Abstract
According to the precepts of the 3-attractor (3-A) water model, effective 2-body water potentials should feature as local minima the bifurcated and inverted water dimers in addition to the well-known linear water dimer global minimum. In order to test the 3-A model, a new pair wise effective intermolecular rigid water potential has been designed. The new potential is part of new class of potentials called BMW (Bushuev-Muguet-Water) which is built by modifying existing empirical potentials. This version (BMW v. 0.1) has been designed by modifying the SPC/E empirical water potential. It is a preliminary version well suited for exploratory Monte-Carlo simulations. The shape of the potential energy surface (PES) around each local minima has been approximated with the help of Gaussian functions. Classical Monte Carlo simulations have been carried out for liquid water in the NPT ensemble for a very wide range of state parameters up to the supercritical water regime. Thermodynamic properties are reported. The radial distributions functions (RDFs) have been computed and are compared with the RDFs obtained from Neutron Scattering experimental data. Our preliminary Monte-Carlo simulations show that the seemingly unconventional hypotheses of the 3-A model are most plausible. The simulation has also uncovered a totally new role for 2-fold H-bonds.
Introduction
Liquid water is not only the most abundant liquid of our lithosphere, but it plays a significant role in biological processes. Liquid water unusual properties are still unexplained [Robinson 96]. Recent explorations of the supercooled [Belissent-Funel 98], [Star 99] and supercritical regions [Tassaing 98], [Belissent-Funel 97], [Tassaing 00] have only increased the awareness of the current lack of theoretical understanding. Strangely enough, the fact that water still constitutes a fascinating and unsolved problem is all but ignored by the general public.
Within the 3-attractor water model ([Muguet 92], [Muguet 95], [Muguet 97], [Muguet 98], [Muguet 99], [Muguet 00]) we postulate that, in the condensed phase, the water-water dimer interaction features two other local minima corresponding to other types of physical interactions, in addition to well known linear H-bond water interaction. The two extra local minima are the inverted water dimer and the bifurcated water dimer.
Since this postulate relates to the dynamics of the water molecules, we prefer to rely on the concepts of attractors. This allows also to distinguish our model from 2-state state models [Robinson 99], which bring with them a notion of thermodynamical phases, and mixtures, which could be misleading. Stricto sensu, this postulate is the only one required to build our model for the liquid dynamics. A stronger postulate, a non-equivalent hypothesis, is that the isolated water dimer does also feature two extra local minima.
Design of the Potential
One ideal theoretical approach would be to be able perform truly ab initio molecular dynamics or even Monte-Carlo simulations, while solving the electronic Schrödinger equation for the whole simulation box. This would take care of all n-body effects. Then, assuming the Born-Oppenheinmer approximation is valid, a quantum dynamics motion of the nuclei would be performed. This approach would take into account the ZPE, that we have shown [Muguet 00] cannot be neglected, and all quantum vibrational and even rotational levels. Of course, the method chosen in order to solve the electronic Schrödinger equation would have be accurate enough to reproduce the potential energy surface of a single dimer. Considering the state of the art (that we are aware of), we set forward the challenging view that there is no current ab initio method, which is good enough to reproduce the water dimer PES. One proof is that current ab initio methods have consistently failed to reproduce the ammonia dimer PES. We have shown that one reason for this failure is the basis set superposition error (BSSE) [Muguet 95]. We proposed a new method to correct the BSSE and it appears that with our new correction method the ammonia dimer PES is improved and in agreement with experimental observations. Yet we believe a convenient and correct method to correct the BSSE has yet to be found. For the ammonia dimer, it is a question of finding the correct global minimum, for the water dimer, it is a question of finding extra local minima, which is much harder and challenging. It must be stressed that performing nuclear classical dynamics, even on a correct n-body ab initio PES would not be sufficient as we have shown the crucial importance of ZPE effects. Some remedies have been proposed to be introduced in a classical MD scheme [Alimi 92]. Another possible approach is to implement path integral representations of the hydrogen atoms, which amounts, in practice, to representing the proton by a necklace. One problem that proves difficult to handle is the quantum correlation between protons and related decorrelation effects [Chatzidimitriou-Dreismann 97]. It is also interesting to notice that the methodical and well-known effort by Clementi and coworkers [Clementi 83] to develop effective water potentials (MCY) with the help of ab initio calculations have not been successful. While waiting to find the correct electronic ab initio PES and then to develop sophisticated MC or MD methods with nuclear quantum dynamics that do not yet exist, a completely different approach is to develop semi-empirical effective 2-body potentials. We are proposing a new class of effective potential called BMW potentials (an acronym for Bushuev-Muguet-Water potentials) that is built by modifying existing empirical potentials. This version 0.1 (BMW v. 0.1) has been designed by modifying the SPC/E empirical water potential. It is a preliminary version suited only for Monte-Carlo simulations.
This alternative empirical approach is less rigorous but it is computationally feasible. We may fit selected parameters of the potential functional while trying to minimize differences between calculated and experimental data. Non-additive interactions may be accounted “effectively”, which means in the sense of a statistical average. This “effective” approach is not necessarily very sensitive to structural details since it implies angular averaging. According to statistical physics principles; macroscopic properties are integral quantities of the Hamiltonian, and a rather weak dependence of liquid properties on the details of the effective dimer PES (Potential Energy Surface) is observed. There are many available potentials for liquid water [Robinson 96] and most of them predict a selected set of experimental properties correctly, but none of them appears to be able to predict correct properties for a wide range of state parameters. Some calculated properties are significantly different from experimental data. It must be stressed also that even the functional form of the potential is unknown.
