Results and Discussion
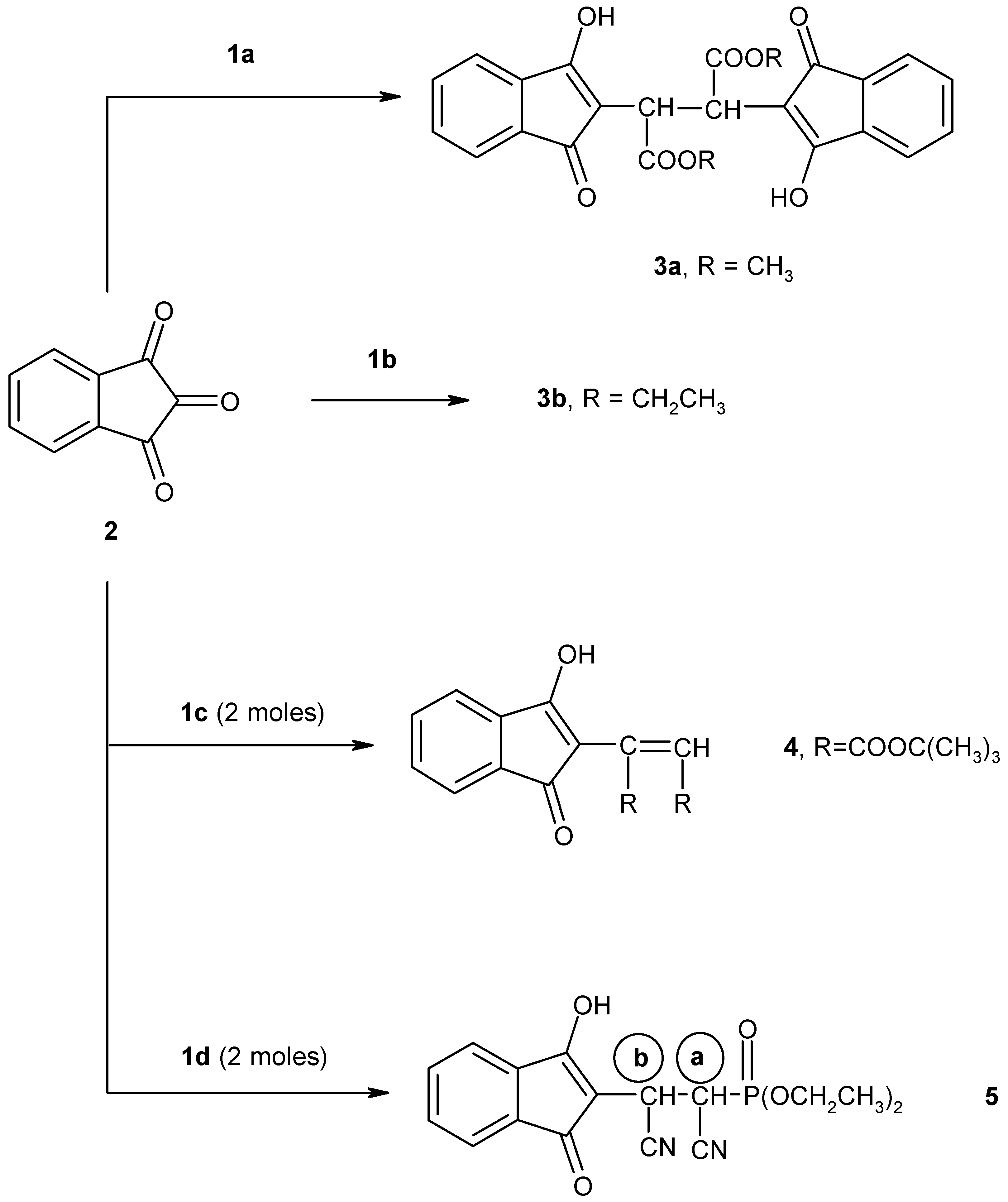
We have found that the reaction of 1,2,3-indantrione (
2) with 1 mol equivalent of methyl diethylphosphonoacetate (
1a), in the presence of alcoholic sodium ethoxide solution, proceeds at room temperature to give a chromatographically pure adduct formulated as dimethyl 2,3-bis(3-hydroxy-1-oxoinden-2-yl)butane-1,4-dioate (
3a) (
Scheme 1).
