 14 | 14 | 12 |
| 11 | 13 | 13 | 13 | 13 | 13, OH | 13 | 13 | 13 |
| 12 | 14 | 14 | 14 | 14 | 14, OH | 14 | 14 | |
| 13 | 11 | 11 | 11 | 11 | 11 | |||
| 14 | 12 | 12 | 12 | 12 | 12 | 12 | 12 | |
| 14a | 11, 13 | 11, 13 | 13 | 13 | 13 | 11, 13 | 11, 13 | 11, 13 |
| 14b | 14 | 14 | 14 | 14 | 14 | 14 | 14 | |
| 14c | ||||||||
| 15a | 2, 4 | 4 | 2, 4 | 2, 4 | 2, 4 | 2, 4 | 4 | 4 |
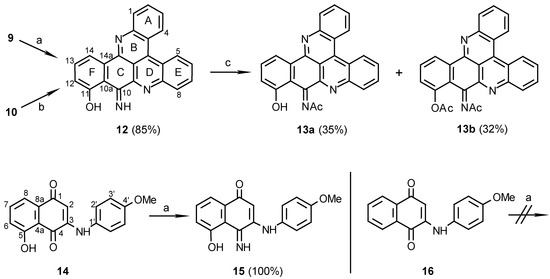
Scheme 3.
Scheme 3.
Reagents and conditions: (a) 25% NH 4OH, MeOH, R.T., 7 days; (b) MeOH / NH 3 , R.T., 14 days; (c) Ac 2O, pyridine, R.T., 24 h.
Quinoneacetimide systems such as ring C in compounds 13a and 13b are stable, e.g. the reported simple acetimides of naphthoquinones and benzoquinones [10]. In contrast, simple quinoneimines are unstable and were seldom isolated [10]. Thus, it was interesting to find that the quinoneimine moiety of compound 12 is stable as was also found to be the case of the natural pyridoacridine calliactine [11] whose structure was determined recently [12]. In both compound 12 and calliactine the hydroxyl group in the β position relative to the imine group seems to stabilize the quinoneimine by a hydrogen- bond. Another example for the latter behaviour was seen in compound 14, synthesized from juglone and p-anisidine (Scheme 3). Compound 14 in methanol with aqueous ammonia, yielded the quinoneimine 15, while compound 16 [13] without the β-OH group, did not form the quinoneimine under the same conditions. These results proved the necessity of a hydroxyl group β to the ketone for the imine formation and also suggest that the quinoneimine ring could be obtained, in the synthesis, before the rings closure of compound 10.
A major target in the present investigation was the study of the electrophilic substitution reactions of pyridoacridines and dihydropyridoacridines for the preparation of derivatives for structure activity relationship studies. It was decided to start with nitration as the nitro groups are easily transformed to other functional groups. Investigating a variety of nitration conditions (HNO3-TFA, HNO3-H2SO4 and NO2BF4 in CH3CN) brought to the best conditions, namely, the use of HNO3-H2SO4, 1:1 vide infra.
The first substrate for nitration was dihydropyridoacridine 17a. Compounds 17a and 17b were obtained in quantitative yields by condensation of compounds 4a or 4b with 1,3-cyclohexanedione (Scheme 4). Nitration of compound 17a gave, after 1 hour, two mononitro isomers 18a and 18b and three dinitroisomers 19a, 19b and 19c after 12 hours of reaction at room temperature.
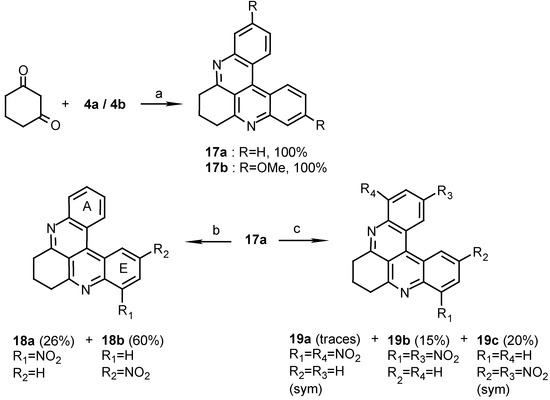
Scheme 4.
Scheme 4.
Reagents and conditions: (a) AcOH / conc. HCl (99.5:0.05), 3-Nitrobenzenesulfonic acid Sodium salt, reflux, 2 h.; (b) HNO 3 / H2SO4 (1:1), R.T., 1 h.; (c) HNO3 / H2SO4 (1:1), R.T., 12 h.
The structures of the different isomers were determined by 1H-NMR and COSY experiments. In all the products the nitro group(s) are in ring A (or E) in positions para or ortho to the nitrogen of the attached pyridine ring (para or ortho positions). The yields of the nitration products indicate that the para position in ring A (and E) is more reactive than the ortho one under the conditions used. Nitration of quinoline with nitric acid in sulphuric acid at 0°C was reported to yield 5- and 8-nitroquinoline in a ratio of 52% to 48% respectively [14]. The nitration experiments of compound 17a show that the para position of ring A (and E) in the dihydropyridoacridine is more reactive than position- 6 of quinoline, under the same conditions [15]. Positions 4 and 5 in the pyridoacridine, which can be compared to the reactive position- 5 of quinoline, are blocked by steric interference and therefore are not substituted.
The nitration of compound 6a afforded a mono-nitro product 20 in 53% yield after 12 hours at room temperature. Because of the absence of long range CH-correlations in the NMR experiments between atoms of rings A or E and F to ring C it was difficult to determine whether the nitro group is attached to ring A or E. However, the nitration position, C-3 on ring A, could be determined from a NOE between H-1 and H-14 (2%), which are ca.3.7 Å apart (see Scheme 5). It was found by 1D and 2D NMR experiments (for HMBC correlations see Table 1) that the nitration went to the para position of ring A as was found for the nitro derivatives of compound 17a which were obtained in higher yields (compounds 18b, 19b and 19c).
Catalytic hydrogenation of compound 20 with 5% Pd-C in AcOH / TFA afforded the amino derivative 21.
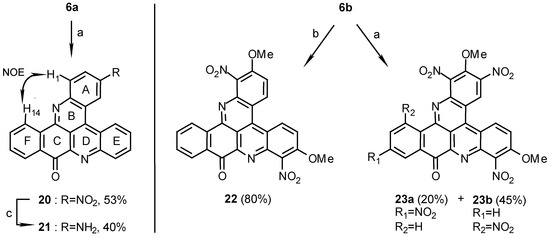
Scheme 5.
Scheme 5.
Reagents and conditions: (a) HNO3 / H2SO4 (1:1), R.T., 12 h.; (b) HNO3 / H2SO4 (1:1), R.T., 1 h.; (c) AcOH / TFA (1:2), 5% Pd/C, H2, 3 Atm, R.T., 1 h.
Nitration of compound 6b, the electron richer 2,7-dimethoxy derivative of 6a, gave a dinitro derivative 22 after 1 hour and two tetra nitro isomers 23a and 23b (Scheme 5) after 12 hours of reaction at room temperature. That the two nitro groups in 22 substituted C-1 and -8, ortho to the quinoline-nitrogen, was clear from the two AB- systems seen in the 1H-NMR spectrum along with the aromatic four- proton system (Experimental).
The structures of 23a and 23b were also determined by 1D and 2D-NMR experiments (for HMBC correlations see Table 1). In compounds 23a and 23b only one of rings A or E was attacked by the electrophile at the para position. The structures of compounds 23a and 23b are tentatively suggested on the basis of the structure of compound 20 as depicted in Scheme 5. Because of the nitro groups at the ortho positions, it was impossible to prove by NOE that the substitution is at the para position of ring A (as in the case of compound 20). In addition to the nitration of rings A and E, ring F in 23a and 23b was also substituted due to long range activation by the methoxyl groups.
The second electrophilic substitution undertaken was bromination. Compounds 6a and 8 did not react with Br2 in CH2Cl2, Br2 in AcOH or NBS in CH2Cl2 and it seems that severe conditions may be needed in order to brominate these compounds. The use of Lewis acids as catalyst precipitated the compound. Quinoneimine 24 [2] afforded the dibromo derivative 26 (Scheme 6) by reaction with Br2 in AcOH; a reaction known for quinones [16]. Compound 26 like other dibromoquinones is expected to afford a variety of derivatives by cycloaddition reactions and by reactions with amines and thiols [13,16,17].
Several of the synthesized new pyridoacridines were tested for in-vitro cytotoxicity against P-388 mouse lymphoma cells (Table 2). It was found that compounds 6a and 8 are more cytotoxic than other reported natural pyridoacridines for which IC50 values of 0.1- 0.4 ug/ml were found [1a]. The cytotoxicity of compound 6a, the electron richer dimethoxy derivative 6b and the electron poorer nitro derivatives 21 and 23b, as well as compound 12 and its acetate derivatives 13a and 13b is lower.
Table 2.
In- vitro cytotoxicity against P-388 mouse lymphoma cells
| Compound | 6a | 6b | 8 | 12 | 13a | 13b | 24 | 25 | 20 | 21 | 23b |
|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (ug/ml) | 0.05 | 10 | 0.1 | >10 | 5 | 10 | 1 | 2.5 | 0.5 | 10 | 2.5 |
Oxidation of compounds 17a and 17b, with cerium ammonium nitrate, afforded benzopyridoacridones 24 and 25, respectively, in high yields (Scheme 6). Amination of the latter quinoacridones (24 and 25) with several primary amines in ethanol afforded two kinds of derivatives; monoamination products (compounds 27a- 32a) and symmetric diamination ones (compounds 27b- 31b and 33b). The diamination products were separated easily from the monoamination products, in each reaction, by silica gel chromatography (eluting with chloroform- methanol mixtures). The diamination products are more polar than the monoamination products and the starting material. Performing the amination in acetonitrile instead of ethanol afforded mainly the monoamination products 27a and 30a with only traces of the diamination products 27b and 30b (Scheme 6). Another derivative, prepared from 24, was compound 32a (Scheme 6), which was derived from 24 by hydrazoic acid in methanol under conditions reported for synthesis of aminonaphthoquinones [18].
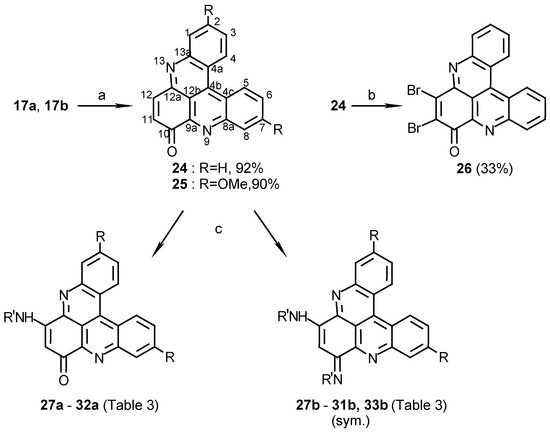
Scheme 6.
Scheme 6.