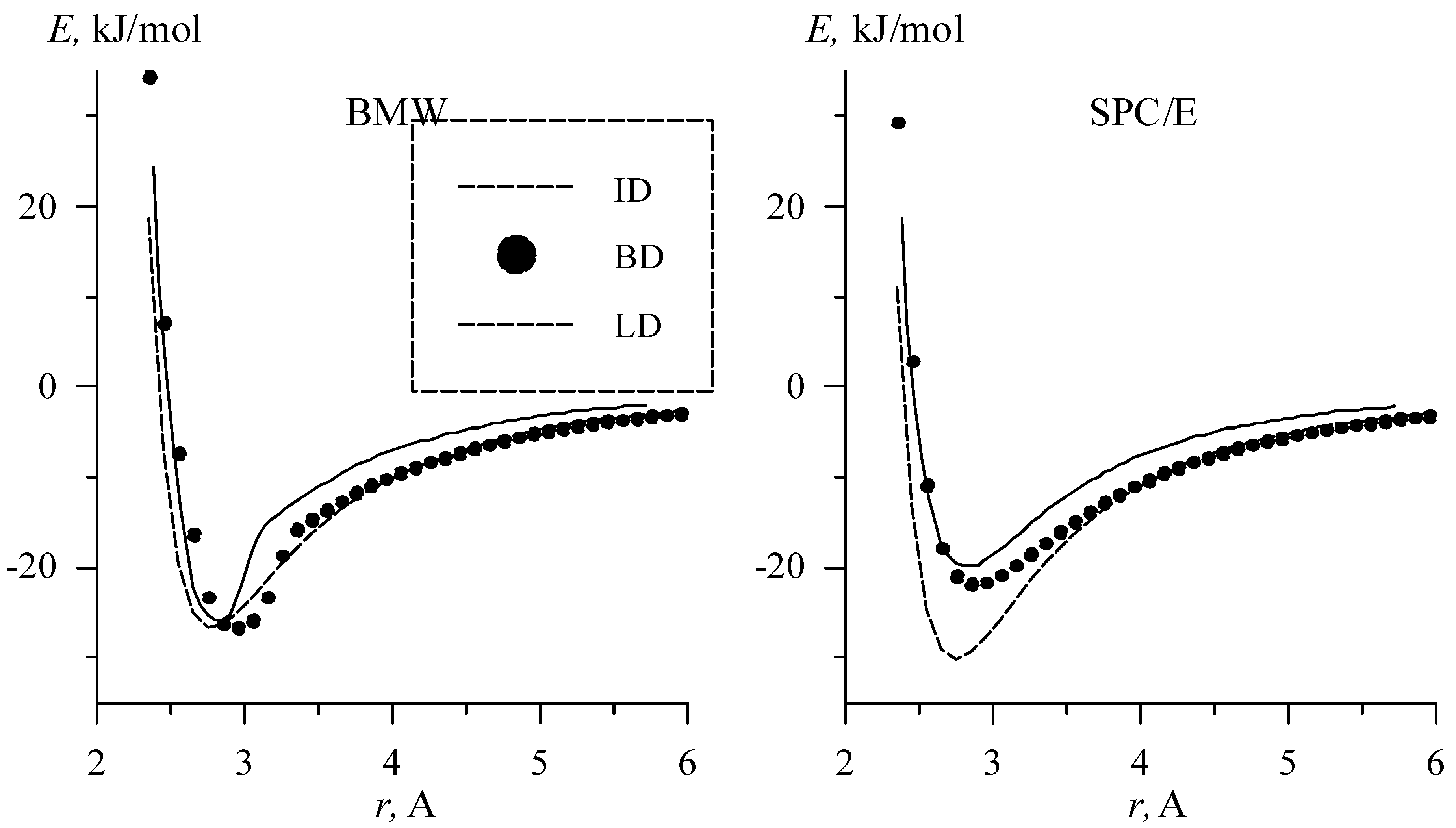
By construction the BMW potentials includes at least two extra local minima in addition to linear dimer (LD) (cf. Fig 1). A first local minima, favored at short intermolecular distances [Muguet 00] is the Inverted Dimer (ID) whose symmetry group is Ci with an inversion center at the mid-point between oxygen atoms. At intermediate intermolecular distances, we find the well-known global minimum linear H-bond dimer (LD) whose symmetry group is Cs with a mirror plane. At larger intermolecular distances become favored the bifurcated dimer (BD). Sometime the term “bifurcated bond” is used for describing a proton engaged in two hydrogen bonds. We prefer to call this situation a twofold bond, in order to avoid any confusion.
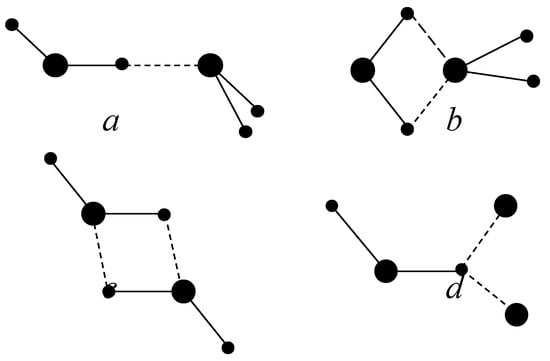
Figure 1.
Schematics of atomic positions. a – linear, b - bifurcated, c - inverted, d – twofold.
Figure 1.
Schematics of atomic positions. a – linear, b - bifurcated, c - inverted, d – twofold.
The ID and BD generates non-tetrahedral symmetry clusters. Experimental results are pointing out that there is a non-perfect random tetrahedral H-bonded network in liquid water. At a first glance, it could appear that the existence of a large number of ID and BD structures might be contradicted by X-ray and neutron scattering data. At near ambient condition radial distribution functions (RDFs) of oxygen atoms has second pick near 4.5 Å that corresponds to the second neighbors from given atom in tetrahedral network. We must, however, remember that by definition
where dloc(r) is local number density of atoms in thin spherical layer on the distance r from fixed atom, and d is mean number density of atoms averaged on whole molecular ensemble. When dloc is computed, we average the density both for angles and distances. Different atomic spatial distributions may give the same radial local density profile. The second peak of RDF goo has much less height than the first one, and therefore we should expect many defects within the tetrahedral network. The non-tetrahedral bonds (ID, BD, twofold bonds) might play a significant role and influence all water properties on micro- and macroscopic levels. One of the goals of this work is to check the consequences of the hypotheses of the 3-A model both in terms of thermodynamical and structural quantities.
