Results and Discussion
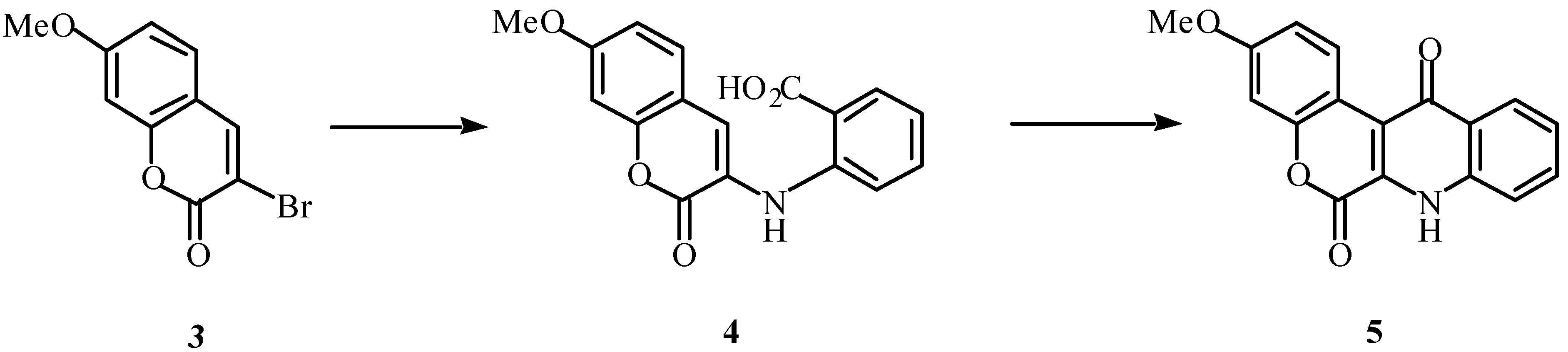
The pathway shown in
Scheme 1 was first explored. Rotenoids are often oxygenated at positions 2 and 3 [
9]. Use of 3-bromo-7-methoxycoumarin (
3) would result in only the monomethoxy rotenonoids, however this material was available in quantity and was seen as a useful prototype for our studies.
We anticipated that Ullmann-type condensation [
10,
11] of
3 with anthranilic acid would afford compound
4, which should undergo intramolecular cyclization to the desired azarotenonoid
5. Reaction of
3 with anthranilic acid in the presence of copper, however, produced acid
6 (42%) after 4 hours, and upon treatment of
6 with polyphosphoric acid (PPA), 3,1-benzoxazin-4-one
7a was obtained (70%,
Scheme 2).
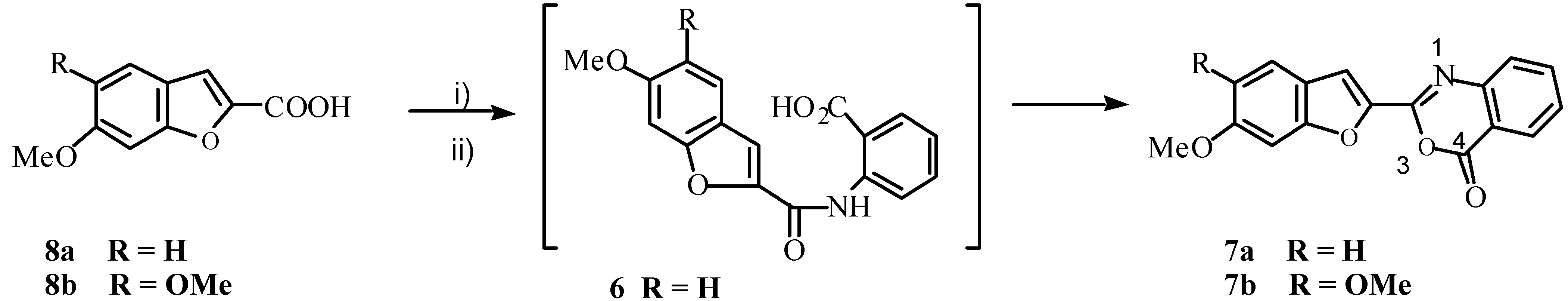
Scheme 2.