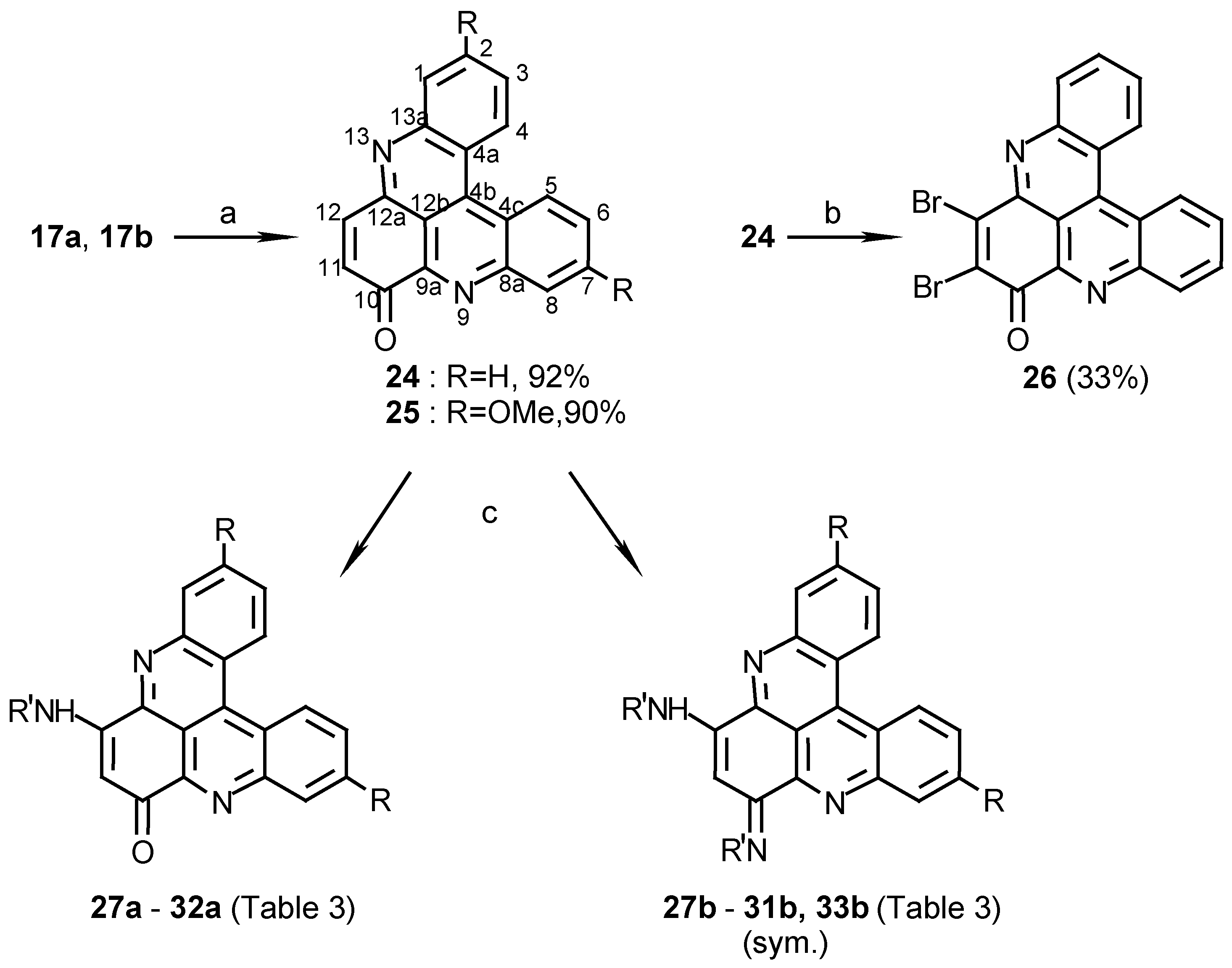
Reagents and conditions: (a) CAN, CH3CN, reflux, 10 min.; (b) Br2, AcOH, 80°C, 2 h.; (c) see Table 3 and Experimental.
The structures of the diamination products were determined by their 1H- and 13C-NMR spectra which indicated symmetric structures (see Experimental). The substitution site in the monoamination product 27a was determined by a HMBC experiment which showed correlations between proton H-11 and carbons C-9a and C-12a and correlations between the substituent NH- proton and carbons C-12a and C-11 (Scheme 7). As seen in Table 3, the symmetric diamination products are more cytotoxic than the monoamination ones and most of the diamination products are more toxic than their parent compounds 24 and 25 (Table 2). Most active are the symmetric derivatives obtained with isobutylamine and methylamine (compounds 27b- 29b) while the more lypophilic derivative obtained with dodecylamine (compound 31a) (as well as 31b) and the more hydrophilic derivative obtained with serinol (compound 33b) are less active.
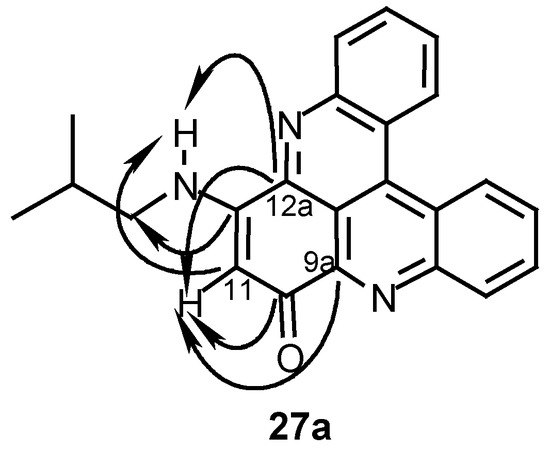
Scheme 7.
CH correlations in HMBC experiment of compound 27a
Scheme 7.
CH correlations in HMBC experiment of compound 27a
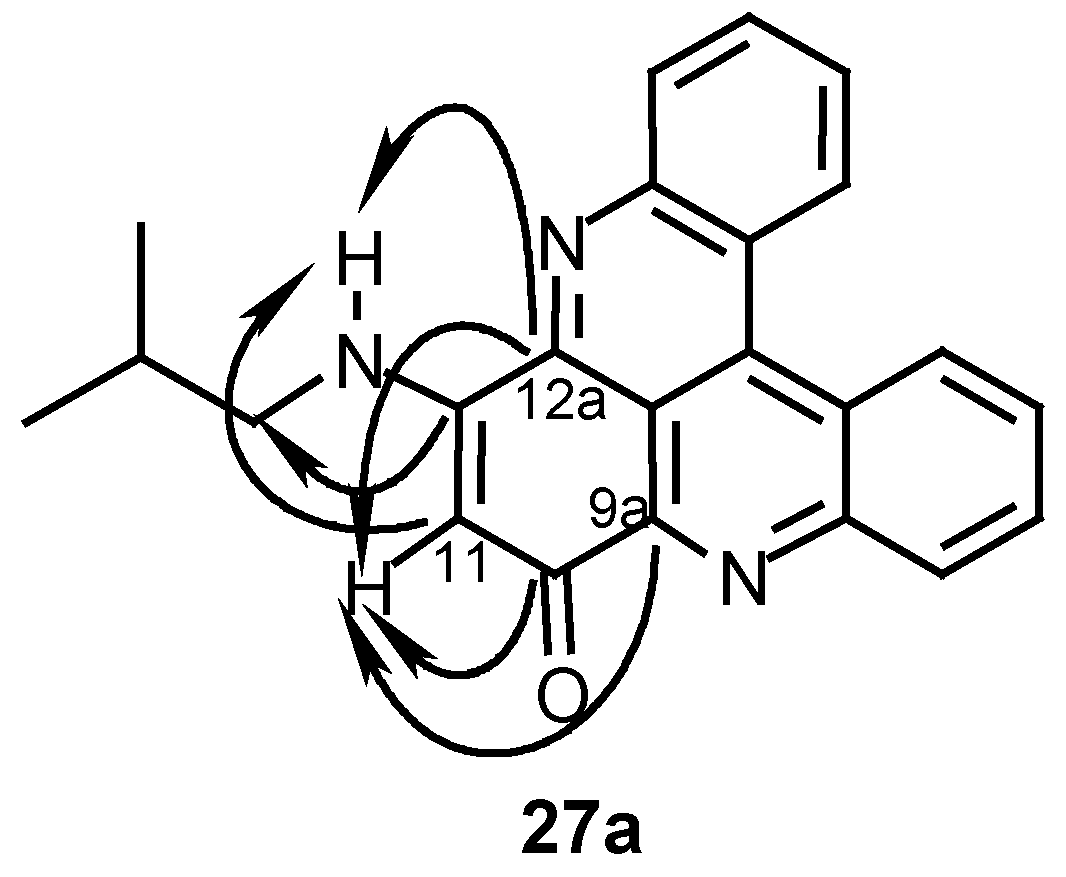
Table 3.
Amination products of compounds 24 and 25 with amines R’NH2 and their in-vitro cytotoxicity against P-388 mouse lymphoma cells.
| Compound | R | R’ | Yield (%) | IC50 (ug/ml) | |||
|---|---|---|---|---|---|---|---|
| a | b | a | b | ||||
| 27 | H | (CH3)2CHCH2 | 36 | 38 | 0.25 | 0.1 | |
| 28 | OCH3 | (CH3)2CHCH2 | 19 | 37 | 1 | 0.1 | |
| 29 | H | CH3 | 46 | 28 | 0.25 | 0.1 | |
| 30 | H | CH3OC6H4 | 55 | 38 | 1 | 0.5 | |
| 31 | H | CH3(CH2)11 | 24 | 18 | 2.5 | 0.5 | |
| 32aa | H | H | 76 | - | 1 | - | |
| 33 | H | (HOCH2)2CH | - | 50 | - | >10 | |
a 32a is the reaction product of 24 with hydrazoic acid (see Experimental).
Experimental
General
Commercially available reagents were purchased from standard chemical suppliers and were used without further purification. 2(N)-(4-methoxyaniline)-1,4-naphthoquinone (16) was synthesized by a literature method [13]. IR spectra (KBr disks) were recorded on a Nicolet 205 FT-IR spectrophotometer. MS and HRMS spectra were recorded on a Fisons Autospec Q instrument. 1H-NMR and 13C-NMR spectra were recorded on a Bruker ARX-500 or a Bruker AMX-360 spectrometer. NOE experiments and 2D NMR spectra (COSY, HMQC and HMBC) were recorded on a Bruker ARX-500 instrument using standard pulse sequences. TLC was performed on Merck precoated Kieselgel 60 F254 plates. Column chromatography was performed using Silica gel 60 H (Merck) unless otherwise stated. The silica was washed with methanol, before use, in a Soxhlet apparatus. In all cases silica gel chromatography was performed with vacuum.
2,2’-Diaminobenzophenone (4a):
Compound 4a was performed from 2,2’-dinitrobenzophenone by hydrogenation, instead of the literature method of reduction with iron powder [19]. 2,2’-Dinitrobenzophenone [19] (250 mg, 0.92 mmol) was dissolved in dichloromethane (40 mL), 5% Pd/C (100 mg) was added and the solution was shaken in a Parr apparatus under H2 (3 atm) for 1 hour. The catalyst was filtered off and the solvent evaporated to afford 4a (195 mg, quantitative yield), yellow crystals (from 80% aqueous methanol), m.p. 134°C (lit [19], 134-135 °C).
2,2’-Diamino-4,4’-dimethoxybenzophenone (4b):
4b was prepared from 2,2’-Dinitro-4,4’-dimethoxybenzophenone [20] (250 mg, 0.75 mmol) under the same conditions as described for the synthesis of 4a. The product was recrystallized from methanol to afford 4b (185 mg, 90%): m.p. 138°C (lit [20], 137-138°C).