g(r) = dloc(r)/d,
Potential and simulation details
So far, to our knowledge, current existing empirical water potentials treat BD and ID as stationary points which are transition states but not local minima. However, we expect [Muguet 00] that MC or MD simulations, even with those potentials, should predict populations of BD and ID simply because the intermolecular Oxygen-Oxygen distance is a temperature and pressure dependent distribution. They should be present, but to our knowledge, nobody has carried out a systematic statistical analysis concerning the presence of ID and BD over a wide range of state parameters. We suspect that the best semi empirical potentials are in fact those who predict, albeit still inaccurately, relatively large populations of ID and BD unknowingly. Consequently we took one of the most popular pair additive potential: the SPC/E potential [Berendsen 87] as the reference potential for further qualitative improvements. The SPC/E potential is able to correctly reproduce vapor pressure, but underestimates the critical temperature of water. On the other hand, the SPC/E model accurately predicts the critical temperature, but underestimate the vapor pressure by more than a factor of two. Our first idea is to approximate the PES near stationary points with the help of additional Gaussian functions. We suppose that the shape of PES in these regions should like a Gaussian-shaped well in a six dimensional space and can be approximated by smooth functions. At this exploratory stage of our investigation, we are not making any further explicit hypotheses concerning the heights of the energy barriers between LD, BD, and ID. A barrier simply results from the intersection of the Gaussian well with the unmodified SPC/E region. We do not use any special functions for barrier approximation.
We underline that this simple procedure might create some discontinuities of the first derivative of the PES. Therefore this preliminary version (v. 0.1) of the BMW potential is suited for MC simulations but not for MD simulations.
We used rigid molecules with geometrical parameters taken from SPC/E model. The lengths of the O-H bonds were 1 Å and the HOH angle was 109.47°. Universal interactions were described by the Lennard-Jones (12-6) potential where ε= 0.6277 kJ/mol, σ = 3.18 Å. These values are only slightly different from original SPC/E ones (ε= 0.649 kJ/mol, σ = 3.166 Å). Point charges are located on H atoms with a -0.405 e charge, and O atoms with a +0.81 e charge (to compare with -0.4238 e and 0.8476 e respectively for SPC/E).
We used the following Gaussian function for the interaction between two molecules m and n
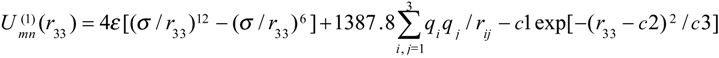 where the adjustable parameters c1=0.29092 kJ/mol, c2=4.4 Å , and c3=0.15 Å2, rij.- are the distances between atoms of two molecules (i=1,2 for atoms H, i=3 for atoms O).
where the adjustable parameters c1=0.29092 kJ/mol, c2=4.4 Å , and c3=0.15 Å2, rij.- are the distances between atoms of two molecules (i=1,2 for atoms H, i=3 for atoms O).
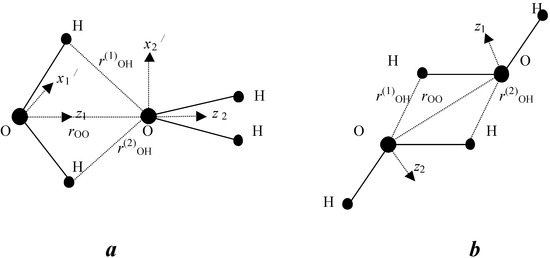
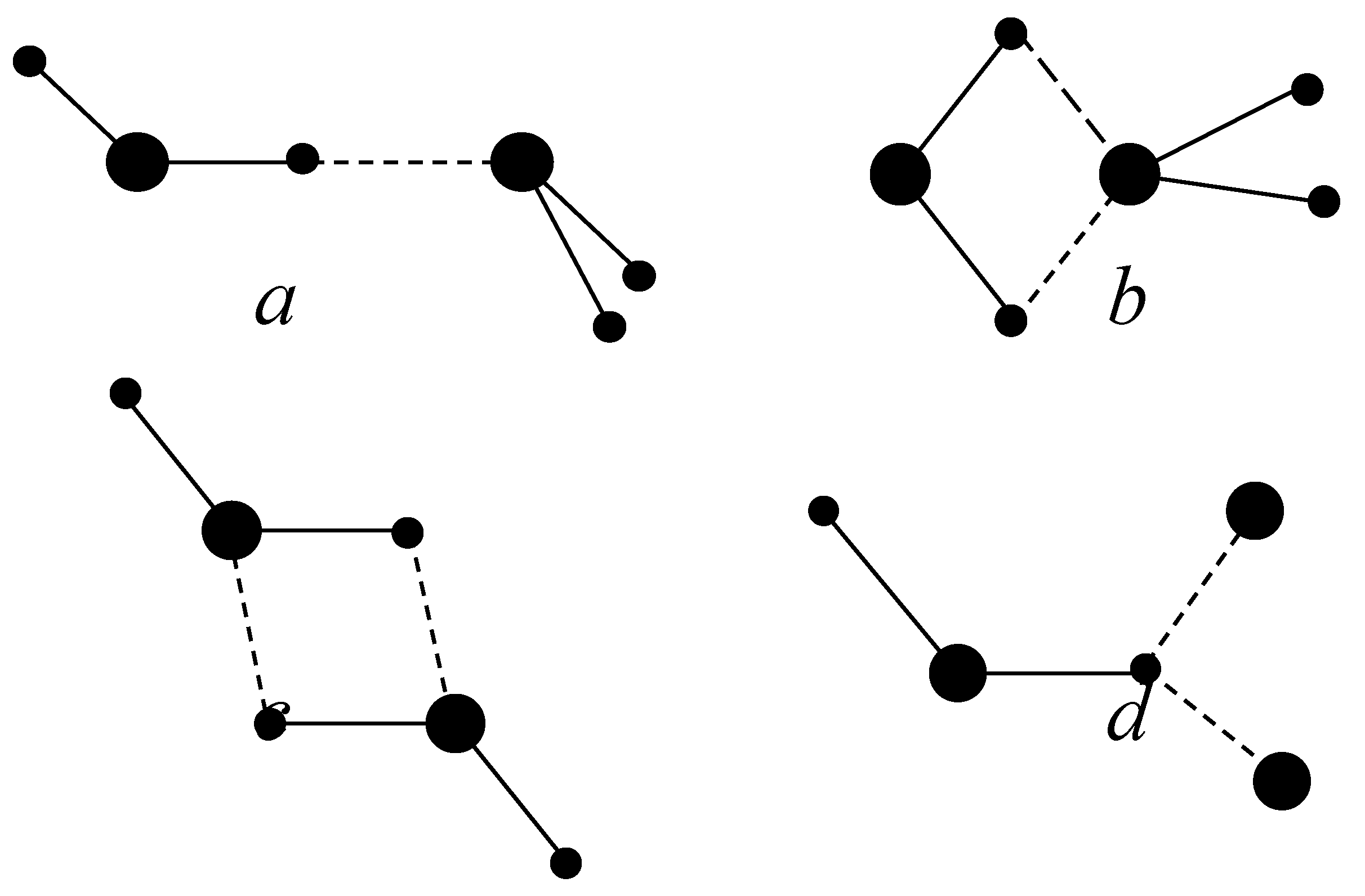
Figure 2.