The structure of
3a was confirmed by its satisfactory IR,
1H-NMR and MS and elemental analyses (cf. Experimental). The
1H-NMR spectrum of
3a showed two proton doublets centered at 4.30 and 4.33 ppm, with
3J
H-H = 16 Hz that suggested a trans configuration for the adduct [
4]. Similarly, triethylphosphonoacetate (
1b) reacted with 1,2,3-indantrione (
2) in the presence of alcoholic sodium ethoxide solution at room temperature for 6 hours to give a crystalline product that was assigned structure
3b (
Scheme 1), on the basis of its IR,
1H-NMR, MS and elemental analyses (cf. Experimental). Formation of compounds
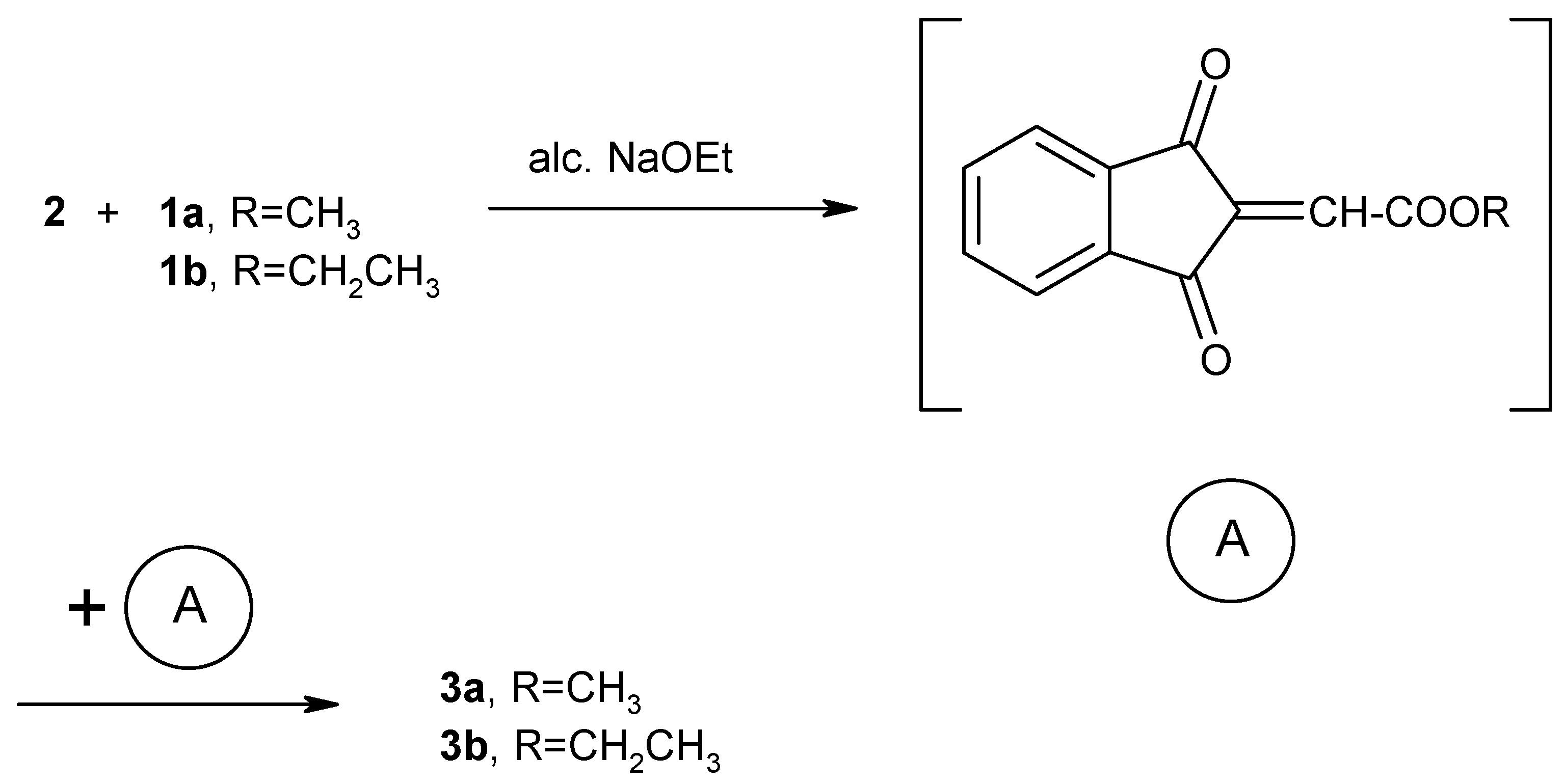
3a and
3b can be explained in terms of carbonyl olefination of
2 by the Wittig-Horner reagents
1a,b to give the reactive intermediate (A). Dimerization of the unstable reactive intermediate (A) results in the formation of the stable dimeric products
3a,b (
Scheme 2).