Reagents: i) antranilic acid, Cu, CuBr, K2CO3, DMF, 140˚C; ii) PPA
Scheme 3.
Reagents: i) PCl5; ii) antranilic acid
Strong IR absorptions at 1635 and 1773 cm
-1, corresponding to the C=N and ester functionality, were observed. Compounds of this type are known to undergo base hydrolysis to produce the corresponding
N-acyl anthranilic acids, which are then readily reconverted to benzoxazinones on treatment with NaOAc/Ac
2O [
12]. Our reaction product exhibited this reactivity, forming compound
6 on treatment with KOH. Subsequent treatment of compound
6 with NaOAc/Ac
2O, caused re-formation of benzoxazinone
7a.
We also obtained benzoxazinone
7a in 40% yield by treatment of acid
8a with PCl
5 and then reacting the resultant acid chloride with anthranilic acid (
Scheme 3). It is clear that under Ullmann reaction conditions, 3-bromo-7-methoxycoumarin (
3) underwent rearrangement to 6-methoxy-benzofuran-2-carboxylic acid (
8a). The rearrangement of such coumarins is known to occur in alkaline media [
13,
14]. We verified our observation by subjecting compound
3 to treatment with potassium carbonate in DMF at 140°C for 4 hours – conditions as obtained for the attempted Ullmann reaction but without copper and anthranilic acid. Quantitative conversion to compound
8a, identical in all respects with an authentic sample [
15], was achieved.
Compounds of type
6, then, would cyclize to yield 3,1-benzoxazin-4-ones more readily than they would to give the desired benzazepines. (Compound
8b on treatment with PCl
5 followed by anthranilic acid gave
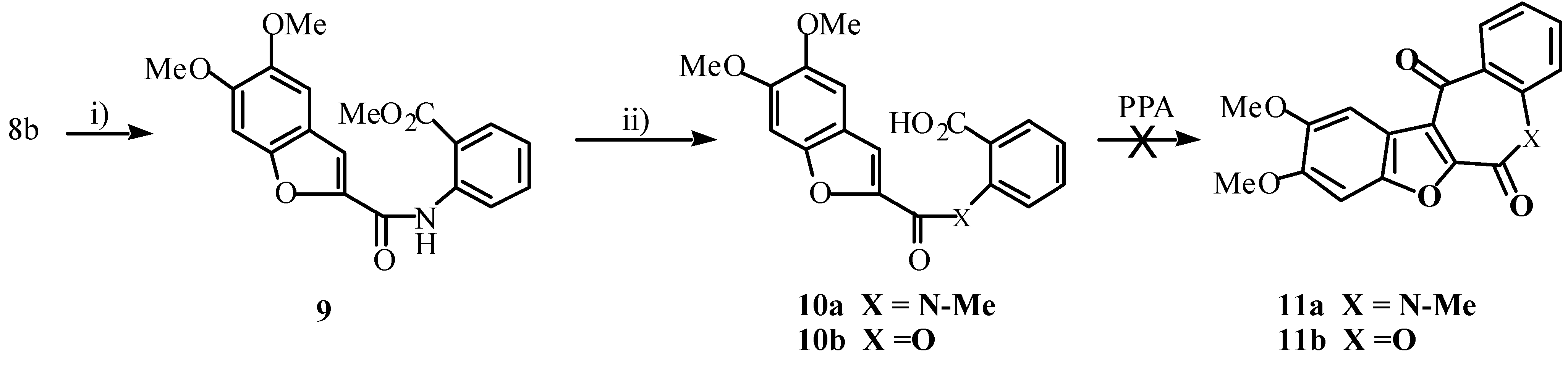
7b.) We considered that absence of the amide proton would disallow benzoxazinone formation, and could augur well for occurrence of the desired cyclization. With this in mind, compound
10a was prepared as shown in
Scheme 4. Treatment of
10a with PPA, however, yielded none of the required
11a and as predicted, none of the corresponding benzoxazinone was formed. Even after heating
10a in PPA at 80-100°C for 24 hours, only starting material was recovered. Compound
10a was also unreactive to Friedel-Crafts conditions. Treatment with PCl
5 followed by SnCl
4 for 23 hours at 95°C afforded only starting material. Our work with the analogous 2-carbophenoxy-5,6-dimethoxybenzofuran-2'-carboxylic acid (
10b, X = O) also shows that it is inert to Friedel-Crafts reaction conditions (using SnCl
4 as Lewis acid) [
16].