Schematic illustration of axis position for BD (a) and ID (b)
Figure 2.
Schematic illustration of axis position for BD (a) and ID (b)
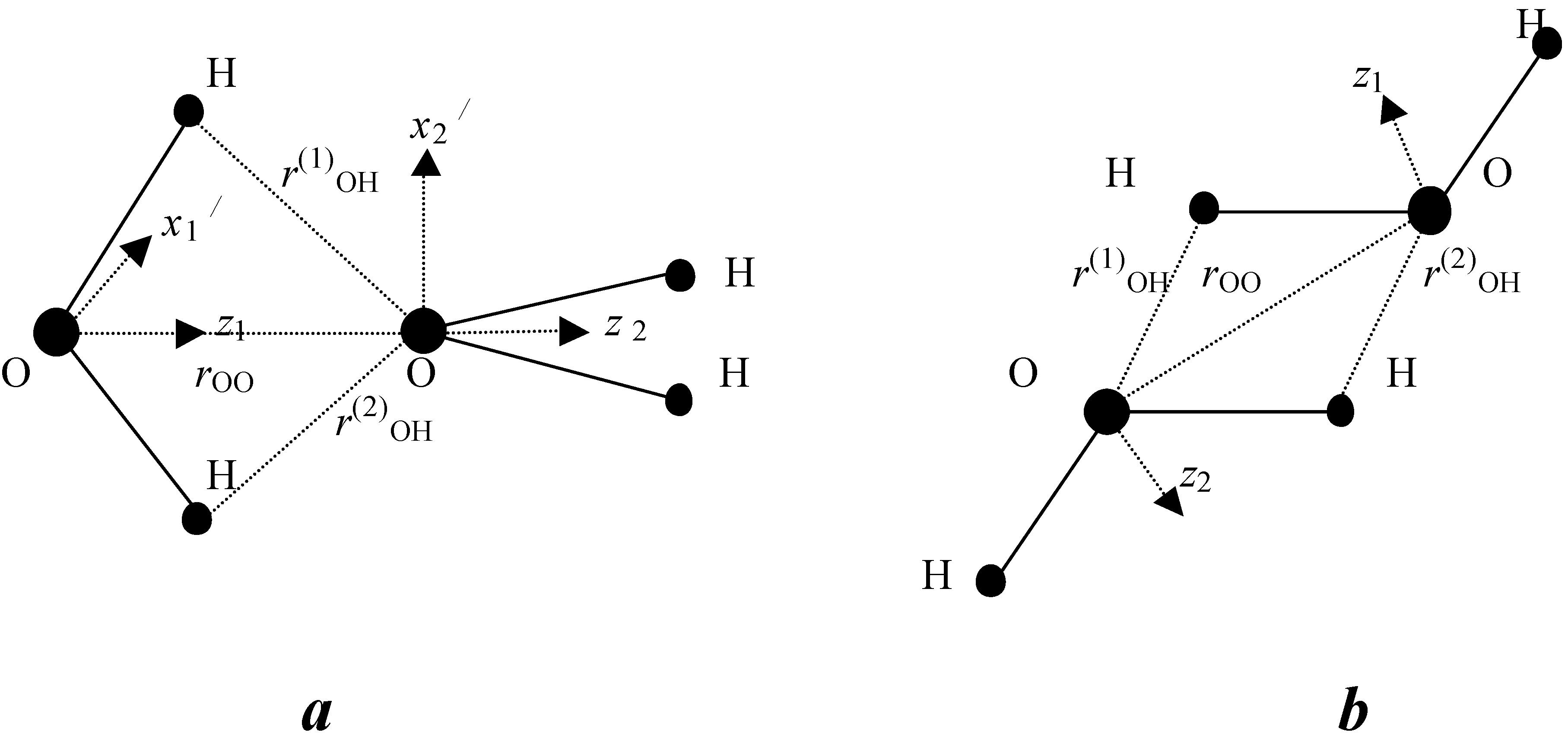
For a BD, the scalar product of dipole moments should be positive. We determined the function
where zi, zj, are unit vectors directed along dipole moments, xi, xj are unit vectors that perpendicular to dipole moments (see Fig. 2). We used geometrical parameters of BD and ID taken from ab initio calculations [Muguet 98], [Muguet 99] r33=2.97 Å , rij=2.47 Å (i=3, j=1,2, or i=1,2, j=3). We proposed that a BD is formed if the product of s and s1 functions is greater than 0.01, where
Energy of BD was calculated using formula
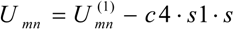 where the other empirical parameter is c4 =7.1128 kJ/mol.
where the other empirical parameter is c4 =7.1128 kJ/mol.
s = exp[−(r33 − 2.97)2 − (zizj − 1)4 / 0.1 − (xixj)8 / 0.003],
s1 = exp[−(r13 − 2.47)4 / 0.01 − r23 − 2.47)4 / 0.01]
For ID, optimal geometrical parameters are r33 = 2.8 Å, r13 and r31 = 2.28 Å. Respectively. Then we compute functions s2 and s3
If the product of this functions was greater than 0.01 we regard that ID exist with the energy
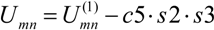 where c5=8.368 kJ/mol.
where c5=8.368 kJ/mol.
s2 = exp[−(r33 − 2.8)2 − (zizj + 1)4 / 0.1]
s3 = exp[−(r13 − 2.28)4 / 0.01 − (r31 − 2.28)4 / 0.01]
The energy dependence on O-O distance for optimal geometry for linear, bifurcated and inverted dimers for BMW and SPC/E potentials are shown in Figure 3.