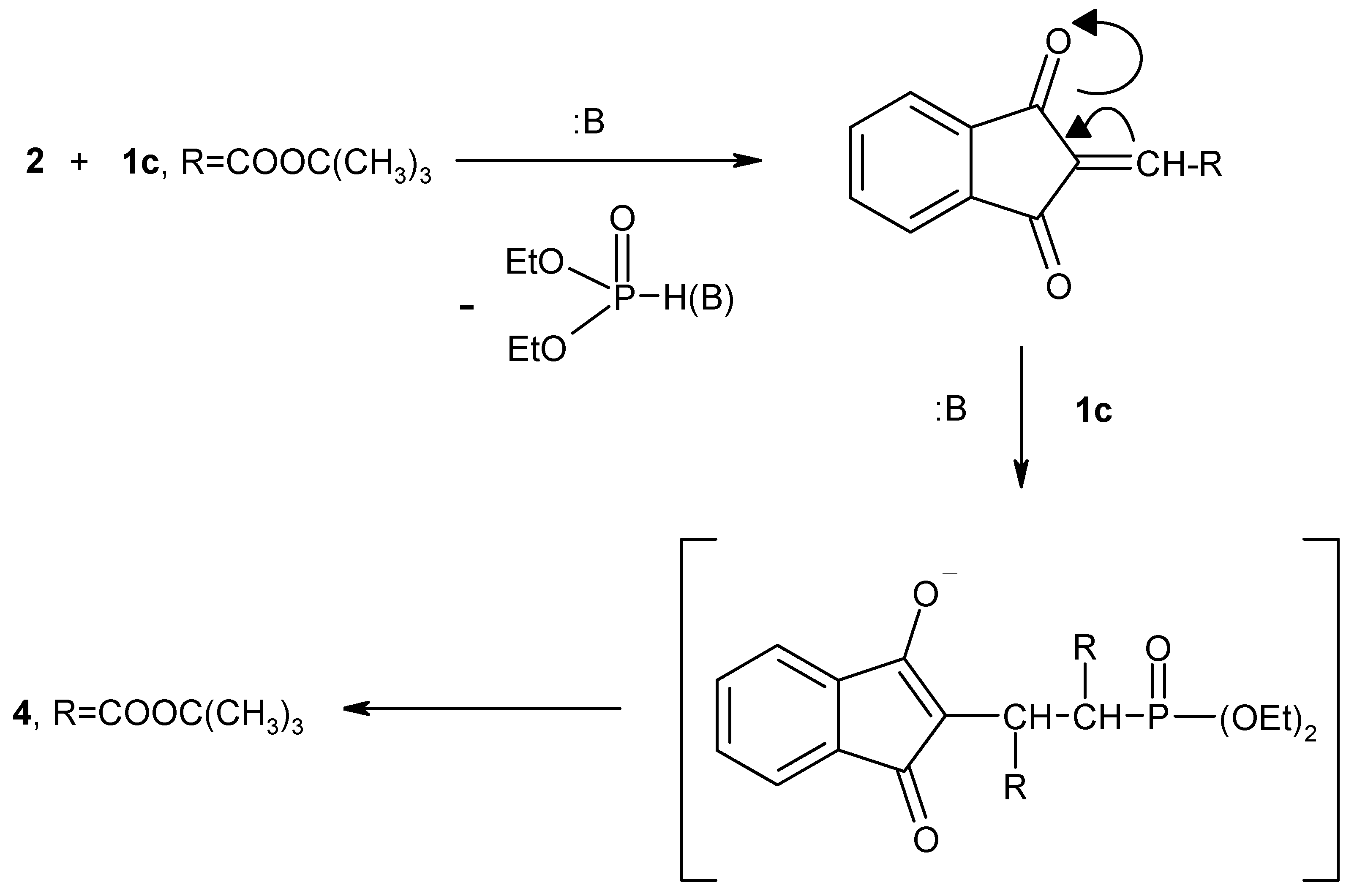
We have also investigated the reaction of compound
2 with
tert-butyl diethylphosphonoacetate (
1c). The reaction of
2 with
1c was performed in alcoholic sodium ethoxide solution in 1:2 molar ratio to give the olefinic product
4 (
Scheme 3).