General procedure for the reaction between arylamines and 1,4-naphthoquinones
The procedure of Pratt [7] was adopted. The corresponding amine (1 equiv.) was dissolved in ethanol, CeCl3·7H2O (0.05 equiv.) was added followed by the naphthoquinone (1.5 equiv.). The resulting red solution was refluxed for 9 h. During this time air saturated with ethanol (prepared by passage of air through hot ethanol to avoid evaporation of the ethanol) was bubbled through the reaction mixture. After cooling, the ethanol was evaporated and the residue purified by chromatography on silica gel (eluting with chloroform/methanol) to afford the desired amination product.
2(N)-(2,2’-diaminobenzophenone)-1,4-naphthoquinone (5a):
4a (450 mg, 2.1 mmol) was dissolved in ethanol (20 mL) and reacted with naphthoquinone (500 mg, 3.2 mmol) by the above described general procedure. The crude mixture containing the product and starting materials was chromatographed by two subsequent silica gel columns (eluting with CHCl3/MeOH, 50:1) to afford 5a (420 mg, 54%), red prisms (from EtOH), m.p. 208°C; MS (EI); m/z: 368 (100) [M+, C23H16N2O3]; IR: = 1667, 1611, 1568, 1512, 1292, 1246 cm-1; 1H-NMR (CDCl3): δ = 6.56 (s, 1 H, 3-H), 6.59 (t, J=7.5 Hz, 1 H, 12’-H), 6.72 (d, J=7.5 Hz, 1 H, 10’-H), 7.21 (t, J=7.5 Hz, 1 H, 11’-H), 7.29 (t, J=7.5 Hz, 1 H, 4’-H), 7.40 (d, J=7.5 Hz, 1 H, 6’-H), 7.52 (d, J=7.5Hz, 1 H, 13’-H), 7.55 (t, J=7.5 Hz, 1 H, 5’-H), 7.63 (d, J=7.5 Hz, 1 H, 3’-H), 7.65 (t, J=7.6 Hz, 1 H, 7-H), 7.75 (t, J=7.6 Hz, 1 H, 6-H), 8.08 (d, J=7.6 Hz, 1 H, 5-H), 8.09 (d, J=7.6 Hz, 1 H, 8-H), 9.30 (br s, 1 H, NH).
2(N)-(2,2’-diamino-4,4’-dimethoxybenzophenone)-1,4-naphthoquinone (5b):
4b (200 mg, 0.74 mmol) was dissolved in ethanol (10 mL) and reacted with naphthoquinone (170 mg, 1.1 mmol) by the general procedure described above. After two subsequent silica gel columns (eluting with CHCl3) compound 5b was isolated (150 mg, 48%): red prisms (from EtOH), m.p. 214°C. – MS (EI); m/z: 428 (100) [M+, C25H20N2O5]; IR: = 1658, 1624, 1576, 1521, 1448, 1244 cm-1; 1H-NMR (CDCl3): δ = 3.81 (s, 3 H, OCH3), 3.90 (s, 3 H, OCH3), 6.14 (s, 1 H, 10’-H), 6.17 (d, J=8.5 Hz, 1 H, 12’-H), 6.62 (s, 1 H, 3-H) , 6.68 (dd, J=8.5, 2.5 Hz, 1 H, 4’-H), 7.11 (d, J=2.5Hz, 1 H, 6’-H), 7.36 (d, J=8.5 Hz, 1 H, 13’-H), 7.44 (d, J=8.5 Hz, 1 H, 3’-H), 7.65 (t, J=7.0 Hz, 1 H, 7-H), 7.74 (t, J=7.0 Hz, 1 H, 6-H), 8.10 (d, J=7.0 Hz, 1 H, 5-H), 8.11 (d, J=7.0 Hz, 1 H, 8-H), 9.90 (br s, 1 H, NH).
10H-benzo[i]quino[2,3,4-kl]acridin-10-one (6a):
5a (400 mg, 1.1 mmol) was added to a solution of 25% aq. ammonia (10mL) in methanol (100mL) and stirred for 7 days at room temperature. During this time the reaction was monitored by TLC, Rf=0.7 and 0.5 for 5a and 6a respectively (petroleum ether/ethyl acetate, 1:1). The reaction mixture was evaporated and the crude product was chromatographed (eluting with CHCl3/MeOH 30:1) to afford 6a (335 mg, 93%), amorphous powder (CHCl3/MeOH, 9:1), m.p. 255°C; Analysis: C23H12N2O (332.1): calcd. C 83.1, H 3.64, N 8.43, found C 82.4, H 3.53, N 8.70; HRMS calcd. for C23H12N2O [M+] 332.0950, found 332.0947; IR: = 1678, 1654, 1562, 1396, 1203 cm-1; 1H-NMR (CDCl3): δ = 7.73 (t, J=7.5 Hz, 1 H, 12-H), 7.83 (t, J=8.0 Hz, 1 H, 3-H), 7.90 (t, J=7.5 Hz, 1 H, 13-H), 7.95 (t, J=8.0 Hz, 1 H, 6-H), 7.95 (t, J=8.0 Hz, 1 H, 2-H), 8.03 (t, J=8.0 Hz, 1 H, 7-H), 8.42 (d, J=8.0 Hz, 1 H, 1-H), 8.52 (d, J=7.5 Hz, 1 H, 11-H), 8.73 (d, J=8.0 Hz, 1 H, 8-H), 9.09 (d, J=8.0 Hz, 1 H, 4-H), 9.11 (d, J=7.5 Hz, 1 H, 14-H), 9.18 (d, J=8.0 Hz, 1 H, 5-H); 13C-NMR (CDCl3): δ = 115.8 (s, C-14c), 122.4 (s, C-4a), 124.2 (s, C-4c), 125.9 (d, C-14) , 127.0 (d, C-5), 127.1 (d, C-4), 127.9 (d, C-11), 128.1 (d, C-3), 129.9 (d, C-6), 130.8 (d, C-2), 130.9 (d, C-7), 131.0 (d, C-1), 131.0 (d, C-12), 132.2 (s, C-10a), 132.4 (d, C-8), 134.7 (d, C-13), 136.0 (s, C-14a), 136.1 (s, C-4b), 145.4 (s, C-9a), 147.2 (s, C-15a), 147.3 (s, C-8a), 147.7 (s, C-14b), 182.2 (s, C-10).
2,7-Dimethoxy-10H-benzo[i]quino[2,3,4-kl]acridin-10-one (6b):
5b (150 mg, 0.35 mmol) was treated with ammonia in methanol by the same procedure described for the synthesis of 6a. The product (6b) was obtained after chromatography (eluting with chloroform/methanol, 30:1) (130 mg, 95%): amorphous powder (CHCl3/MeOH, 8:2), m.p. 296°C; Analysis: C25H16N2O3 (392.4): calcd. C 76.5, H 4.11, N 7.14, found C 76.4, H 3.90, N, 7.84; MS (EI); m/z: 392 (100) [M+], 377 (5) [M+- CH3], 361 (14) [(M+- CH3O]; IR: = 1676, 1611, 1587, 1564, 1415, 1239, 1218 cm-1; 1H-NMR (CDCl3): δ = 4.08 (s, 3 H, OCH3), 4.10 (s, 3 H, OCH3), 7.40 (dd, 1 H, J=9.5, 2.5 Hz, 3-H), 7.54 (dd, 1 H, J=9.5, 2.5 Hz, 6-H), 7.64 (t, 1 H, J=8.0 Hz, 12-H), 7.77 (d, 1 H, J=2.5 Hz, 1-H), 7.88 (t, 1 H, J=8.0 Hz, 13-H), 8.04 (d, 1 H, J=2.5 Hz, 8-H), 8.49 (d, 1 H, J=8.0 Hz, 11-H), 8.90 (d, 1 H, J=9.5 Hz, 4-H), 8.98 (d, 1 H, J=9.5 Hz, 5-H), 9.07 (d, 1 H, J=8.0 Hz, 14-H); 13C-NMR (CDCl3): δ = 55.5 (OCH3), 55.9 (OCH3), 108.7 (d, C-8), 110.3 (d, C-1), 113.1 (s, C-14c), 116.0 (s, C-4a), 118.7 (s, C-4c), 119.8 (d, C-3), 122.5 (d, C-6), 126.0 (d, C-14), 128.0 (d, C-11), 128.2 (d, C-5), 128.4 (d, C-4), 131.4 (d, C-12), 131.4 (s, C-10a), 135.0 (d, C-13), 135.3 (s, C-14a), 137.0 (s, C-4b), 143.7 (s, C-9a), 147.6 (s, C-14b), 148.0 (s, C-8a), 149.0 (s, C-15a), 159.0 (s, C-7), 159.4 (s, C-2), 182.3 (s, C-10).
9H-benzo[i]pyrido[2,3,4-kl]acridin-9-one (8):
TFA-kynuramine (7)[4] (500 mg, 1.9 mmol), naphthoquinone (330 mg, 2.1 mmol) and CeCl3·7H2O (35 mg, 0.094 mmol) were dissolved in ethanol (25 mL). The reaction mixture was refluxed for 9 h. while air was bubbled through it as described above in the general procedure. After evaporation of the ethanol and chromatography (eluting with CHCl3/MeOH, 50:1) the crude product was added to a solution of 25% aq. ammonia (10 mL) in methanol (100 mL) and was stirred at room temperature for 7 days. The reaction mixture was evaporated and chromatographed (eluting with chloroform/methanol, 30:1) to afford 8 (70mg, 13%), amorphous powder (chloroform/methanol, 9:1), mp 258°C (lit [8], 260-2620C).
3(N)-(2,2’-diaminobenzophenone)-5-hydroxy-1,4-naphthoquinone (9):
4a (100 mg, 0.47 mmol) and CeCl3 ·7H2O (186 mg, 0.50 mmol) were dissolved in ethanol (10 mL) and 5-Hydroxy-1,4-naphthoquinone (juglone) (87 mg, 0.50 mmol) was added. The color of the solution changed immediately from yellow to red. The reaction mixture was stirred at room temperature, with bubbled air, for 3 days. The solvent was then evaporated and the red product purified by chromatography (eluting with CHCl3/MeOH, 100:1) (144mg, 80%): red prisms (from EtOH), m.p. 221°C; MS (EI); m/z: 384 (100) [M+, C23H16N2O4]; IR: = 3450, 1625, 1606, 1572, 1513, 1448, 1273, 1242 cm-1; 1H-NMR (CDCl3): δ = 6.2 (br s, 2 H, NH2), 6.52 (s, 1 H, H-2), 6.58 (t, J=7.5 Hz, 1 H, 12’-H), 6.72 (d, J=7.5Hz, 1 H, 10’-H), 7.16 (dd, J=7.5, 2.0 Hz, 1 H, 6-H), 7.21 (t, J=7.5 Hz, 1 H, 11’-H), 7.30 (t, J=7.5 Hz, 1 H, 4’-H), 7.40 (d, J=7.5 Hz, 1 H, 6’-H), 7.51 (d, J=7.5 Hz, 1 H, 13’-H), 7.54 (t, J=7.5 Hz, 1 H, 5’-H), 7.60 (d, J=7.5 Hz, 1 H, 8-H), 7.61 (d, J=7.5 Hz, 1 H, 3’-H), 7.62 (t, J=7.5 Hz, 1 H, 7-H), 9.45 (br s, 1 H, NH), 11.60 (s, 1 H, OH).