Monte Carlo simulations for bulk liquid water include 512 molecules in a cubic elementary cell. Periodic boundary conditions are applied in all three directions. The simulations are carried out in an isobaric-isothermal (N,p,T) ensemble in a wide range parameters of state. Standard Metropolis sampling was performed. A potential cutoff of 10 Å is chosen to reduce computational effort. Long-range correction term for the Lennard – Jones interaction was less than 0.3 kJ/mol and was not included in the total energy. We suppose that second order corrections should give a systematic shift on thermodynamic functions and are weakly dependent on state points.
New configurations were generated by randomly translating selected monomers in all three Cartesian directions and by random rotations. At 298 K the ranges were 0.15 Å for the translation and 0.15 radians for the rotation. These choices yielded acceptance rates of 45%. Volume moves were made every 1024 accepted configurations and involved scaling all the intermolecular distances. The maximum length of cell change was 0.12 Å that gave acceptance rates of 20%. The lengths of Markov’s chain were exceeded 200 millions of configurations.
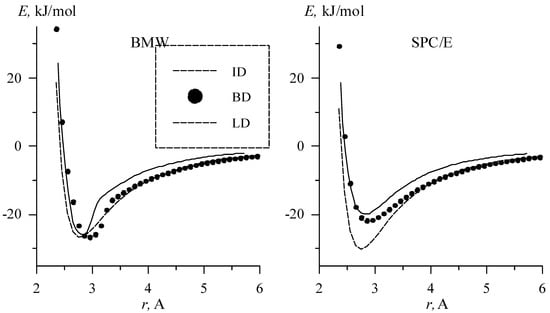
Figure 3.
Energy dependences on distance for inverted, bifurcated, and linear dimmers
Figure 3.
Energy dependences on distance for inverted, bifurcated, and linear dimmers
Results and Discussion
We have chosen the same series of thermodynamical points as those studied in the Neutron Scattering experiments by Soper and coworkers [Soper 97], [Botti 98], [Soper 00]. These papers present different sets of radial distribution functions (RDF). For the most part, however, RDFs are obtained from the same experimental data but analyzed with two different methods. Pair correlation curves from the 1997-1998 and 2000 papers have been established from almost the same set of S(q) experimental data which correspond to the spectra in the momentum space. There is a little more data in the 2000 paper. The transformation from the momentum space to the coordinate space is not as simple, as one would imagine at a first glance, because of truncation. A simple Fourier Transformation does not suffice. We are not going to delve here with this highly technical topic. What matters is that the pair correlation functions were not reconstructed in the same way. For the 1997 set of data, the pair function curves were constructed by a "maximum entropy" fitting method. While for the 2000 set of data, a new EPSR (Empirical Potential Structure Refinement) method was used. In this new method, an "empirical potential" is added to the "reference potential" in order to reproduce the s(q) momentum spectra. The SPC/E empirical potential was chosen as a reference potential. The respective Fourier transform RDFs reconstructions are differing in many respect.