Compound
4 was obtained in a sharp-melting chromatographically pure form. Its elemental and mass spectral analyses corresponded to a molecular formula of C
21H
24O
6, and the structure of the adduct was further confirmed by its IR,
1H-NMR and mass spectral data (cf. Experimental). A possible explanation of the course of the reaction of
tert-butyl diethylphosphonoacetate (
1c) with 1,2,3-indantrione (
2) is shown in
Scheme 3. In the presence of NaOEt solution trione
2 reacts with two moles of
tert-butyl diethylphosphonoacetate (
1c) to give the stable olefinic compound
4. Next, when 1,2,3-indantrione (
2) was treated with one mole equivalent of diethyl(cyanomethyl)phosphonate (
1d) in the presence of alcoholic sodium ethoxide solution at room temperature for 10 hours, adduct
5 together with some unchanged trione
2 were isolated. Carrying out the reaction using two moles of
1d instead of one led to the formation of adduct
5 in a good yield (
Scheme 1). The proposed structure of compound
5 was confirmed by correct elemental analyses, molecular weight determination by MS, IR spectrum,
31P NMR spectrum which indicates a phosphonate structure [
6,
7,
8] and
1H NMR spectrum.
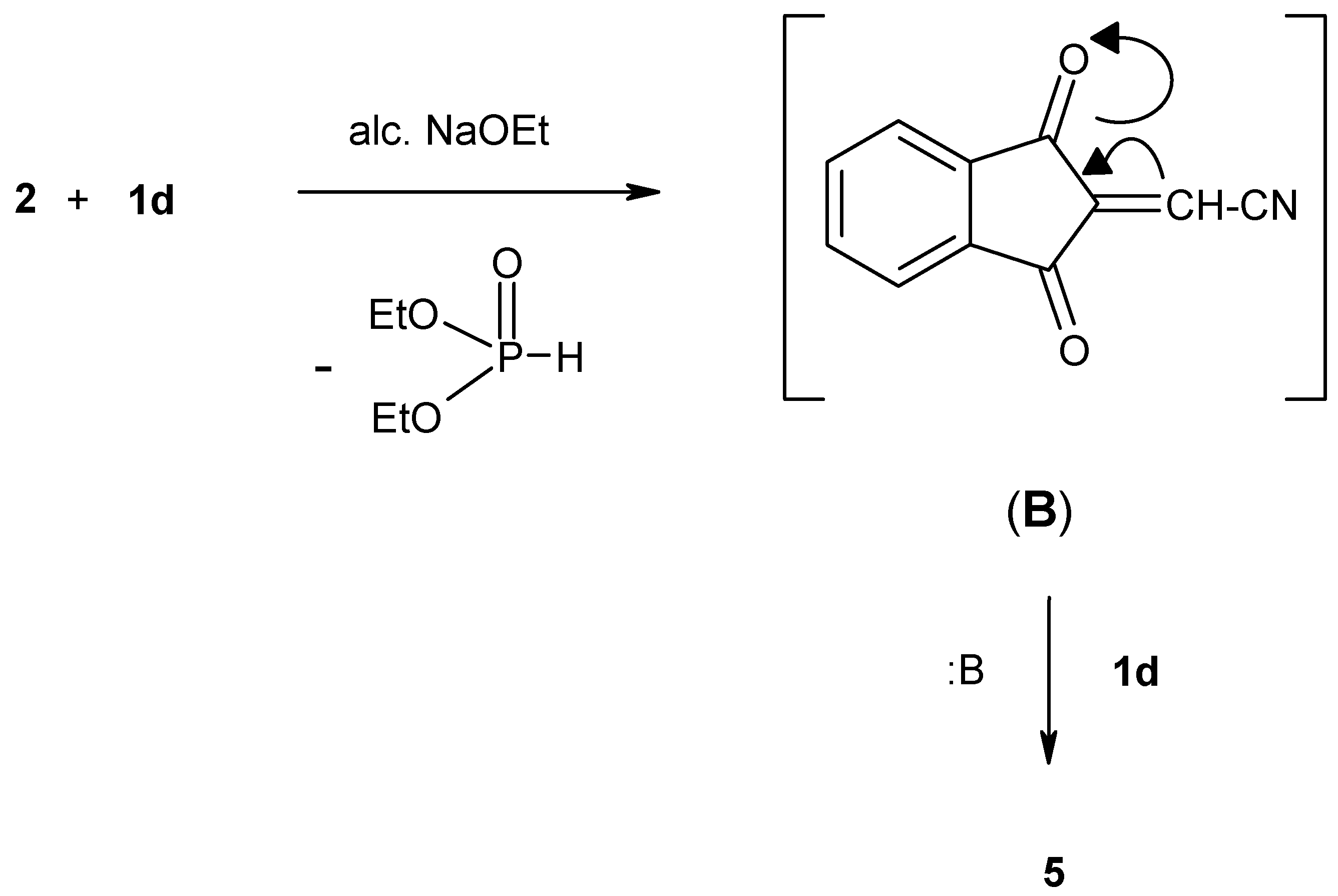
Apparently compound