11-Hydroxy-10H-benzo[i]quino[2,3,4-kl]acridin-10-one (10):
9 (70 mg, 0.18 mmol) was added to a solution of Et3N (2 mL) in methanol (20 mL) and the reaction mixture was stirred at room temperature for 10 days. The solution was evaporated and the residual solid chromatographed (eluting with CHCl3/MeOH, 100:1) to afford 10 (61 mg, 98%), yellow needles (CHCl3-MeOH 50:1), m.p. 292°C; HRMS calcd. for C23H12N2O2 [M+] 348.0899, found 348.0899; IR: = 3441, 2924, 1639, 1611, 1567, 1458, 1402, 1277 cm-1; 1H-NMR (CDCl3): δ = 7.30 (d, J=8.0 Hz, 1 H, 12-H), 7.83 (t, J=8.0 Hz, 1 H, 13-H), 7.88 (t, J=8.5 Hz, 1 H, 3-H), 8.00 (t, J=8.5 Hz, 1 H, 6-H), 8.00 (t, J=8.5 Hz, 1 H, 2-H), 8.08 (t, J=8.5 Hz, 1 H, 7-H), 8.67 (d, J=8.5 Hz, 1 H, 1-H), 8.73 (d, J=8.0 Hz, 1 H, 14-H), 8.83 (d, J=8.5 Hz, 1 H, 8-H), 9.08 (d, J=8.5 Hz, 1 H, 4-H), 9.17 (d, J=8.5 Hz, 1 H, 5-H), 12.90 (s, 1 H, OH); 13C-NMR (CDCl3/CD3OD, 50:1): δ = 116.9 (s, C-10a), 117.7 (d, C-14), 120.3 (d, C-12), 122.7 (s, C-4a), 124.6 (s, C-4c), 127.2 (d, C-5), 127.3 (d, C-4), 128.4 (d, C-3), 130.4 (d, C-6), 131.1 (d, C-7), 131.2 (d, C-2), 131.3 (d, C-1), 132.7 (d, C-8), 136.1 (s, C-4b), 136.4 (s, C-14a), 137.8 (d, C-13), 145.3 (s, C-9a), 147.5 (s, C-14b), 147.5 (s, C-8a), 147.5 (s, C-15a), 163.9 (s, C-11), 183.6 (s, C-10); C-14c could not be seen due to a long relaxation time.
11-Acetoxy-10H-benzo[i]quino[2,3,4-kl]acridin-10-one (11):
10 (10 mg, 0.029 mmol) was acetylated with acetic anhydride-pyridine, 1:1 (1 mL), at room temperature for 24 h. The reaction mixture was evaporated and chromatographed (eluting with CHCl3/MeOH, 40:1) to give 11 (11 mg, 95%), yellow needles (CHCl3/MeOH, 100:1), m.p. 222oC; MS (EI); m/z: 390 (5) [M+, C25H14N2O3], 348 (100) [M+-CH2CO]; IR: = 2924, 1765, 1677, 1563, 1401, 1193 cm-1; 1H-NMR (CDCl3/CD3OD, 50:1): δ = 2.52 (s, 3 H, COCH3), 7.28 (d, J=8.0 Hz, 1 H, 12-H), 7.69 (t, J=8.0 Hz, 1 H, 13-H), 7.80 (t, J=8.0 Hz, 1 H, 3-H), 7.80 (t, J=8.0 Hz, 1 H, 2-H), 7.85 (t, J=8.0 Hz, 1 H, 6-H), 7.93 (t, J=8.0 Hz, 1 H, 7-H), 8.23 (d, J=8.0 Hz, 1 H, 1-H), 8.57 (d, J=8.0 Hz, 1 H, 14-H), 8.89 (d, J=8.0 Hz, 1 H, 8-H), 8.96 (d, J=8.0 Hz, 1 H, 4-H), 9.00 (d, J=8.0 Hz, 1 H, 5-H).
11-Hydroxy-10-imino-10H-benzo[i]quino[2,3,4-kl]acridine (12):
Method A: 9 (110 mg, 0.28 mmol), was added to a mixture of 25% aq. ammonia (2 ml) and methanol (20 mL) and stirred at room temperature for 7 days. The reaction mixture was then evaporated and the residue chromatographed (eluting with CHCl3/MeOH, 30:1) to afford 12 (83 mg, 85%).
Method B: 10 (10 mg, 0.029 mmol) was stirred in a saturated ammonia/methanol solution (2 mL) for 14 days. The solvent was then evaporated and the residue chromatographed using CHCl3/MeOH, 30:1, as eluant to afford 12 (7 mg, 70%), dark-green needles (CHCl3/MeOH, 20:1), m.p. 266 C; HRMS calcd. for C23H13N3O [M+] 347.10586, found 347.10590; IR: = 3384, 1615, 1565, 1488, 1472, 1405, 1124, 1048 cm-1; 1H-NMR (CDCl3): δ = 7.11 (d, J=8.0 Hz, 1 H, 12-H), 7.57 (d, J=8.0Hz, 1 H, 13-H), 7.77 (t, J=8.0 Hz, 1 H, 3-H), 7.88 (t, J=8.0 Hz, 1 H, 6-H), 7.89 (t, J=8.0 Hz, 1 H, 2-H), 7.97 (t, J=8.0 Hz, 1 H, 7-H), 8.32 (d, J=8.0 Hz, 1 H, 1-H), 8.37 (d, J=7.5 Hz, 1 H, 14-H), 8.40 (d, J=8.0 Hz, 1 H, 8-H), 9.00 (d, J=8.0 Hz, 1 H, 4-H), 9.10 (d, J=8.0 Hz, 1 H, 5-H), 10.81 (br s, 1 H), 14.8 (br s, 1 H); 13C-NMR (CDCl3): δ = 113.5 (s, C-10a), 115.9 (d, C-14), 122.4 (s, C-4a), 123.6 (d, C-12), 124.3 (s, C-4c), 127.1 (d, C-4), 127.3 (d, C-5), 127.6 (d, C-3), 129.2 (d, C-6), 130.8 (d, C-7), 130.8 (d, C-2), 131.2 (d, C-8), 131.3 (d, C-1), 133.3 (s, C-14a), 135.2 (d, C-13), 136.1 (s, C-4b), 143.2 (s, C-9a), 146.7 (s, C-8a), 147.9 (s, C-15a), 148.8 (s, C-14b), 162.1 (s, C-10), 171.0 (s, C-11); C-14c could not be seen due to a long relaxation time.
Acetylation of compound 12 to afford compounds 13a and 13b:
12 (10 mg, 0.029 mmol) was added to a mixture of acetic anhydride/pyridine, 1:1 (1 mL), and the solution was stirred at room temperature for 24 h. The reaction mixture was evaporated and chromatographed. Elution with CHCl3/MeOH, 200:1, afforded the mono N-acetylated product 13a (4 mg, 35%) and further elution with CHCl3/MeOH, 100:1, afforded the diacetylated product 13b (4 mg, 32%). Acetylation of 13a (1 mg, 2.6 µmol) by the same procedure gave the O,N-diacetate derivative 13b (1 mg). 13a: MS (EI); m/z: 389 (14) [M+, C25H15N3O2], 373 (66) [M+- O], 347 (100) [M+- CH2CO]; IR: = 3430, 1698, 1615, 1568, 1404, 1241, 1175 cm-1; 1H-NMR (CDCl3): δ = 2.72 (s, 3 H, NCOCH3), 7.22 (d, J=8.5 Hz, 1 H, 12-H), 7.66 (t, J=8.5 Hz, 1 H, 13-H), 7.76 (t, J=8.5 Hz, 1 H, 3-H), 7.89 (t, J=8.5 Hz, 1 H, 2-H), 7.91 (t, J=8.0 Hz, 1 H, 6-H), 7.97 (t, J=8.0 Hz, 1 H, 7-H), 8.31 (d, J=8.5 Hz, 1 H, 1-H), 8.37 (d, J=8.0 Hz, 1 H, 8-H), 8.61 (d, J=8.5 Hz, 1 H, 14-H), 8.98 (d, J=8.5 Hz, 1 H, 4-H), 9.09 (d, J=8.5 Hz, 1 H, 5-H); 13C-NMR (CDCl3): δ = 26.6 (NCOCH3), 114.4 (s, C-14c), 114.8 (s, C-10a), 117.8 (d, C-14), 120.3 (d, C-12), 122.5 (s, C-4a), 123.8 (s, C-4c), 127.1 (d, C-4), 127.3 (d, C-5), 128.0 (d, C-3), 129.8 (d, C-6), 131.0 (d, C-7), 131.0 (d, C-2), 131.2 (d, C-8), 131.2 (d, C-1), 134.6 (d, C-13), 135.0 (s, C-14a), 136.5 (s, C-4b), 141.8 (s, C-9a), 146.1 (s, C-8a), 147.8 (s, C-15a), 148.4 (s, C-14b), 153.8 (s, C-10), 161.6 (s, C-11), 183.8 (NCOCH3). 13b: MS (EI); m/z: 433 (17) [(M+2)+, C27H19N3O3], 389 (15) [M+-CH2CO], 373 (100) [(M+2)+-CH3CO2H], 347 (82) [M+- 2CH2CO]; IR: = 3430, 1767, 1676, 1640, 1569, 1204 cm-1; 1H-NMR (CDCl3): δ = 2.47 (s, 3 H, OCOCH3), 2.60 (s, 3 H, NCOCH3), 7.35 (dd, J=8.0, 1.5 Hz, 1 H, 12-H), 7.77 (t, J=8.0 Hz, 1 H, 13-H), 7.79 (t, J=8.0 Hz, 1 H, 6-H), 7.88 (t, J=8.0 Hz, 1 H, 3-H), 7.89 (t, J=8.0 Hz, 1 H, 7-H), 7.95 (dt, J=8.0, 1.5 Hz, 1 H, 2-H), 8.34 (d, J=8.0 Hz, 1 H, 8-H), 8.38 (dd, J=8.0, 1.5 Hz, 1 H, 1-H), 8.98 (d, J=8.0 Hz, 1 H, 14-H), 9.08 (d, J=8.0 Hz, 1 H, 4-H), 9.10 (dd, J=8.0, 1.5 Hz, 1 H, 5-H).
3N-(4-methoxyaniline)-5-hydroxy-1,4-naphthoquinone (14):
p-Anisidine (40 mg, 0.32 mmol) was reacted with juglone (57 mg, 0.33 mmol) in the present of CeCl3 ·7H2O (122 mg, 0.33 mmol) by the procedure described for the preparation of compound 9. The red product was purified by chromatography (eluting with CHCl3/MeOH, 100:1) (65 mg, 69%): red crystals (ethanol), m.p. 211°C. – MS (EI); m/z: 295 (100) [M+, C17H13NO4]; IR: = 3274, 1627, 1590, 1572, 1516, 1240 cm-1; 1H-NMR (CD3SOCD3): δ = 3.78 (s, 3 H, OCH3), 5.87 (s, 1 H, 2-H), 7.01 (d, J=8.5 Hz, 2 H, 3’-H), 7.24 (d, J=8.5 Hz, 1 H, 6-H), 7.28 (d, J=8.5 Hz, 2 H, 2’-H), 7.44 (d, J=8.5 Hz, 1 H, 8-H), 7.72 (t, J=8.5 Hz, 1 H, 7-H), 9.18 (s, 1 H, NH), 11.54 (s, 1 H, OH); 13C-NMR (CD3SOCD3): δ = 55.3 (q, OCH3), 101.3 (d, C-2), 114.3 (s, C-4a), 114.6 (d, C-3’), 117.6 (d, C-8), 122.1 (d, C-6), 125.8 (d, C-2’), 130.5 (s, C-1’), 133.1 (s, C-8a), 137.6 (d, C-7), 146.9 (s, C-3), 157.1 (s, C-4’), 160.5 (s, C-5), 181.6 (s, C-1), 185.7 (s, C-4).