The 1997 curves are more structured than the 2000 ones. One must stress that the SPC/E potential features only the linear water dimer as a mininum and that a significant empirical potential had to be added to the reference SPC/E potential in the EPSR procedure which shows that the reference potential is far from being adequate. We hypothesize that the inadequacy and the bias brought by the reference potential is going to artificially smear out interesting curves features. Extreme caution must be exercised when comparing “experimental” radial distribution functions.

Table 1.
| Run Label | Temperature (°K) | Pressure (Bar) | 1997-98 | 2000 |
|---|---|---|---|---|
| 268a | 268 | 270 | No | Yes |
| 268b | 268 | 2100 | No | Yes |
| 268c | 268 | 4000 | No | Yes |
| 298a | 298 | 1 | Yes | Yes |
| 298b | 298 | 2100 | No | Yes |
| 423a | 423 | 100 | Yes | Yes |
| 423b | 423 | 1900 | Yes | Yes |
| 573a | 573 | 100 | Yes | Yes |
| 573b | 573 | 100 | Yes | Yes |
| 573c | 573 | 500 | Yes | Yes |
| 573d | 573 | 1100 | Yes | Yes |
| 573e | 573 | 1970 | Yes | Yes |
| 573f | 573 | 2800 | Yes | Yes |
| 673a | 673 | 500 | Yes | Yes |
| 673b | 673 | 800 | Yes | Yes |
| 673c | 673 | 1300 | Yes | Yes |
| 673d | 673 | 3400 | Yes | Yes |
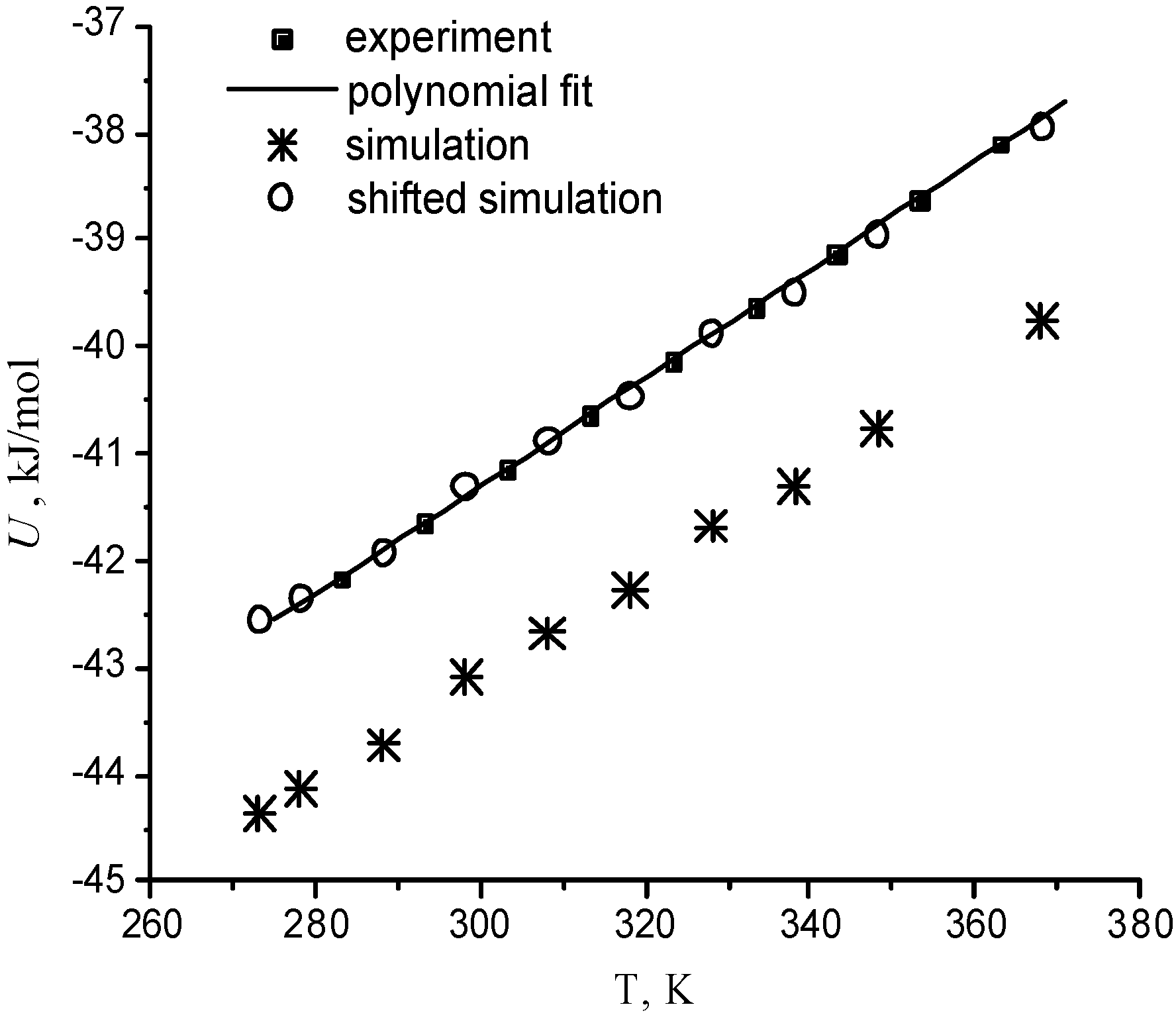
For one supercritical condition (Run 673b), it is also possible to compare with the results from the experiments of Bellissent-Funel and coworkers [Tassaing 98], which were obtained in very similar experimental conditions (380°C instead of 400°C). The temperature dependence of the molecular number density and potential energy is compared with the corresponding experimental curves in Figure 4 and Figure 5 at P=0.1 MPa. The standard deviations are 3·10-4 Å-3 for number densities and 0.3 kJ/mol for potential energies. Computed water properties agree very well with experimental data. Only small little systematic shifts are observed. The relative error does not exceed 3% for the density and 4.5% for the energy. The ability of the BMW potential in reproducing correctly experimental densities for some state points can be checked in Table 2. It means that calculated thermal expansion coefficient, heat capacity, isothermal compressibility coefficient are quite close to experimental values in a wide temperature range.
Table 2.
Number density of liquid water at different parameters of state.
| Run label | Temp. (K) | Pressure (MPa) | Density (molecules per Å3) | ||
| calculated | correcteda | Experimental [Soper 97] | |||
| 268a | 268 | 27 | 0.0347 | 0.0337 | 0.0338 |
| 268b | 268 | 210 | 0.0372 | 0.0362 | 0.0362 |
| 268c | 268 | 400 | 0.0391 | 0.0381 | 0.0381 |
| 298a | 298 | 0.1 | 0.0341 | 0.0331 | 0.0334 |
| 298b | 298 | 210 | 0.0369 | 0.0359 | 0.0357 |
a obtained by subtraction of 0.001 Å-3 from calculated densities.
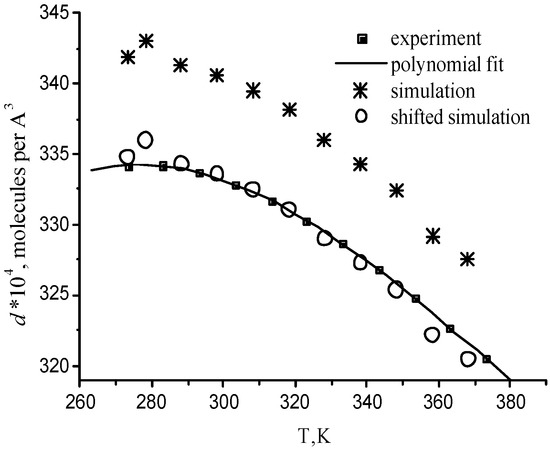
Figure 4.
Temperature dependence of water number. The shifted simulation curve is shifted down by 0.0007 Å-3
Figure 4.