2 reacts with one mole equivalent of diethyl (cyanomethyl)phosphonate (
1d) in the presence of sodium ethoxide solution to give the unstable olefinic intermediate (B) which then reacts with another mole of
1d, in the presence of NaOEt solution, to give the stable phosphonate adduct
5 (
Scheme 4).
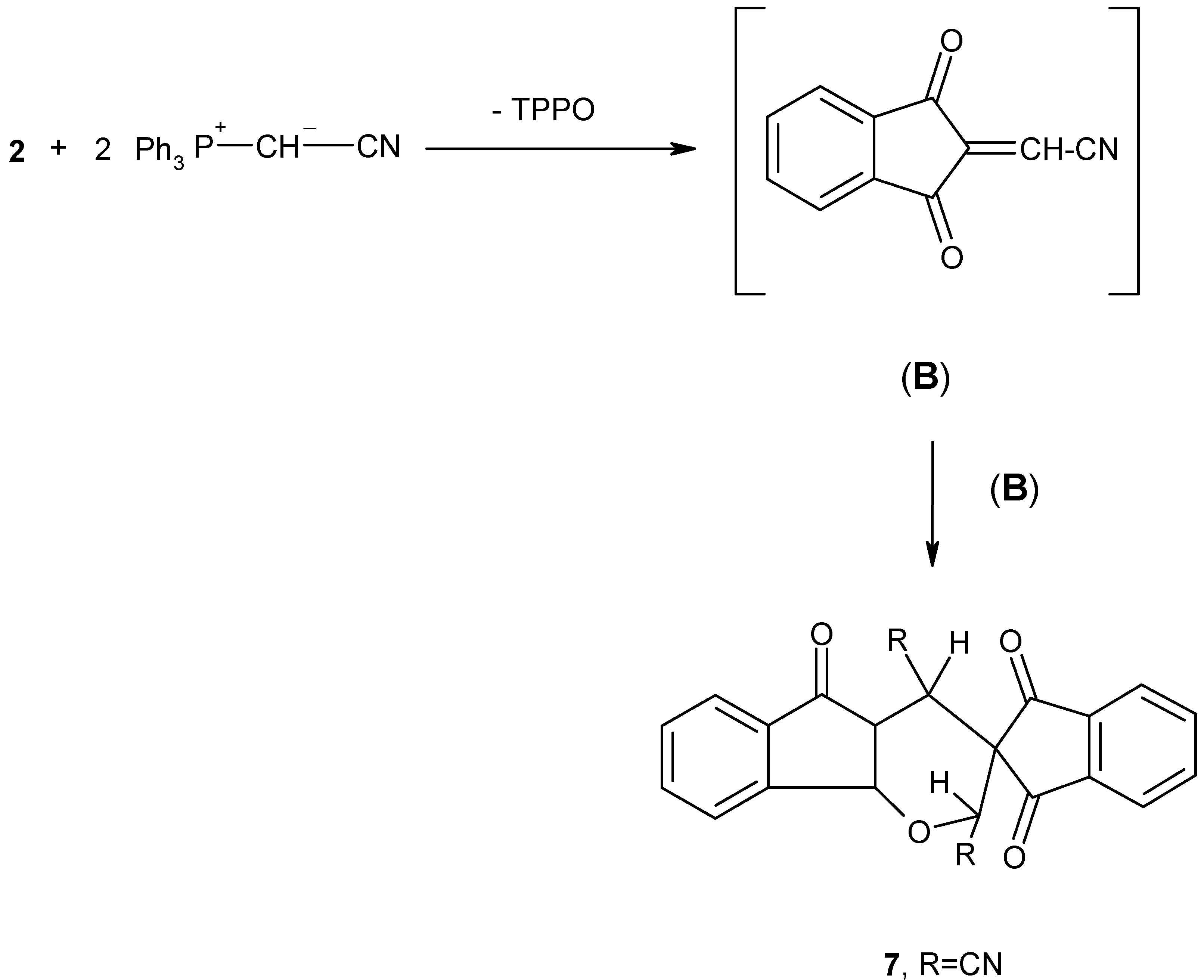
Finally, this study has been extended to include the reaction of 2 with cyanomethylene-triphenylphosphorane (Ph3P+--CHCN) (6) to establish whether it would behave in a similar manner. Thus, 1,2,3-indantrione (2) was treated with two mole equivalents of phosphonium ylide 6 in dry benzene at room temperature for 5 hours.
The reaction mixture was separated by column chromatography on silica gel, whereby, triphenylphosphine oxide, as well as a crystalline product
7 were isolated (
Scheme 5). Compound
7 was obtained as colourless crystals (60% yield) and was formulated as 2,4-dihydrospiro(indan-2,3-(2H)indeno{[1,2-b]pyran}-1,3,5(4H)-trione dicyanate.
The structure of the new spiro compound
7 was established from its elemental analyses and spectral properties (cf. Experimental). Adduct
7 can be obtained
via carbonyl olefination with one molecule of ylide
6 to give the reactive intermediate (B), with expulsion of triphenylphosphine oxide. Dimerization of (B) by a Diels-Alder type reaction [