3(N)-(4-methoxyaniline)-5-hydroxy-1,4-naphthoquinon-4-imine (15):
14 (20 mg, 0.068 mmol) was added to a mixture of 25% aq. ammonia (2 mL) and methanol (20 mL) and stirred at room temperature for 7 days. Evaporation of the solvent afforded compound 15 (20 mg, 100%); HRMS calcd. for C17H14N2O3 [M+] 394.1004, found 294.1008; IR: = 3300, 1571, 1535, 1513, 1257 cm-1; 1H-NMR (CD3SOCD3): δ = 3.77 (s, 3 H, OCH3), 5.66 (s, 1 H, 2-H), 7.04 (d, J=9.0 Hz, 2 H, 3’-H), 7.14 (d, J=8.0 Hz, 1 H, 6-H), 7.26 (d, J=9.0 Hz, 2 H, 2’-H), 7.39 (d, J=8.0 Hz, 1 H, 8-H), 7.49 (t, J=8.0 Hz, 1 H, 7-H); 13C NMR (CD3SOCD3): δ = 54.9 (q, OCH3), 100.5 (d, C-2), 113.4 (s, C-4a), 114.3 (d, C-3’), 115.5 (d, C-8), 121.8 (d, C-6), 125.5 (d, C-2’), 130.2 (s, C-1’), 131.6 (s, C-8a), 132.7 (d, C-7), 147.1 (s, C-3), 156.6 (s, C-4’), 161.5 (s, C-5), 161.6 (s, C-4), 181.6 (s, C-1).
10H,11H,12H-dihydroquino[2,3,4-kl]acridine (17a):
To a stirred solution of 4a (400 mg, 1.9 mmol) in AcOH (50 mL) and conc.HCl (0.25 mL), 1,3- cyclohexanedione (425 mg, 3.8 mmol) and sodium m-nitrophenylsulfonate (1.3 g, 5.7 mmol) were added and the reaction mixture was refluxed for 2 h. After cooling, the mixture was poured onto ice (100 g), the solution brought to pH 8 with 25% ammonia and then extracted with chloroform (3 x 30 mL). The chloroform solution was washed with water (2 x 50 mL) and evaporated to afford 17a (515 mg, quantitative), amorphous powder (CHCl /MeOH, 20:1), m.p. 186°C; HRMS calcd. for C H N [M+] 270.1157, found 270.1157; IR: = 2940, 1585, 1572, 1488, 1400, 1389 cm-1; 1H-NMR (CDCl3): δ = 2.42 (quintet, J=6.0 Hz, 2 H, 11-H), 3.50 (t, J=6.0 Hz, 4 H, 10-, 12-H), 7.76 (t, J=7.0 Hz, 2 H, 2-, 7-H), 7.89 (t, J=7.0 Hz, 2 H, 3-, 6-H), 8.28 (d, J=7.0 Hz, 2 H, 1-, 8-H), 9.06 (d, J=7.0 Hz, 2 H, 4-, 5-H); 13C-NMR (CDCl3): δ = 22.3 (t, C-11), 34.4 (t, C-10), 116.5 (s, C-12b), 122.4 (s, C-4a), 126.1 (d, C-3), 127.0 (d, C-4), 129.2 (d, C-1), 130.0 (d, C-2), 135.9 (s, C-4b), 146.6 (s, C-8a), 159.5 (s, C- 9a).
2,7-Dimethoxy-10H,11H,12H-dihydroquino[2,3,4-kl]acridine (17b):
Reacting 4b (520 mg, 1.9 mmol) with 1,3-cyclohexanedione (425 mg, 3.8 mmol) by the same procedure described for the synthesis of 17a afforded 17b (625mg, quantitative), amorphous powder (CHCl3/MeOH, 20:1), mp 218°C; HRMS calcd. for C21H18N2O2 [M+] 330.1368, found 330.1368; IR: = 2950, 1612, 1583, 1413, 1219, 1033 cm-1; 1H-NMR (CDCl3): δ = 2.57 (quintet, J=6.5 Hz, 2 H, 11-H), 3.90 (t, J=6.5 Hz, 4 H, 10-, 12-H), 4.16 (s, 6 H, OMe), 7.65 (dd, J=9.0, 2.5 Hz, 2 H, 3-, 6-H), 8.15 (d, J=2.5 Hz, 2 H, 1-, 8-H), 8.95 (d, J=9.0Hz, 2 H, 4-, 5-H).
General procedure for nitration of compounds 6a, 6b and 17a:
The pyridoacridine (10 mg) was added to a 1:1 mixture of conc.H2SO4/fuming HNO3 (2 mL) at 0°C. The reaction mixture was then allowed to warm up to room temperature and after 1 h. or 12 h. it was poured onto ice (10 g). The solution was neutralized with 10% NaOH and extracted with chloroform (4 x 10 mL). The chloroform solution was evaporated and the residue was chromatographed.
8-Nitro-10H,11H,12H-dihydroquino[2,3,4-kl]acridine (18a) and 6-Nitro-10H,11H,12H- dihydroquino [2,3,4-kl]acridine (18b):
Reaction of 17a (10 mg, 0.037 mmol) by the above described procedure for 1 h. gave two products that were separated by chromatography. Elution with dichloromethane afforded the less polar isomer 18a (3 mg, 26%) and further elution with CH2Cl2/MeOH, 30:1, afforded the more polar isomer 18b (7 mg, 60%). 18a: MS (EI); m/z: 315 (100) [M+, C19H13N3O2], 285 (37) [M+- NO], 269 (30) [M+- NO2]; IR: = 2925, 1591, 1374, 1139 cm-1; 1H-NMR (CDCl3/CD3OD, 50:1): δ = 2.43 (quintet, J=6.0 Hz, 2 H, 11-H), 3.51 (t, J=6.0 Hz, 2 H, 12a-H), 3.75 (t, J=6.0 Hz, 2 H, 10a-H), 7.94 (t, J=8.0 Hz, 1 H, 6-H), 8.05 (t, J=8.0 Hz, 1 H, 2-H), 8.18 (t, J=8.0 Hz, 1 H, 3-H), 8.25 (d, J=8.0 Hz, 1 H, 7-H), 9.06 (d, J=8.0 Hz, 1 H, 1-H), 9.07 (d, J=8.0 Hz, 1 H, 4-H), 9.21 (d, J=8.0 Hz, 1 H, 5-H). 18b: HRMS calcd. for C19H13N3O2 [M+] 315.1008, found 315.1006; MS (EI); m/z: 315 (100) [M+], 269 (33) [M+- NO2]; IR: = 2927, 1588, 1339 cm-1; 1H-NMR (CDCl3/CD3OD, 50:1): δ = 2.42 (quintet, J=6.0 Hz, 2 H, 11-H), 3.50 (t, J=6.0 Hz, 2 H, 12-H, interchangeable), 3.54 (t, J=6.0 Hz, 2 H, 10a-H), 7.87 (t, J=7.5 Hz, 1 H, 3-H), 7.97 (t, J=7.5 Hz, 1 H, 2-H), 8.32 (d, J=7.5 Hz, 1 H, 1-H), 8.36 (d, J=7.5 Hz, 1 H, 8-H), 8.63 (dd, J=7.5, 2.5 Hz, 1 H, 7-H), 8.95 (d, J=7.5 Hz, 1 H, 4-H), 9.93 (d, J=2.5 Hz, 1 H, 5-H).
1,8-Dinitro-10H,11H,12H-dihydroquino[2,3,4-kl]acridine (19a), 3,8-Dinitro-10H,11H, 12H-dihydro- quino[2,3,4-kl]acridine (19b) and 3,6-Dinitro-10H,11H,12H-dihydroquino [2,3,4-kl]acridine (19c):
Reaction of 17a (10 mg, 0.037mmol) in conc.H2SO4/fuming HNO3 , 1:1 (2ml), by the general above procedure for 12 h. gave three products that were separated by chromatography. Elution with dichloromethane/petroleum ether, 4:1, afforded isomer 19a (0.5 mg). Elution with dichloromethane afforded isomer 19b (2 mg, 15%) and isomer 19c (3 mg, 20%). 19a: MS (EI); m/z: 360 (47) [M+, C19H12N4O4], 330 (60) [M+- NO], 300 (100) [M+- 2NO]; IR: = °2925, 1620, 1583, 1534 cm-1; 1H-NMR (CDCl3): δ = 2.39 (quintet, J=7.0 Hz, 2 H, 11-H), 3.50 (t, J=7.0 Hz, 4 H, 10-, 12-H), 7.83 (t, J=7.5 Hz, 2 H, 3-, 6-H), 8.13 (d, J=7.5 Hz, 2 H, 2-, 7-H), 9.10 (d, J=7.5 Hz, 2 H, 4-, 5-H). 19b: HRMS calcd. for C19H12N4O4 [M+] 360.0859, found 360.0858; MS (EI); m/z: 360 (100) [M+], 330 (23) [M+- NO], 302 (29) [(M+2)+- 2NO], 267 (31) [(M-1)+- 2NO2]; IR: = 2925, 1618, 1589, 1535, 1341 cm-1; 1H-NMR (CDCl3): δ = 2.43 (quintet, J=7.0 Hz, 2 H, 11-H), 3.52 (t, J=7.0 Hz, 2 H, 12 -H), 3.59 (t, J=7.0 Hz, 2 H, 10-H, interchangeable), 7.95 (t, J=8.0 Hz, 1 H, 6-H), 8.19 (d, J=8.0 Hz, 1 H, 7-H), 8.55 (d, J=8.0 Hz, 1 H, 1-H), 8.73 (dd, J=8.0, 2.0 Hz, 1 H, 2-H), 9.16 (d, J=8.0 Hz, 1 H, 5-H), 9.90 (d, J=2.0 Hz, 1 H, 4-H). 19c: MS (EI); m/z: 360 (100) [M+, C19H12N4O4], 315 (49) [(M+1)+- NO2], 267 (53) [(M-1)+- 2NO2]; IR: = 2925, 1600, 1588, 1506, 1346 cm-1; 1H-NMR (CDCl3): δ = 2.54 (quintet, J=6.0 Hz, 2 H, 11-H), 3.67 (t, J=6.0 Hz, 4 H, 10-, 12-H), 8.60 (d, J=8.0 Hz, 2 H, 1-, 8-H), 8.79 (dd, J=8.0, 2.0 Hz, 2 H, 2-, 7-H), 9.96 (d, J=2.0 Hz, 2 H, 4-, 5-H).