Scheme 4.
Reagents: I) methyl anthranilate, DCC, DMAP, CH2Cl2, rt; ii) NaH, MeI, THF
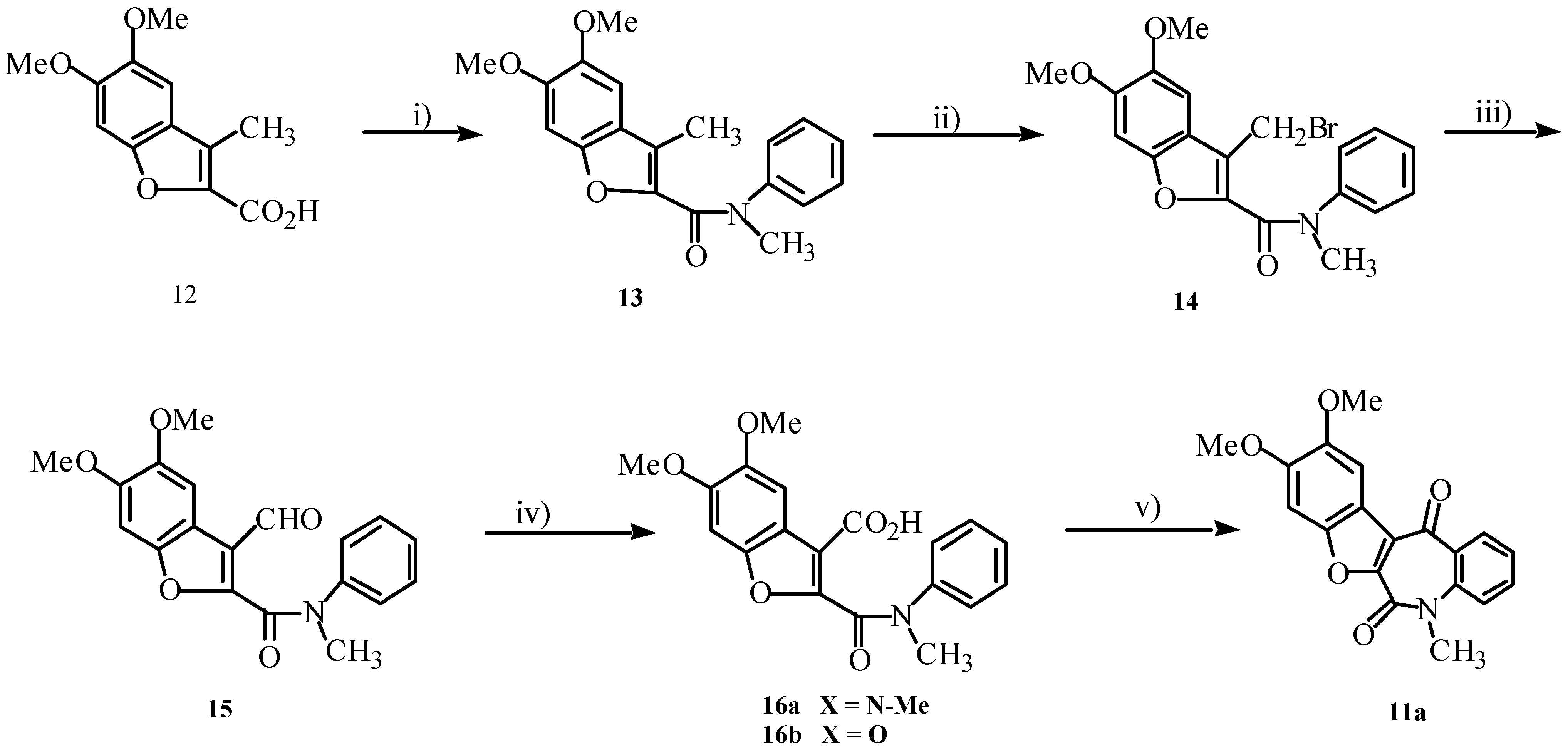
Synthesis of compound
11a was eventually achieved by the pathway shown in
Scheme 5. Attempts at direct oxidation of
13 to
16a using KMnO
4 were unsuccessful. Oxidation of
14 usingtrimethylamine-
N-oxide (TMANO), however, produced aldehyde
15 (50%), which on further oxidation yielded
16a (90%) [
17,
18,
19]. Heating
16a in PPA at 90
0C for 24 hours effected intramolecular cyclization to
11a in 25% yield.