10], afforded the final product (
Scheme 5). From the above results, it is evident that 1,2,3-indantrione (
2) behaves toward cyanomethylene-triphenylphosphorane
6 in a manner quite similar to that previously reported for the reaction of 1,2,3-indantrione with alkoxycarbonylmethylenetriphenylphosphoranes which produce the spiro compound
7 (R = COOCH
3 or COOC
2H
5) almost exclusively (
Scheme 5) [
10].
Experimental
General
All melting points are uncorrected. Wittig-Horner reagents
1a-d were prepared by the Michaelis-Arbuzov reaction [
12,
13]. Cyanomethylenetriphenylphosphorane was prepared by a published procedure [
14]. The IR spectra were measured in KBr disks with a Perkin Elmer Infracord Model 157 Grating Spectrophotometer. The
1H-NMR spectra were run on Varian Spectrophotometer at 200 MHz, using TMS as an internal reference. The
31P-NMR spectra were recorded in CDCl
3 (vs., 85% H
3PO
4 as an external standard) with a JNM-PS-100Fa Spectrometer. The mass spectra were run at 70 eV with Kratos MS equipment and/or a Varian MAT 311 A Spectrometer.
Reaction of 1,2,3-indantrione (2) with methyl diethylphosphonoacetate (1a): Preparation of dimethyl 2,3-bis(3-hydroxy-1-oxoinden-2-yl)butane-1,4-dioate (3a).