Temperature dependence of water number. The shifted simulation curve is shifted down by 0.0007 Å-3
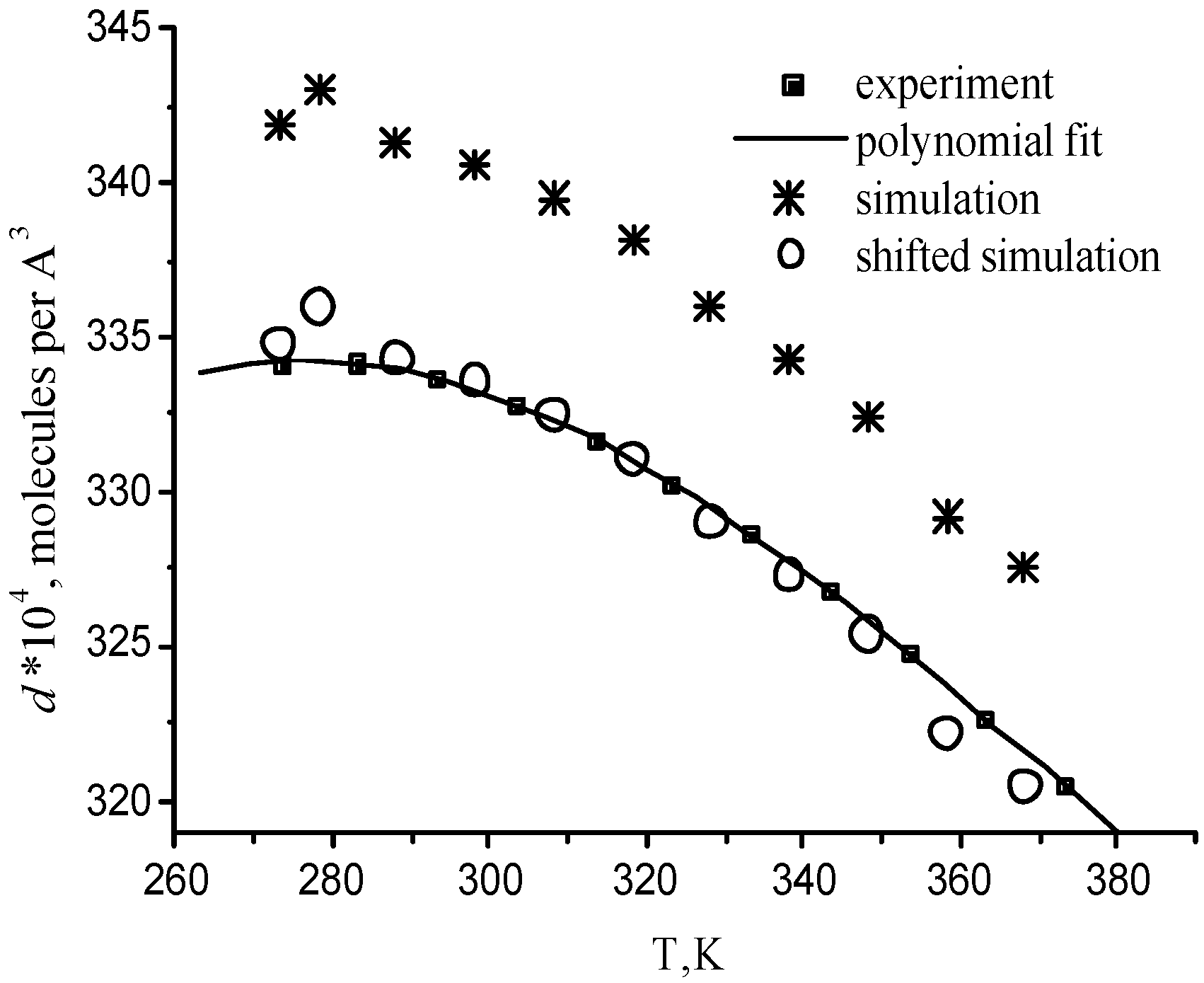
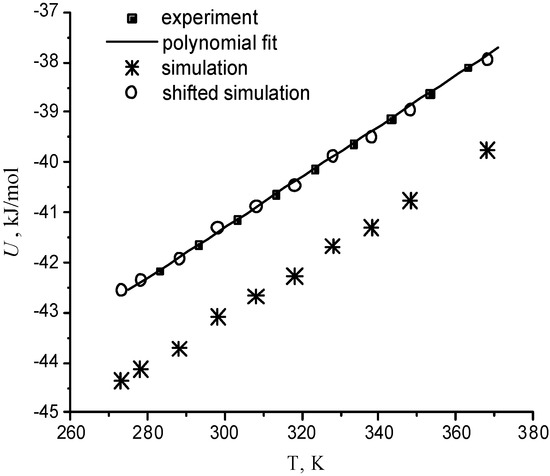
Figure 5.
Temperature dependence of the density configurational potential energy of water The shifted simulation curve is shifted up by 1.8 kJ/mol.
Figure 5.
Temperature dependence of the density configurational potential energy of water The shifted simulation curve is shifted up by 1.8 kJ/mol.
We have compared BMW thermodynamic predictions with data obtained from a comparative study [Jedlovsky 99] that used a set of polarizable and non-polarizable potentials. The data are summarized in Table 3. Polarizable potentials are not predicting accurate densities (d) for all state points, especially for 573b and 673b. Flexible potentials based on rigid SPC and TIPS models give errors ~10 - 30 % for state points close to liquid-vapor coexisting curve. At ambient conditions, configurational potential energies are close to experimental data for all models. Polarizable potentials predict energies that are too high at other state points. Among the non-polarizable potentials, BMW is the most effective in reproducing densities. The BMW potential predicts the best thermodynamical quantities for almost all points.
We hypothesize that polarizable models are exaggerating the strength of the linear H-bond through cooperativity and therefore they severely quenches the populations of ID and BD in an even more radical fashion than conventional non-polarizable models.
Table 3.
Thermodynamic properties of the BMW water model. Experimental data and properties of five tested water models as obtained from (N,p,T) Monte Carlo simulations [Jedlovsky 99] are shown for comparisons
| Model | BSVa | CCb | DCc | TIP4P | SPC/E | BMW | Experiment |
| 298a, T=298 K, p=0.1 MPa | |||||||
| U, kJ mol-1 d*104 , A-3 | -42.72 362.2 | -39.38 295.6 | -41.27 354.2 | -42.52 364.4 | -47.48 352 | -43.68 341 | -41.53 334 |
| 573e, T=573 K, p=197 MPa | |||||||
| U, kJ mol-1 d*104 , A-3 | -26.57 277.6 | -22.54 230.4 | -24.97 260.8 | -29.96 288.7 | -33.98 296 | -30.88 296 | -30.05 296 |
| 573b, T=573 K, p=10 MPa | |||||||
| U, kJ mol-1 d*104 , A-3 | -2.96 15.2 | -2.23 14 | -2.76 16.4 | -8.21 242 | -29.82 222 | -27.11 230 | -26.72 240 |
| 673b, T=673 K, p=80 MPa | |||||||
| U, kJ mol-1 d*104 A-3 | -16.22 164.7 | -12.17 120.7 | -15.07 153.9 | -20.55 185.7 | -26.53 213.8 | -24.04 219 | -23.09 221 |
Polarizable models: aBrodholt-Sampoli-Vallauri, bChialvo-Cummings, cDang-Chang (DC). Nonpolarizable models: SPC/E, TIP4P, BMW
In the supercritical regime and its vicinity, computed and experimental densities of water with corresponding run labels are reported in Table 4. Significant differences with experimental data are only noticed for the first three points. The maximum deviation is observed for state point 573b, which is very close to the experimental liquid – vapor coexistence curve. It is only on 11 K lower then boiling temperature at 10 MPa. For other states, deviations are minimal
Table 4.