Scheme 5.
Reagents: I) N-methylaniline, DCC, DMAP, CH2Cl2, rt; ii) NBS, CCl4, hυ;
iii) TMANO, DMSO, rt; iv) NBS, CCl4, hυ, then H2O; v) PPA
The fact that we achieved hydrolysis of amide 13 in refluxing NaOH (40% aqueous) prompted us to investigate the possibility of similar reaction with our compound 11a in hope of possible rearrangement to the azarotenonoid. Compound 11a, however, proved resistant to treatment with NaOH.
Experimental
General
All melting points are uncorrected. IR spectra (KBr disks) were obtained on a Perkin Elmer 735B model or a Perkin Elmer 1600 FT-IR spectrometer. NMR spectra were recorded were determined in CDCl3 solutions on a Bruker 200 MHz spectrometer. The resonances are reported in δ units downfield from TMS; J values are given in Hz. For ease of reporting 1H-NMR data, the protons on the benzofuran side of the molecule have been labelled as H and those on the phenyl ring as H'. Elemental analyses were carried out by MEDAC Ltd., Egham, Surrey, UK.
2-[(6-Methoxybenzofuran-2-carbonyl)-amino]benzoic acid (6)
To a 25 mL round bottomed flask fitted with a reflux condenser and a calcium chloride guard tube, were added 7-methoxy 3-bromocoumarin (3) (0.50 g, 1.97 mmol) and N, N-dimethylformamide (10 mL). Anthranilic acid (0.27 g, 1.97 mmol), potassium carbonate (0.875 g, 6.30 mmol), copper powder (5.0 mg) and copper (I) bromide (8 mg) were added with stirring to this solution and the mixture was heated at 140°C for 4 hours. The mixture was then allowed to cool, diluted with water (40 mL), and treated with decolorizing carbon (0.12 g). After filtration the filtrate was acidified with hydrochloric acid (3N) and the resulting precipitate was collected and washed with water (25 mL). Recrystallization from ethanol gave 6 (0.26 g, 42%); mp 209-212°C; IR cm-1: 1624, 1675, 3350 (br); 1H-NMR δH: 3.92 (3H, s, -OCH3), 5.95 (1H, br, -N-H), 6.85 (1H, dd, J = 2, 8, H-5), 7.15 (2H, m, H-7, 4'), 7.50 (3H, m, H-3, 4, 5'), 8.15 (1H, dd, J = 1, 8, H-6'), 8.80 (1H, dd, J = 1, 8, H-3'), 12.5 (1H, s, COOH); 13C-NMR δC: 55.6, 95.8, 111.1, 113.4, 116.0, 120.1, 120.7, 122.7, 122.7, 131.5, 134.1, 141.1, 148.2, 156.2, 157.1, 159.9,170.3; MS m/e (relative intensity) 311.03 (M+, 82), 293.02 (14), 192.03 (22), 175.04 (100), 119.06 (46). HRMS calcd. for C17H13NO5: 311.0794. Found: 311.0794.