3-Nitro-10H-benzo[i]quino [2,3,4-kl]acridin-10-one (20):
Reaction of 6a (80 mg, 0.24 mmol) in conc.H2SO4/fuming HNO3, 1:1 (5mL), for 12 h. by the above general procedure followed by crystallization of the crude product from pyridine afforded 20 (48 mg, 53%), yellow needles, m.p. 339°C; MS (EI); m/z: 377 (100) [M+, C23H11N3O3], 347 (36) [(M+- NO], 330 (41) [(M-1)+- NO2]; IR: = 1682, 1513, 1404, 1388, 1340, 1276 cm-1; 1H-NMR (CDCl3): δ =7.79 (t, J=8.0 Hz, 1 H, 12-H), 7.93 (t, J=8.0 Hz, 1 H, 13-H), 8.12 (t, J=8.0 Hz, 1 H, 7-H), 8.12 (t, J=8.0 Hz, 1 H, 6-H), 8.50 (d, J=8.0 Hz, 1 H, 11-H), 8.54 (d, J=9.0 Hz, 1 H, 1-H), 8.71 (d, J=8.0 Hz, 1 H, 8-H), 8.72 (dd, J=9.0, 2.5 Hz, 1 H, 2-H), 9.09 (d, J=8.0 Hz, 1 H, 14-H), 9.11 (d, J=8.0 Hz, 1 H, 5-H), 10.01 (d, J=2.5 Hz, 1 H, 4-H); 13C-NMR (CDCl3/CF3CO2D, 100:1): δ = 116.5 (s, C-14c), 121.6 (s, C-4a), 123.6 (d, C-4),123.7 (s, C-4c), 124.7 (d, C-2), 126.4 (d, C-5), 126.7 (d, C-14), 128.4 (d, C-11), 131.5 (d, C-6), 131.8 (d, C-7), 131.9 (d, C-8), 132.2 (s, C-10a), 132.3 (d, C-12), 132.6 (d, C-1), 134.8 (s, C-14a), 135.3 (d, C-13), 136.8 (s, C-4b), 146.0 (s, C-3), 146.7 (s, C-8a), 149.0 (s, C-9a), 149.8 (s, C-15a), 151.3 (s, C-14b), 181.1 (s, C-10).
3-Amino-10H-benzo[i]quino[2,3,4-kl]acridin-10-one (21):
20 (40 mg, 0.11 mmol) was dissolved in AcOH (3 mL) and TFA (6mL), 5% Pd-C (25 mg) was added and the reaction mixture was shaken in a Parr apparatus under H2 (3 atm) for 1 h. The catalyst was then filtered off, the solution poured onto ice (10 g), brought to pH 8 with 25% ammonia and then extracted with chloroform (3 x 20 mL). After evaporation of the solvent the residue was chromatographed (eluting with CHCl3/MeOH, 30:1) to afford 21 (15 mg, 40%); HRMS calcd. for C23H13N3O [M+] 347.1059, found 347.1058; IR: = 1677, 1646, 1540, 1515 cm-1; 1H-NMR (CD3SOCD3): δ = 7.42 (d, J=8.0 Hz, 1 H, 2-H), 7.71 (t, J=8.0 Hz, 1 H, 12-H), 7.93 (t, J=8.0 Hz, 1 H, 13-H), 8.05 (t, J=8.0 Hz, 1 H, 6-H), 8.09 (t, J=8.0 Hz, 1 H, 7-H), 8.13 (d, J=8.0 Hz, 1 H, 1-H), 8.28 (d, J=2.0 Hz, 1 H, 4-H), 8.30 (d, J=8.0 Hz, 1 H, 11-H), 8.53 (d, J=8.0 Hz, 1 H, 8-H), 8.91 (d, J=8.0 Hz, 1 H, 14-H), 9.31 (d, J=8.0 Hz, 1 H, 5-H).
2,7-Dimethoxy-1,8-dinitro-10H-benzo[i]quino[2,3,4-kl]acridin-10-one (22):
Reaction of 6b (10 mg, 0.026 mmol) in conc.H2SO4/fuming HNO3, 1:1 (2ml), by the above general procedure for 1 hr afforded 22 (10 mg, 80%); HRMS calcd. for C25H14N4O7 [M+] 482.0862, found 482.0855, 452 (100) [(M+- NO]; MS (EI); m/z: 482 (79) [M+], 452 (100) [M+-NO]; IR: = 1687, 1617, 1540, 1375, 1290 cm-1; 1H-NMR (CD3SOCD3): δ = 4.21 (s, 3 H, OCH3), 4.24 (s, 3H, OCH3), 7.85 (t, J=8.5 Hz, 1 H, 12-H), 7.97 (d, J=10.0 Hz, 1 H, 3-H), 7.99 (t, J=8.5 Hz, 1 H, 13-H), 8.13 (d, J=10.0 Hz, 1 H, 6-H), 8.27 (d, J=8.5 Hz, 1 H, 11-H), 8.66 (d, J=8.5 Hz, 1 H, 14-H), 9.28 (d, J=10.0 Hz, 1 H, 4-H), 9.36 (d, J=10.0 Hz, 1 H, 5-H).
2,7- Dimethoxy-1,3,8,12-tetrainitro-10H-benzo[i]quino[2,3,4-kl]acridin-10-one (23a) and 2,7-di- methoxy-1,3,8,14-tetranitro-10H-benzo[i]quino[2,3,4-kl]acridin-10-one (23b):
Reaction of 6b (10 mg, 0.026 mmol) in conc.H2SO4/fuming HNO3, 1:1 (2 ml), by the above general procedure for 12 hr gave two products that were separated by chromatography. Elution with dichloromethane afforded first the less polar isomer 23a (3 mg, 20%) and then the more polar isomer 23b (7 mg, 45%). 23a: MS (EI); m/z: 572 (100) [M+, C25H12N6O11], 542 (45) [M+- NO], 482 (78) [M+-3NO], 452 (32) [M+- 4NO]; IR: = 2925, 1706, 1619, 1547, 1375 cm-1; 1H-NMR (CD3SOCD3/CF3CO2D, 100:1): δ = 4.18 (s, 3 H, OCH3), 4.29 (s, 3 H, OCH3), 8.28 (d, J=10.0 Hz, 1 H, 6-H), 8.82 (dd, J=8.5, 2.5 Hz, 1 H, 13-H), 8.90 (d, J=8.5 Hz, 1 H, 14-H), 8.90 (d, J=2.5 Hz, 1 H, 11-H), 9.48 (d, J=10.0 Hz, 1 H, 5-H), 9.86 (s, 1 H, 4-H); 1H-NMR (CD3CN): δ = 4.30 (s, 3 H, OCH3), 4.32 (s, 3 H, OCH3), 8.15 (d, J=10.0 Hz, 1 H, 6-H), 8.70 (dd, J=8.5, 2.0 Hz, 1 H, 13-H), 8.99 (d, J=8.5 Hz, 1 H, 14-H), 9.05 (d, J=2.0 Hz, 1 H, 11-H), 9.30 (d, J=10.0Hz, 1 H, 5-H), 9.75 (s, 1 H, 4-H). 23b: HRMS calcd. for C25H12N6O11 [M+] 572.0564, found 572.0562; IR: = 2925, 1692, 1629, 1557, 1547, 1376 cm-1; 1H-NMR (CD3SOCD3): δ = 4.14 (s, 3 H, OCH3), 4.27 (s, 3 H, OCH3), 8.06 (t, J=8.0 Hz, 1 H, 12-H), 8.22 (d, J=8.0 Hz, 1 H, 13-H), 8.26 (d, J=9.5 Hz, 1 H, 6-H), 8.54 (d, J=8.0 Hz, 1 H, 11-H), 9.43 (d, J=9.5 Hz, 1 H, 5-H), 9.79 (s, 1 H, 4-H).
10H-quino[2,3,4-kl]acridin-10-one (24):
To a solution of 17a (300 mg, 1.1 mmol) in acetonitrile (120 mL) cerium ammonium nitrate (2.4 g, 4.4 mmol) was added and the reaction mixture was refluxed for 10 min. The acetonitrile was evaporated and the residue dissolved in chloroform (100 mL), washed with 0.1% aq. ammonia (2 x 100 mL) and evaporated to afford 24 (290 mg, 92%), amorphous powder (CHCl3/MeOH, 20:1), m.p. 254°C; HRMS calcd. for C19H10N2O [M+] 282.0793, found 282.0799; IR: = 1663, 1565, 1493, 1280 cm-1; 1H-NMR (CDCl3): δ = 7.11 (d, J=11.0 Hz, 1 H, 11-H), 7.90 (t, J=8.5 Hz, 1 H, 3-H), 7.98 (t, J=8.5 Hz, 1 H, 2-H), 7.98 (t, J=8.5 Hz, 1 H, 6-H), 7.99 (d, J=11.0 Hz, 1 H, 12-H), 8.05 (t, J=8.5 Hz, 1 H, 7-H), 8.43 (d, J=8.5 Hz, 1 H, 1-H), 8.73 (d, J=8.5 Hz, 1 H, 8-H), 9.14 (d, J=8.5 Hz, 1 H, 4-H), 9.19 (d, J=8.5 Hz, 1 H, 5-H).
2,7-Dimethoxy-10H-quino[2,3,4-kl]acridin-10-one (25):
Oxidation of 17b (360 mg, 1.1 mmol) by the same procedure described for the synthesis of 24 afforded 25 (340 mg, 90%), amorphous powder (CHCl3/MeOH, 20:1), m.p. 291°C; HRMS calcd. for C21H14N2O3 [M+] 342.1004, found 342.1004; IR: = 1666, 1608, 1415, 1259, 1225 cm-1; 1H-NMR (CDCl3/CD3OD, 50:1): δ = 4.03 (s, 3 H, OCH3), 4.04 (s, 3 H, OCH3), 7.02 (d, J=10 Hz, 1 H, 11-H), 7.43 (dd, J=8.5, 2.5 Hz, 1 H, 3-H), 7.51 (dd, J=8.5, 2.5 Hz, 1 H, 6-H), 7.72 (d, J=2.5 Hz, 1 H, 1-H), 7.93 (d, J=10 Hz, 1 H, 12-H), 7.94 (s, 1 H, 8-H), 8.88 (d, J=8.5 Hz, 1 H, 4-H), 8.92 (d, J=8.5 Hz, 1 H, 5-H).
11,12-Dibromo-10H-quino[2,3,4-kl]acridin-10-one (26):
24 (30 mg, 0.11 mmol) was dissolved in acetic acid (5 mL) and Br2(0.2 mL, 3.9 mmol) was added at room temperature. The mixture was stirred at 80°C for 2 h. and then allowed to cool to room temperature. Water (25 mL) and chloroform (25 mL) were added. The organic phase was extracted with 5% NaHSO3 (10 mL) and evaporated. The crude product was chromatographed (eluting with chloroform) to afford 26 (16 mg, 33%); HRMS calcd. for C19H8Br2N2O [M+] 439.8988, found 439.9000; IR: = 3425, 1669, 1491, 1394, 1242, 767 cm-1; 1H-NMR (CDCl3): δ = 7.86 (t, J=8.0 Hz, 1 H, 3-H), 7.93 (t, J=8.0 Hz, 1 H, 2-H), 7.93 (t, J=8.0 Hz, 1 H, 6-H), 7.99 (t, J=8.0 Hz, 1 H, 7-H), 8.39 (d, J=8.0 Hz, 1 H, 1-H), 8.56 (d, J=8.0 Hz, 1 H, 8-H), 8.97 (d, J=8.0 Hz, 1 H, 4-H), 9.04 (d, J=8.0 Hz, 1 H, 5-H).