A solution of 1 mol of sodium ethoxide in absolute ethanol was treated with an equimolar amount of phosphonate
1a (0.21 g, 0.001 mol). After 5 min. 1 mol of trione
2 was added and the resulting mixture was stirred at room temperature for 6 hr. The reaction mixture was poured onto a small amount of water, extracted with ethyl acetate, dried, and the extracts were evaporated under vacuum. The residue was purified on a silica gel column using 40-60°C pet. ether- ethyl acetate (v:v) as eluent to give compound
3a, which was recrystallized from benzene (yield 78%); m.p. 182-183°C; Analysis Calcd. for C
24H
18O
8 (434.408), C, 66.36; H, 4.18; Found, C, 66.45; H, 4.22; Mol. wt (MS) = 434 (M
+, 75%); IR (cm
-1): 1720, 1675;
1H-NMR (ppm): 3.30 (3H, s, OCH
3); 3.50 (3H, s, OCH
3), 8.20 (s, 1H, D
2O exchangeable, OH) and 8.35 ppm (s, 1H, D
2O exchangeable, OH); signals at 4.30 ppm (d, 2H,
3J
HH = 16 Hz) and 4.33 ppm (d, 2H,
3J
HH = 16 Hz) suggest the trans configuration of adduct
3a [
4]; 6.80-8.05 (complex m, 8H, aromatic protons).
Reaction of 1,2,3-indantrione (2) with triethylphosphonoacetate (1b): Preparation of diethyl 2,3-bis-(3-hydroxy-1-oxoinden-2-yl)butane-1,4-dioate (3b).
A solution of 1 mol of sodium ethoxide in absolute ethanol was treated with an equimolar amount of phosphonate 1b (0.22 g, 0.001 mol), After 5 min. 1 mol of trione 2 was added and the resulting mixture was stirred at room temperature for 4 hr. The reaction mixture was poured onto a small amount of water, extracted with ethyl acetate and the filtered extracts were evaporated under vacuum after drying over anhydrous sodium sulfate. The residual material was purified on a silica gel column using 40/60°C pet. ether - ethyl acetate (v:v) as an eluent to give 3b as pale orange crystals (yield 80%); m.p. 159-160°C from benzene; Analysis Calcd. for C26H22O8 (462.462): C,67.53; H, 4.80; Found, C, 67.6; H, 4.83; Mol. wt (MS) = 462. (M+, 65%); IR (cm-1): 3500 (OH), 1740 (C=O, ester), 1680 cm-1 (C=O, indene); 1H-NMR (δ): 0.6 (t, 3H, ethoxy-CH3), 1.25 (t, 3H, ethoxy-CH3), 3.45 (q, 2H, ethoxy-CH2), 3.7 (q, 2H, ethoxy-CH2), 4.5 (2H, t, 2JHH = 16 Hz, CH-CH) and 6.8-8 (m, 8H, Ar), . 8.2 (s, 1H, D2O exchangeable proton, OH), 8.35 (s, 1H, D2O exchangeable proton, OH).
Reaction of tert-butyl diethylphosphonoacetate (1c) with 1,2,3-indantrione (2): Preparation of di-tert-butyl (Z)-2-(3-hydroxy-1-oxoinden-2-yl)but-2-ene-1,4-dioate (4).
The reaction of phosphonate 1c (0.002 mol) with trione 2 (0.001 mol) was carried out in alcoholic sodium ethoxide solution (0.002 mol of Na in 20 mL C2H5OH). The mixture was maintained under stirring at room temperature over night. The reaction mixture was poured onto water and extracted with chloroform. The extracts were evaporated under vacuum after drying. The residue was purified on a silica gel column using 40/60°C pet. ether - ethyl acetate (v:v) as an eluent to give compound 4 (yield 76%); M.p. 98-100°C (from cyclohexane); Analysis, Calcd. for C21H24O6 (372.423): C, 67.73; H, 6.50; Found, C, 67.65; H, 6.55; mol. wt. (MS) = 372. (M+, 70%); IR (cm-1): 1715 (C=O, indene), 1740 (C=O, ester) and 3524 (OH); 1H-NMR: 1.85 ppm (s, 9H, C (CH3)3) and 1.90 ppm (s, 9H, C(CH3)3), 6.95 (s, 1H, methine), 7.35-7.60 (m, 4H, Ar) and 9.40 (s, 1H, D2O exchangeable proton, OH).