2-(6’-Methoxybenzofuran)-3,1-benzoxazin-4-one (7a)
a) Compound 6 (2.0 g, 6.43 mmol) was added to well-stirred PPA (20 mL) which had been preheated to 90°C. This mixture was stirred at 110°C for 2 h, then poured onto ice (200 g), neutralized with 6N ammonia, and the resultant precipitate filtered and washed with saturated sodium bicarbonate. The solid was air-dried and purified by chromatography (neutral alumina, 4:1 hexane-EtOAc) to give 7a (1.3 g, 70%), mp 210-212°C (MeOH); IR cm-1: 1773, 1635, 1599; 1H-NMR δH: 3.90 (3H, s, -O-CH3 ), 6.95-7.00 (1H, dd, J = 2, 8, 5'-H), 7.15 (1H, d, J = 2, H-7'), 7.50-7.65 (3H, m, H-6, H-8, H-3'), 7.65-7.95 (2H, m, H-7, H-4'), 8.20-8.26 (1H, dd, J = 1, 8, H-5); 13C-NMR δC: 55.5, 95.6, 113.5, 113.9, 116.7, 120.4, 122.8, 127.0, 128.3, 128.6, 136.7, 144.3, 146.4, 157.5, 160.5. Anal. Calcd. for C17H11NO4: C, 69.61; H, 3.78; N, 4.78; Found: C, 69.46. H, 3.74; N, 4.76%.
b) PCl
5 (0.63 g, 3.04 mmol) was added, with stirring, to a mixture of 6-methoxybenzofuran-2- carboxylic acid (
8a, 0.38 g, 1.99 mmol) [
13] in benzene (5mL). This was heated at reflux for 2 h, after which time it was cooled to room temperature and then in an ice-bath. Anthranilic acid (0.28 g, 2.01 mmol was dissolved in pyridine (1 mL) and this solution was added dropwise to the reaction mixture with cooling and stirring. Upon completion of addition the ice-bath was removed and the reaction mixture was stirred at room temperature for 3 h. Benzene was then evaporated
in vacuo and water added to the resultant crude brown precipitate. This was then collected by filtration and air dried. Recrystallisation from methanol yielded
7a as pale yellow crystals (0.235 g, 40%).
Alkaline hydrolysis of compound 7a.
Compound 7a (51.6 mg, 0.18 mmol) was added to ethanol (2 mL) with stirring. To this mixture potassium hydroxide (50 mg, 0.89 mmol) was added and the mixture heated at reflux for 2 h. The reaction mixture was then concentrated in vacuo and the crude product dissolved in the minimum amount of water and acidified with conc. HCl. The product was collected by filtration, washed with cold water and dried at the pump, to yield 6 as a white powder (51.4 mg, 94%), mp 209-212ºC.
Conversion of 6 to 7a using sodium acetate and acetic anhydride.
A mixture of 6 (38.7 mg, 0.12 mmol), acetic anhydride (0.65 mL) and sodium acetate (204 mg) was heated to 140ºC with stirring for 1 h. The reaction mixture was then cooled in an ice-bath and immediately solidified to give a pale yellow precipitate. Water (5 mL) was added to this precipitate and the solution extracted with dichloromethane. The organic extract was then concentrated in vacuo and the crude product recrystallised from methanol to yield 7a as a pale yellow powder (20.3 mg, 56%), mp 210-213ºC.
2-(5',6'-Dimethoxybenzofuran)-3,1-benzoxazin-4-one (7b).