12-Isobutylamino-10H-quino[2,3,4-kl]acridin-10-one (27a):
24 (40 mg, 0.14 mmol) and isobutylamine (0.12 mL, 1.2 mmol) in acetonitrile (10 mL) were stirred at room temperature for 48 h. The reaction mixture was then evaporated and the residue chromatographed by a silica gel column (eluting with CHCl3/MeOH, 30:1) to afford 27a (18 mg, 36%); HRMS calcd. for C23H19N3O [M+] 353.1528, found 353.1525; MS (EI); m/z: 353 (20) [M+], 310 (100) [M+- (CH3)2CH]; IR: = 2925, 1609, 1514, 1466, 1258, 761 cm-1; 1H-NMR (CDCl3): δ = 1.15 (d, J=7.0 Hz, 6 H, 3’-H), 2.20 (quintet, J=7.0 Hz, 1 H, 2’-H), 3.31 (t, J=7.0 Hz, 2 H, 1’-H), 6.09 (s, 1 H, 11-H), 7.49 (br s, 1 H, NH), 7.76 (t, J=8.0 Hz, 1 H, 3-H), 7.79 (t, J=8.0 Hz, 1 H, 6-H), 7.86 (t, J=8.0 Hz, 1 H, 2-H), 7.89 (t, J=8.0 Hz, 1 H, 7-H), 8.21 (d, J=8.0 Hz, 1 H, 1-H), 8.60 (d, J=8.0 Hz, 1 H, 8-H), 8.91 (d, J=8.0 Hz, 1 H, 5-H), 8.93 (d, J=8.0 Hz, 1 H, 4-H); 13C-NMR (CDCl3): δ = 20.5 (q, C-3’), 28.0 (d, C-2’), 50.5 (t, C-1’), 100.7 (d, C-11), 115.3 (s, C-12b), 123.5 (s, C-4c), 123.8 (s, C-4a), 126.8 (d, C-5), 127.3 (d, C-4), 129.0 (d, C-3), 129.3 (d, C-6), 130.7 (d, C-2), 130.8 (d, C-7), 131.0 (d, C-1), 132.9 (d, C-8), 134.9 (s, C-4b), 144.6 (s, C-9a), 145.7 (s, C-12a), 147.0 (s, C-13a), 147.6 (s, C- 8a), 152.1 (s, C-12), 180.4 (s, C-10).
10,12-Di(isobutylamino)quino[2,3,4-kl]acridine (27b):
24 (100 mg, 0.36 mmol) and isobutylamine (0.30 mL, 3.0 mmol) in ethanol (25 mL) were stirred at room temperature for 18 h. The reaction mixture was evaporated and the residue chromatographed by two subsequent silica gel columns (eluting with CHCl3/MeOH/trifluoroacetic acid, 20:1:0.01) to afford 27b (56 mg, 38%); HRMS calcd. for C27H26N4 [(M - 2)+] 406.2157, found 406.2156; IR: = 3445, 1611, 1559, 1462, 1386, 1125, 766 cm-1; 1H-NMR (CDCl3/CD3OD, 20:1): δ = 1.13 (d, J=7.0 Hz, 12 H, 3’-H), 2.25 (quintet, J=7.0 Hz, 2 H, 2’-H), 3.59 (d, J=7.0 Hz, 4 H, 1’-H), 6.19 (s, 1 H, 11-H), 7.99 (t, J=8.0 Hz, 2 H, 3-, 6-H), 8.05 (t, J=8.0 Hz, 2 H, 2-, 7-H), 8.46 (d, J=8.0 Hz, 2 H, 1-, 8-H), 9.16 (d, J=8.0 Hz, 2 H, 4-, 5-H), 9.49 (br s, 1 H, NH); 13C-NMR (CDCl3/CD3OD, 20:1): δ = 20.1 (q, C-3’), 28.4 (d, C-2’), 51.1 (t, C-1’), 87.6 (d, C-11), 113.0 (s, C-12b), 124.2 (s, C-4a), 127.4 (d, C-4), 131.1 (d, C-3), 132.0 (d, C-2), 132.2 (d, C-1), 136.1 (s, C-4b), 141.1 (s, C-9a), 146.2 (s, C-8a), 156.0 (s, C-10).
2,7-Dimethoxy-12-isobutylamino-10H-quino[2,3,4-kl]acridin-10-one (28a) and 10,12-Di (isobutyl- amino)-2,7-dimethoxyquino[2,3,4-kl]acridine (28b):
25 (50 mg, 0.15 mmol) was reacted with isobutylamine (0.20 mL, 2.0 mmol) by the described procedure for the synthesis of 27b. Two obtained compounds were separated by chromatography; elution with CHCl3/MeOH, 30:1, afforded the monoamination product (28a) (12 mg, 19%) and further elution with CHCl3/MeOH, 10:1, afforded the diamination product (28b) (25mg, 37%). 28a: MS (EI); m/z: 413 (29) [M+, C25H23O3N3], 370 (100) [M+- (CH3)2CH]; IR: = 2959, 1658, 1612, 1562, 1467, 1450, 1422, 1223, 1134 cm-1; 1H-NMR (CDCl3): δ = 1.11 (d, J=7.0 Hz, 6 H, 3’-H), 2.17 (quintet, J=7.0 Hz, 1 H, 2’-H), 3.25 (t, J=7.0 Hz, 2 H, 1’-H), 4.03 (s, 3 H, OCH3), 4.05 (s, 3 H, OCH3), 6.07 (s, 1 H, 11-H), 7.40 (d, J=10.0 Hz, 1 H, 3-H), 7.42 (d, J=10.0 Hz, 1 H, 6-H), 7.50 (br s, 1 H, NH), 7.58 (s, 1 H, 1-H), 8.00 (s, 1 H, 8-H), 8.83 (d, J=10.0 Hz, 1 H, 5-H), 8.86 (d, J=10.0 Hz, 1 H, 4-H). 28b: MS (EI); m/z: 466 (61) [(M-2)+, C29H30N4O2], 423 (100) [(M-2)+- (CH3)2CH]; IR: = 2924, 1658, 1612, 1564, 1467, 1412, 1252, 1219, 669 cm-1; 1H-NMR (CDCl3): δ = 1.14 (d, J=7.0 Hz, 12 H, 3’-H), 2.28 (quintet, J=7.0 Hz, 2 H, 2’-H), 3.67 (d, J=7.0 Hz, 4 H, 1’-H), 4.06 (s, 6 H, OCH3), 5.89 (s, 1 H, 11-H), 7.36 (dd, J=9.0, 1.5 Hz, 2 H, 3-, 6-H), 7.70 (d, J=1.5 Hz, 2 H, 1-, 8-H), 8.58 (d, J=9.0 Hz, 2 H, 4-, 5-H), 9.78 (br s, 1 H, NH); 13C-NMR (CDCl3): δ = 20.6 (q, C-3’), 28.7 (d, C-2’), 51.4 (t, C-1’), 56.2 (OCH3), 87.3 (d, C-11), 110.4 (s, C-12b), 111.3 (d, C-1), 117.6 (s, C-4a), 121.9 (d, C-3), 128.0 (d, C-4), 134.8 (s, C-4b), 141.5 (s, C-9a), 148.1 (s, C-8a), 155.3 (s, C-10), 161.7 (s, C-2).
12-Methylamino-10H-quino[2,3,4-kl]acridin-10-one (29a):
24 (40 mg, 0.14 mmol), methylamine (0.20 mL of 33% methylamine in ethanol, 1.6 mmol) and CeCl3·7H2O (52 mg, 0.14 mmol) were mixed together in ethanol (10 mL).The reaction mixture was stirred at room temperature for 15 min. then evaporated and chromatographed by two subsequent silica gel columns (eluting with CHCl3/MeOH, 30:1) to afford 29a (20 mg, 46%); MS (EI); m/z: 311 (100) [M+, C20H13N3O]; IR: = 3430, 1610, 1560, 1419 cm-1; 1H-NMR (CDCl3/CD3OD, 20:1): δ = 3.11 (s, 3 H, NCH3), 5.97 (s, 1 H, 11-H), 7.74 (t, J=8.0 Hz, 1 H, 3-H), 7.78 (t, J=8.0 Hz, 1 H, 6-H), 7.82 (t, J=8.0 Hz, 1 H, 2-H), 7.89 (t, J=8.0 Hz, 1 H, 7-H), 8.14 (d, J=8.0 Hz, 1 H, 1-H), 8.51 (d, J=8.0 Hz, 1 H, 8-H), 8.90 (d, J=8.0 Hz, 1 H, 5-H), 8.92 (d, J=8.0 Hz, 1 H, 4-H).
10,12-Di(methylamino)quino[2,3,4-kl]acridine (29b):
24 (30 mg, 0.11 mmol) was reacted with methylamine (0.20 mL of 33% methylamine in ethanol, 1.6 mmol) in ethanol (10 mL) at room temperature for 24 h. The product (29b) was purified by silica gel column chromatography eluting with CHCl3/MeOH, 10:1 (10 mg, 28%); MS (EI); m/z: 322 (100) [(M-2)+, C21H14N4], 294 (44) [(M-2)+- CH2N]; IR: = 3405, 1618, 1564, 1419 cm-1; 1H-NMR (CDCl3/CD3OD, 20:1): δ = 3.38 (s, 6 H, NCH3), 5.83 (s, 1H, 11-H), 7.83 (t, J=8.0 Hz, 2 H, 3-, 6-H), 7.90 (t, J=8.0 Hz, 2 H, 2-, 7-H), 8.21 (d, J=8.0 Hz, 2 H, 1-, 8-H), 8.79 (d, J=8.0 Hz, 2 H, 4-, 5-H).
12-(4-methoxyanilino)-10H-quino[2,3,4-kl]acridin-10-one (30a):
24 (10 mg, 0.036 mmol) and p-anisidine (5 mg, 0.041 mmol) were refluxed in acetonitrile for 48 h. The solvent was evaporated and the residue chromatographed on a silica gel column (eluting with CHCl3/MeOH, 50:1) to afford 30a (8 mg, 55%); MS (EI); m/z: 403 (100) [M+, C26H17N3O2], 372 (36) [M+- CH3O]; IR: = 3448, 1614, 1556, 1512, 1245 cm-1; 1H-NMR (CDCl3): δ = 3.87 (s, 3 H, OCH3), 6.54 (s, 1 H, 11-H), 7.00 (d, J=9.0 Hz, 2 H, 3’-H), 7.38 (d, J=9.0 Hz, 2 H, 2’-H), 7.88 (t, J=8.5 Hz, 1 H, 3-H), 7.88 (t, J=8.5 Hz, 1 H, 6-H), 7.96 (t, J=8.5 Hz, 1 H, 2-H), 7.96 (t, J=8.5 Hz, 1 H, 7-H), 8.37 (d, J=8.5 Hz, 1 H, 1-H), 8.67 (d, J=8.5 Hz, 1 H, 8-H), 9.07 (d, J=8.5 Hz, 1 H, 5-H), 9.10 (d, J=8.5 Hz, 1 H, 4-H).
10,12-Di(4-methoxyanilino)quino[2,3,4-kl]acridine (30b):
24 (10 mg, 0.036 mmol) and p-anisidine (10 mg, 0.081 mmol) in ethanol (4 mL) were stirred at 50°C for 12 h. The ethanol was then evaporated and the residue chromatographed (eluting with CHCl3/MeOH, 10:1) to afford 30b (7 mg, 38%); MS (EI); m/z: 509 (100) [(M+1)+], 508 (99) [M+, C33H24N4O2]; IR: = 3440, 2925, 1607, 1548, 1506, 1460, 1253, 1171, 1105, 1025 cm-1; 1H-NMR (CDCl3/CD3OD, 20:1): δ = 3.89 (s, 6 H, OCH3), 6.71 (s, 1 H, 11-H), 7.00 (d, J=8.0 Hz, 4 H, 3’-H), 7.47 (d, J=8.0 Hz, 4 H, 2’-H), 7.91 (t, J=8.0 Hz, 2 H, 3-, 6-H), 7.99 (t, J=8.0 Hz, 2 H, 2-, 7-H), 8.53 (d, J=8.0 Hz, 2 H, 1-, 8-H), 8.99 (d, J=8.0 Hz, 2 H, 4-, 5-H).