Reaction of diethyl(cyanomethyl)phosphonate (1d) with 1,2,3-indantrione (2): Preparation of [1,2-dicyano-2-(3-hydroxy-1-oxo-1H-inden-2-yl)-ethyl]-phosphonic acid diethyl ester (5).
A solution of two moles of sodium ethoxide in absolute ethanol was treated with equimolar amounts of phosphonate
1d and trione
2 and the resulting mixture was stirred at room temperature for 10 hr. The reaction mixture was poured onto a small amount of water, extracted with ethyl acetate and the extract was dryed over anhydrous sodium sulfate then evaporated under reduced pressure. The residual material was applied to a silica gel column chromatography using 40/60°C pet. ether – ethyl acetate (v:v) as an eluent to give 5 as pale yellow crystals in 75% yield; m.p. 176-177°C from benzene; Analysis, calcd. for C
17H
17N
2O
5P (360.311): C, 56.67; H, 4.76; N, 7.77; P, 8.60%. Found: C, 54.6; H2, 4.8; N, 7.4; P, 8.27%; mol. wt. (MS) = 360 (M
+, 90%). IR (cm
-1): 3502 (OH), 1230 (P-O-bonded)[
5], 1047 (P-O-C
2H
5) [
5] and 2208 cm
-1 (CN).
31P-NMR: δ = 19.95 ppm indicates a phosphonate structure [
6,
7,
8].
1H-NMR δ (ppm): 1.25 (6H, t) and 3.95 ppm (4H, q) (P-OCH
2CH
3) [
9]; 3.70 ppm (d, 1H, with
3J
HP = 12 Hz, J
HH = 7.5 Hz) and 3.65 ppm (
2J
HP = 10 Hz, J
HH = 7.5 Hz) for the two methine protons b and a, respectively (
Scheme 1); 7.35-7.64 ppm (4H, m, aromatic protons); 9.4 (s, D
2O exchangeable, OH).
Reaction of 1,2,3-indantrione (2) with cyanomethylene triphenylphosphorane (6): Preparation of 2,4-dihydrospiro(indan-2,3-(2H)indeno{[1,2-b]pyran}-1,3,5(4H)-trione dicyanate (7).
To a suspension of trione 2 (0.16g, 0.001 mol) in dry benzene (25 mL), was added a solution of the ylide 6 (0.6g, 0.002 mol) in absolute benzene (25 mL). The reaction mixture was stirred at room temperature for 5 h, then evaporated under reduced pressure. The residual substance was dissolved in CH2Cl2 (100 mL) and evaporated till dryness in the presence of silica gel (5 g). The reaction mixture was then added to a column previously loaded with silica gel in petroleum ether. The column was developed with petroleum ether (60/80°C) containing increasing amounts of CHCl3. A fraction eluted with 9:1 v/v petroleum ether/CHCl3 gave a substance that was recrystallized from benzene to give compound 7 as colourless crystals (60% yield); m.p. 168-169°C; Anal. Calcd. for C22H10N2O4 (366.336): C, 72.13, H, 2.75; N, 7.64; Found : C, 72.3; H, 2.8; N, 7.66%; Mol. wt (MS) = 366 (M+, 60%); IR (cm-1): 1745 (C=O, cyclopentanone), 1715 (C=O, 1,3-dicarbonyl), 2226 (CN) and 3100 (CH, aromatic). 1H-NMR (ppm) 4.21 and 4.23 (two s, 2H, dihydropyran ring), 7.47 (dd, 1H, JHH = 8.4 Hz, JHH = 1.1 Hz), 7.68 (m, 1H), 7.43 (m, 1H), 8.21 (dd, 1H, JHH = 8.4 Hz, JHH = 1.1 Hz), 7.8 (m, 4H, Ar). A fraction eluted by petroleum ether/CHCl3 (7:3 v/v) yielded colourless needles, shown to be triphenylphosphine oxide by m.p., mixed m.p., and comparison of the respective IR and MS spectra.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