PCl
5 (0.76 g, 3.63 mmol) was added, with stirring, to a mixture of 5,6-dimethoxybenzofuran-2-carboxylic acid (
8b, 0.50 g, 2.26 mmol) [
20] in benzene (8 mL). This was heated at reflux for 2 h, then cooled to room temperature and then in an ice-bath. A solution of anthranilic acid (0.33 g, 2.41 mmol) in pyridine (1 mL) was added dropwise to the reaction mixture with cooling and stirring. Upon completion of addition the ice-bath was removed and the reaction mixture was stirred at room temperature for 3 h. Benzene was then evaporated
in vacuo and water added to the resultant crude precipitate which was collected by filtration and air dried. The crude was purified by chromatography (SiO
2, dichloromethane) to yield
7b as yellow crystals (0.28 g, 39%), mp 231-232°C (MeOH); IR cm
-1 1751, 1632, 1598;
1H-NMR δ
H 3.90 (3H, s, O-CH
3), 4.00 (3H, s, -O-CH
3), 7.00 (1H, s, H-3'), 7.12 (1H, s, H-7'), 7.40-7.55 (1H, m, H-7), 7.60 (1H, s, H-4'), 7.70-7.90 (2H, m, H-6, H-8), 8.20-8.30 (1H, dd,
J = 2, 8, H-5);
13C-NMR δ
C 56.2, 56.3, 95.2, 95.2, 102.4, 113.8, 119.4,127.2, 128.2, 128.7, 128.7, 136.7, 144.4, 146.7, 147.7, 151.1, 151.9, 158.6. Anal. Calcd. for C
18H
13NO
5: C, 66.86; H, 4.06; N, 4.33 %; Found: C, 66.60; H, 4.03; N, 4.31.
2-[(5,6-Dimethoxybenzofuran-2-carbonyl)-amino]benzoic acid methyl ester (9).
5,6-Dimethoxybenzofuran-2-carboxylic acid [
20] (0.513 g, 2.437 mmol) was added, with stirring to a solution of methyl anthranilate (0.303 g, 2.00 mmol) in dichloromethane (10 mL). To this mixture was added 4-(
N,
N-dimethylamino)pyridine (DMAP) (0.036 g, 0.294 mmol) and 1,3-dicyclo-hexylcarbodiimide (DCC) (0.506 g, 2.45 mmol) and the whole left to stir at room temperature for 24 h. The reaction mixture was then filtered under vacuum and the filtrate concentrated
in vacuo. The crude product was recrystallised from CH
2Cl
2/MeOH to yield compound
9 as feathery brown crystals (0.530 g, 77%), mp 210-211°C; IR cm
-1: 1695, 1670, 1568;
1H-NMR δ
H: 3.90 (3H, s, -O-CH
3), 4.00 (3H, s, -O-CH
3), 4.10 (3H, s, -CO
2-CH
3), 7.10-7.20 (3H, m, H-4, H-7, H-4'), 7.50-7.65 (2H, m, H-3, H-5'), 8.10 (1H, d,
J = 8, H-6'), 8.90 (1H, d,
J = 8, H-3');
13C-NMR δ
C: 52.4, 56.2, 56.2, 95.3, 102.5, 111.8, 115.2, 119.5, 120.5, 122.7, 130.9, 134.6, 141.1, 147.3, 148.1, 150.3, 150.5, 157.2, 168.6. Anal. Calcd. for C
19H
17NO
6: C, 64.22; H, 4.82; N, 3.94. Found: C, 63.91; H, 4.87; N, 4.11%.
2-[(5,6-Dimethoxybenzofuran-2-carbonyl)-methylamino]benzoic acid (10a).
Sodium hydride (0.093 g, 3.87 mmol) was stirred with pre-dried THF (6 mL) in an atmosphere of nitrogen. Compound 9 (0.20 g, 0.58 mmol) was added and the mixture was stirred at room temperature for 30 minutes. Iodomethane (0.11 mL, 1.72 mmol) was added and the reaction mixture was then heated at reflux for 6 h. After this time, ethanol was added to the reaction mixture, solvent was removed in vacuo and water (20 mL) added to the resultant precipitate. The solution formed was then extracted exhaustively with CH2Cl2, the extract concentrated in vacuo, and the crude product recrystallised from hexane to yield 10a as light brown shiny crystals (0.102 g, 70%), mp 194-196°C; IR cm-1: 1727, 1678, 1621; 1H-NMR δH: 3.45 (3H, s, -NH-CH3), 3.85 (3H, s, -O-CH3), 3.90 (3H, s, -O-CH3), 6.05 (1H, s, H-7), 6.80-6.90 (2H, m, H-4, H-4'), 7.29-7.38 (1H, dd, J = 1, 8, H-5'), 7.50-7.70 (2H, m, H-3, H-6'), 8.09-8.18 (1H, dd, J = 1, 8, H-3'); 13C-NMR δC: 39.2, 55.7, 55.7, 94.5, 102.1, 111.6, 118.4, 128.0, 129.1, 129.4, 131.7, 132.8, 143.3, 146.5, 147.0, 149.2, 149.5, 159.1, 166.4; MS m/e (relative intensity) 355 (M+, 46), 310 (12), 205 (100), 149 (40). HRMS calcd. for C19H17NO6: 355.1056. Found: 355.1071.