12-Dodecylamino-10H-quino[2,3,4-kl]acridin-10-one (31a) and 10,12-Di(dodecylamino)quino- [2,3,4-kl]acridine (31b):
24 (10 mg, 0.036 mmol) and dodecylamine (0.030 mL, 0.13 mmol) were stirred in ethanol (4 mL) at 50°C for 12 h. After evaporation of the ethanol, the two products were separated by silica gel chromatography; elution with CHCl3/MeOH, 30:1, to afford the monoamination product (31a) (4 mg, 24%) and by elution with CHCl3/MeOH, 10:1, the diamination product (31b) which was further purified on a Sephadex LH-20 column (eluting with CHCl3/MeOH/petroleum ether, 1:1:2) (4 mg, 18%). 31a: MS (EI); m/z: 465 (64) [M+, C31H35N3O], 310 (100) [M+- (CH2)10CH3]; IR: = 2923, 2852, 1610, 1561, 1466 cm-1; 1H-NMR (CDCl3): δ = 0.90 (t, J=7.5 Hz, 3 H, 12’-H), 1.35 (br m, 14 H, 5’-H - 11’-H), 1.46 (m, 2 H, 4’-H), 1.54 (quintet, J=7.5 Hz, 2 H, 3’-H), 1.91 (quintet, J=7.5 Hz, 2 H, 2’-H), 3.45 (m, 2 H, 1’-H), 6.25 (s, 1 H, 11-H), 7.63 (br s, 1 H, NH), 7.87 (t, J=8.0 Hz, 1 H, 3-H), 7.87 (t, J=8.0 Hz, 1 H, 6-H), 7.96 (t, J=8.0 Hz, 1 H, 2-H), 7.98 (t, J=8.0 Hz, 1 H, 7-H), 8.35 (d, J=8.0 Hz, 1 H, 1-H), 8.68 (d, J=8.0 Hz, 1 H, 8-H), 9.06 (d, J=8.0 Hz, 1 H, 5-H), 9.09 (d, J=8.0 Hz, 1 H, 4-H). 31b: MS (EI); m/z: 630 (100) [(M- 2)+, C43H58N4], 475 (33) [(M- 2)+- (CH2)10CH3]; IR: = 2922, 2851, 1640, 1611, 1563, 1467, 1442, 1408 cm-1; 1H-NMR (CDCl3): δ = 0.85 (t, J=7.0 Hz, 6 H, 12’-H), 1.2 (br m, 28 H, 5’-H - 11’-H), 1.38 (m, 4 H, 4’-H), 1.55 (m, 4 H, 3’-H), 1.96 (m, 4 H, 2’-H), 3.91 (m, 4 H, 1’-H), 6.22 (s, 1 H, 11-H), 7.92 (t, J=8.0 Hz, 2 H, 3-, 6-H), 8.00 (t, J=8.0 Hz, 2 H, 2-, 7-H), 8.55 (d, J=8.0 Hz, 2 H, 1-, 8-H), 8.99 (d, J=8.0 Hz, 2 H, 4-, 5-H), 9.83 (br s, 1 H, NH).
12-Amino-10H-quino[2,3,4-kl]acridin-10-one (32a):
The procedure of Couladouros [17] was adopted. To a solution of 24 (10 mg, 0.036 mmol) in methanol (2 mL), under nitrogen, was added a solution of sodium azide (14 mg, 0.22 mmol) in water (0.5 mL) and the solution was acidified to pH 4 with 1N HCl. After stirring at room temperture for 15 h. the reaction mixture was extracted with chloroform (2 x 10 mL) and the combined organic layer was washed with water (20 ml) and evaporated to afford 32a (8 mg, 76%); HRMS calcd. for C19H11N3O [M+] 297.0902, found 297.0901; IR: = 3425, 1644, 1616, 1546, 1515 cm-1; 1H-NMR (CDCl3/CD3OD, 20:1): δ = 6.23 (s, 1 H, 11-H), 7.77 (t, J=8.0 Hz, 1 H, 3-H), 7.80 (t, J=8.0 Hz, 1 H, 6-H), 7.86 (t, J=8.0 Hz, 1 H, 2-H), 7.89 (t, J=8.0 Hz, 1 H, 7-H), 8.20 (d, J=8.0 Hz, 1 H, 1-H), 8.49 (d, J=8.0 Hz, 1 H, 8-H), 8.94 (d, J=8.0 Hz, 1 H, 5-H), 8.96 (d, J=8.0 Hz, 1 H, 4-H).
10,12(N,N)-Di(2-amino-1,3-propanediol)quino[2,3,4-kl]acridine (33b):
24 (10 mg, 0.036 mmol) and 2-amino-1,3-propanediol (serinol) (10 mg, 0.11 mmol) were reacted in ethanol (5 mL) to afford 33b (8mg, 50%), by the procedure described for the synthesis and purification of 30b. MS (FAB); m/z: 445 (100) [(M+1)+, C25H25N4O4]; IR: = 3332, 1613, 1559, 1415, 1390, 1050 cm-1; 1H-NMR (CD3SOCD3): δ = 3.84 (m, 8 H, 2’-H), 4.45 (quintet, J=5.0 Hz, 2 H, 1’-H), 5.37 (t, J=5.0 Hz, 4 H, OH), 6.91 (s, 1 H, 11-H), 8.08 (t, J=7.5 Hz, 2 H, 3-, 6-H), 8.16 (t, J=7.5 Hz, 2 H, 2, 7-H), 8.47 (d, J=7.5 Hz, 2 H, 1-, 8-H), 9.28 (d, J=7.5 Hz, 2 H, 4-, 5-H).
References
- Molinski, T. F. Marine pyridoacridine alkaloids: structure, synthesis and biological chemistry. Chem.Rev. 1993, 93, 1825–1838. [Google Scholar] [CrossRef] Faulkner, D.J. Marine natural products. Nat. Prod. Rep. 2001, 18, 1–49, and earlier reports in this series. [Google Scholar]
- Gellerman, G.; Rudi, A.; Kashman, Y. Biomimetic synthesis of pyrido[2,3,4-kl]acridines. Tetrahedron Lett. 1993, 34, 1823–1826. [Google Scholar] [CrossRef]
- Gellerman, G.; Babad, M.; Kashman, Y. A two step biomimetic total synthesis of Eilatin. Tetrahedron Lett. 1993, 34, 1827–1830. [Google Scholar] [CrossRef]
- Gellerman, G.; Rudi, A.; Kashman, Y. Biomimetic synthesis of ascididemin and derivatives. Syn. 1994, 239–241. [Google Scholar] [CrossRef]
- Gellerman, G.; Rudi, A.; Kashman, Y. The biomimetic synthesis of marine alkaloid related pyrido- and pyrrolo[2,3,4-kl]acridines. Tetrahedron 1994, 50, 12959–12972. [Google Scholar] [CrossRef]
- Schurman, J. V.; Becker, E. I. Synthesis of naphthylaminonaphthoquinones. J. Org. Chem. 1953, 18, 211–217. [Google Scholar] [CrossRef]
- Pratt, Y. T. Quinolinequinones. VI. Reactions with aromatic amines. J. Org. Chem. 1962, 27, 3905–3910. [Google Scholar] [CrossRef]
- Peterson, J. R.; Zjawiony, J. K.; Liu, S.; Hufford, C. D.; Clark, A. M.; Rogers, R. D. Copyrine alkaloids: synthesis, spectroscopic characterization, and antimycotic/ antimycobacterial activity of A- and B-ring-functionalized sampangines. J. Med. Chem. 1992, 35, 4069–4077. [Google Scholar] [CrossRef] [PubMed]
- Macleod, J. W.; Thomson, R. H. Studies in the juglone series. VI. The addition of aniline and toluene-p-thiol to 5-substituted 1,4-naphthoquinones. J. Org. Chem. 1960, 25, 36–42. [Google Scholar] [CrossRef]
- (a)Patai, S.; Rappoport, Z. The chemistry of the quinonoid compounds. Vol. 2, 1988; 1249. [Google Scholar] Patai, S. The chemistry of the carbon- nitrogen double bond. 1970, 663. [Google Scholar]
- Cimino, G.; Crispino, A.; De Rosa, S.; De Stefano, S.; Gavagnin, M.; Sodano, G. Studies on the structure of calliactine, the zoochrome of the sea anemone calliactis parasitica. Tetrahedron 1987, 43, 4023–4030. [Google Scholar] [CrossRef] Schmitz, F. J.; DeGuzman, F. S.; Hossain, M. B.; van der Helm, D. Cytotoxic aromatic alkaloids from the ascidian amphicarpa meridiana and leptoclinides sp.: meridine and 11- hydroxyascididemin. J. Org. Chem. 1991, 56, 804–808. [Google Scholar]
- Kitahara, Y.; Nakahara, S.; Yonezawa, T.; Nagatsu, M.; Shibano, Y.; Kubo, A. Synthetic studies on pentacyclic aromatic alkaloids, kuanoniamine A, 11-hydroxyascididemin, and neocalliactine acetate. Tetrahedron 1997, 53, 17029–17038. [Google Scholar] [CrossRef]
- Luo, Y. L.; Chou, T. C.; Cheng, C. C. Design of antineoplastic agents on the basis of the “2- phenyl-naphthalene-type” structural pattern. 3. synthesis and biological activity evaluation of 5H- benzo[b]naphtho[2,3-d]pyrrole-6,11-dione derivatives. J. Heterocyclic Chem. 1996, 33, 113–117. [Google Scholar] [CrossRef]
- Dewar, M. J. S.; Maitlis, P. M. Electrophilic substitution. Part XI. Nitration of some six-membered nitrogen-heterocyclic compounds in sulphuric acid. J. Chem. Soc. 1957, 2521–2528. [Google Scholar] [CrossRef]
- Crout, D. H. G.; Penton, J. R.; Schofield, K. Heteroaromatic reactivity. Part V. A quantitative study of the products of nitration of quinolinium ion in sulphuric acid. J. Chem. Soc. (B) 1971, 1254–1256. [Google Scholar] [CrossRef]
- Thomson, R. H. Studies in the juglone series. I. Some halogen derivatives and their reaction with aniline. J. Org. Chem. 1948, 13, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Shin, K. J.; Kim, D. C.; Yoo, K. H.; Kim, D. J.; Park, S. W. A new synthetic route to 6,7- dichloro-5,8-quinoxaline-dione and synthesis of its derivatives. Heterocycles 1996, 43, 2495–2502. [Google Scholar]
- Couladouros, E. A.; Plyta, Z. F.; Haroutounian, S. A. Efficient synthesis of aminonaphthoquinones and azidobenzohydroquinones: mechanistic considerations of the reaction of hydrazoic acid with quinones. An overview. J. Org. Chem. 1997, 62, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Partridge, M. W.; Vipond, H. J. 3,6-phenanthrolines derived from 2,2’-diaminobenzophenone. J. Chem. Soc. (C) 1962, 632–635. [Google Scholar] [CrossRef]
- Gorvin, J. H.; Whalley, D. P. Aromatic nitro-group displacement reactions. Part 1. A novel route to substituted 10-phenylacridones. J. Chem. Soc. Perkin Trans. 1979, 1, 1364–1370. [Google Scholar] [CrossRef]
- Sample availability: Samples not available.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.