5,6-Dimethoxy-3-methylbenzofuran-2-carboxylic acid methyl phenyl amide (13).
To a mixture of
N-methylaniline (0.462 g, 4.31 mmol) in CH
2Cl
2 (25 mL), 3-methyl-5,6-dimethoxybenzofuran-2-carboxylic acid [
21] (1.01 g, 4.27 mmol) was added with stirring. DMAP (0.090 g, 0.44 mmol) was then added, followed by DCC (0.934 g, 4.53 mmol). The reaction mixture was left to stir at room temperature for 23 h. After this time the mixture was filtered and the filtrate was washed successively with water (2 x 10 mL), 5% acetic acid (2 x 10 mL) and again with water (2 x 10 mL). The organic solution was then dried (Na
2SO
4) and concentrated
in vacuo. The crude product was recrystallised from hexane to yield
13 as brown diamond shaped crystals (0.852 g, 62%), mp 102-103°C; IR cm
-1: 1653, 1620, 1597;
1H-NMR δ
H: 2.40 (3H, s, Ar-CH
3), 3.50 (3H, s, -N-CH
3), 3.80 (3H, s, -O-CH
3), 3.85 (3H, s, -O-CH
3), 6.56 (1H, s, H-7), 6.85 (1H, s, H-4), 7.10-7.35 (5H, m, -N-Ar);
13C-NMR δ
C: 9.2, 39.2, 56.2, 56.2, 94.7, 100.8, 120.7, 122.6, 126.0, 126.3, 128.9, 143.3, 144.5, 146.7, 148.4, 149.8, 161.6. Anal. Calcd. for C
19H
19NO
4: C, 70.14; H, 5.89; N, 4.31. Found: C, 69.81; H, 6.04; N, 4.58%.
3-Bromomethyl-5,6-dimethoxybenzofuran-2-carboxylic acid methyl phenyl amide (14).
To a mixture of the amide 13 (1.66 g, 5.10 mmol) in CCl4 (100 mL), N-bromosuccinimide (NBS) (1.11 g, 6.24 mmol) was added with stirring. The reaction was then enclosed in a dark box and irradiated with a 100 W light bulb which allowed gentle reflux. After 24 h the mixture was filtered, the filtrate was concentrated in vacuo, and the resulting crude product was triturated with hot hexane to yield 14 as a tan powder (1.50 g, 73%), mp 113-115°C; IR cm-1: 1702, 1602, 1595; 1H-NMR δH: 3.50 (3H, s, -N-CH3), 3.80 (3H, s, -O-CH3), 3.90 (3H, s, -O-CH3), 5.00 (2H, s, -CH2-Br), 6.50 (1H, s, H-7), 7.05 (1H, s, H-4), 7.15-7.36 (5H, m, -N-Ar); 13C-NMR δC: 22.2, 39.3, 56.3, 56.3, 94.6, 100.8, 118.5, 123.24, 126.0, 126.1, 126.8, 128.9, 129.1, 143.9, 144.0, 147.1, 148.5, 150.3, 160.4. Anal. Calcd. for C19H18BrNO4: C, 56.45; H, 4.49; N, 3.46. Found: C, 56.19; H, 4.57; N, 3.86. %.
5,6-Dimethoxy-3-formylbenzofuran-2-carboxylic acid methyl phenyl amide (15).
To 14 (1.41 g, 3.50 mmol) was added a solution of TMANO (0.40 g, 5.31 mmol) in DMSO (15 mL) and the mixture was stirred at room temperature for 1h, then poured into 5% saline solution (200 mL) with swirling. The resultant yellow precipitate was collected by filtration. Recrystallisation from hexane/ethyl acetate yielded 15 as pale yellow crystals (0.60 g, 50%), mp 124-125°C; IR cm-1: 1673, 1639, 1619, 1594; 1H-NMR δH: 3.50 (3H, s, -N-CH3), 3.80 (3H, s, -O-CH3), 3.90 (3H, s, -O-CH3), 6.60 (1H, s, H-7), 7.10-7.45 (5H, m, -N-Ar), 7.60 (1H, s, H-4), 10.65 (1H, s, -CHO); 13C-NMR δC: 38.0, 55.8, 55.8, 93.8, 102.5, 115.1, 123.2, 125.7, 127.0, 128.9, 142.8, 148.0, 148.2, 150.0, 153.3, 158.8, 187.6. Anal. Calcd. for C19H17NO5 : C, 67.25; H, 5.05; N, 4.13. Found : C, 67.03; H, 5.06; N, 4.28%.
5,6-Dimethoxy-2-(methyl phenylcarbamoyl)-benzofuran-3-carboxylic acid (16a).
Compound 15 (0.39 g, 1.15 mmol) was dissolved in CCl4 (47 mL) and NBS (0.26 g, 1.46 mmol) was added with stirring. The reaction was enclosed in a dark box and irradiated with a 100 W light bulb for 24 h. The reaction mixture was then filtered and the filtrate concentrated in vacuo. To this brown residue, water (20 mL) was added and the mixture was left to stand overnight. The water was decanted and the resultant brown solid was dissolved in ethyl acetate. The solution was dried (Na2SO4) and concentrated in vacuo . The crude product was recrystallised from ethanol to yield 16a as a tan powder (0.37g, 90%); mp 170-172°C; IR cm-1: 1705, 1638, 1619, 1549; 1H-NMR δH: 3.55 (3H, s, -N-CH3), 3.80 (3H, s, -O-CH3), 3.95 (3H, s, -O-CH3) 7.20-7.55 (6H, m, -N-Ar, H-7), 7.70 (1H, s, H-4); 13C-NMR δC: 39.2, 56.1, 56.1, 93.4, 103.1, 117.9, 125.9, 127.8, 129.1, 142.7, 148.0, 148.3, 150.7, 161.0, 162.6; MS m/e (relative intensity) 355 (M+, 14), 337 (24), 311 (100), 249 (67). HRMS calcd. for C19H17NO6: 355.1056. Found: 355.1054.
2,3-Dimethoxy-7-methyl-7, 12-dihydro-6H-[1]-benzofuro-[2,3-c]-[1]-benzazepin-6,12-dione (11a).
To PPA (2 mL) which had been pre-heated to 900C, compound 16a (0.100 g, 0.28 mmol) was added with stirring over 15 min. The reaction mixture was then heated at 90°C for 24 h, poured into water/crushed ice (100mL) and was extracted exhaustively with ethyl acetate. The organic extract was dried (Na2SO4) and concentrated in vacuo and the crude product was chromatographed (SiO2, 2:1 dichloromethane-ethyl acetate) to yield 11a as bright yellow needles (0.024 g, 25%); mp 227-229°C; IR cm-1: 1637, 1617, 1563; 1H-NMR δH: 3.70 (3H, s, -N-CH3), 3.95 (3H, s, -O-CH3), 4.00 (3H, s, -O-CH3), 7.19 (1H, s, H-4), 7.35-7.50 (2H, m, H-1, H-10), 7.60-7.70 (2H, m, H-9, H-8), 8.15 (1H, dd, J = 1, 8, H-11); 13C-NMR δC: 39.0, 56.3, 56.3, 94.8, 103.0, 116.3, 122.0, 125.2, 125.7, 130.2, 132.8, 132.9, 140.0, 147.5, 148.7, 150.7, 151.7, 157.7, 184.6. Anal. Calcd. for C19H15NO5: C, 67.65; H, 4.48; N, 4.15. Found: C, 67.15; H, 4.51; N, 4.35%.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