Density BMW potential
| Run
label | T, K | p, MPa | Number densitya (molecules per Å3 *104 ) | ||
| Experiment [Soper 97] | BMW | error, % | |||
| 423a | 423 | 10 | 308 | 317 (5) | 3.0 |
a Standard deviations are given in parentheses.
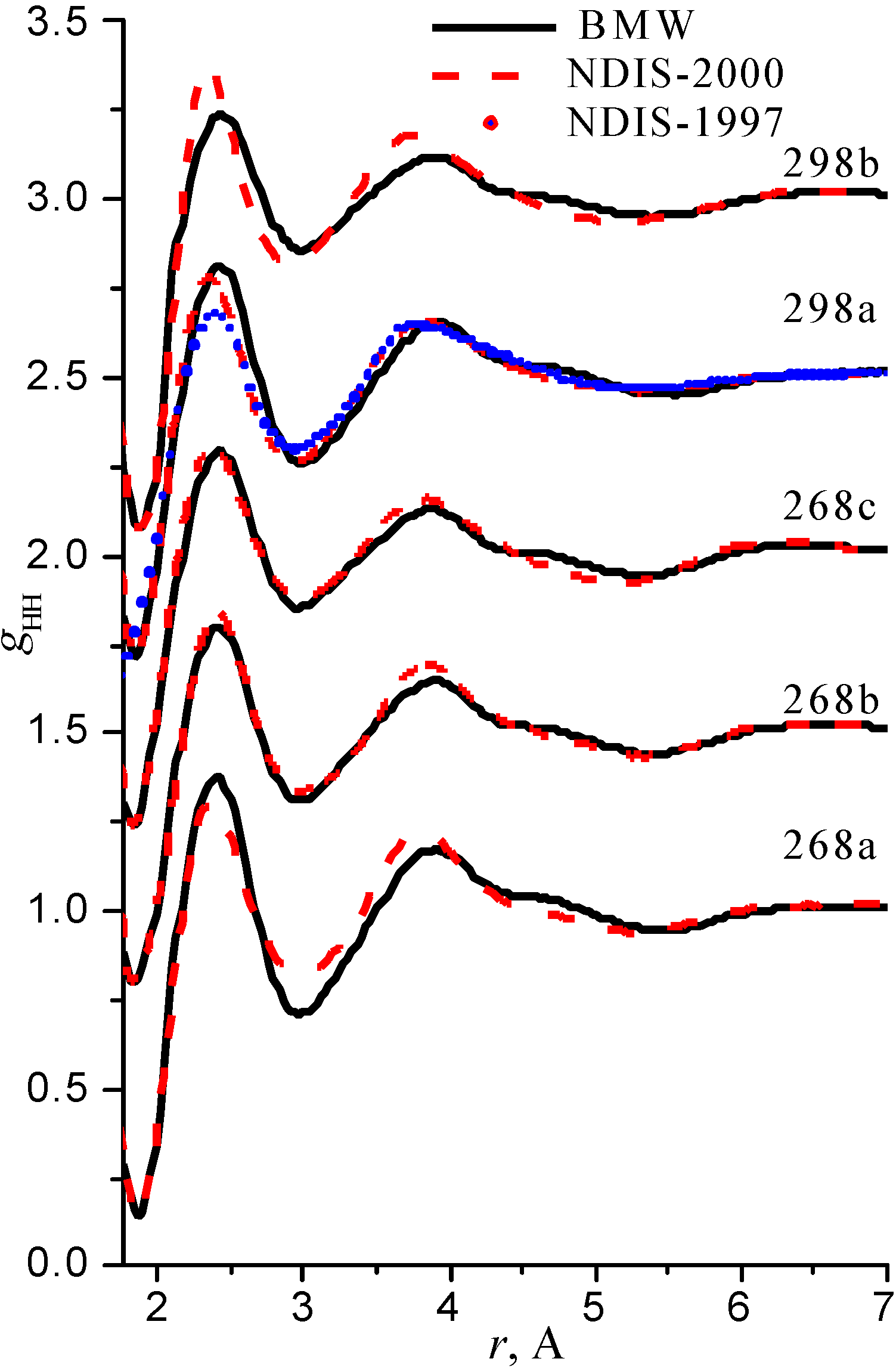
We have used the RDFs reported in the neutron scattering studies NDIS by Soper and coworkers [Soper 97], [Botti 98], [Soper 00]. NDIS-1997 and NDIS-2000 were used for comparison with computed RDFs. The atom-atom RDFs (gij(r)) for five points are shown in Figure 6, Figure 7 and Figure 8 concerning the low temperature regime (graphics are shifted along the y-axis for clarity).
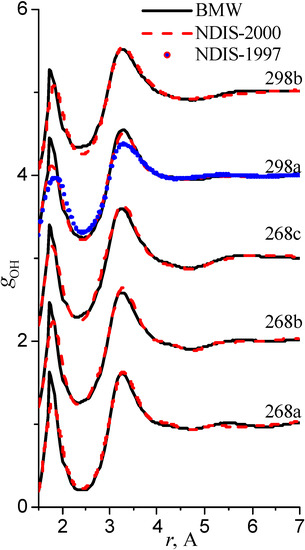
Figure 6.
Radial distribution functions gOH
Figure 6.
Radial distribution functions gOH
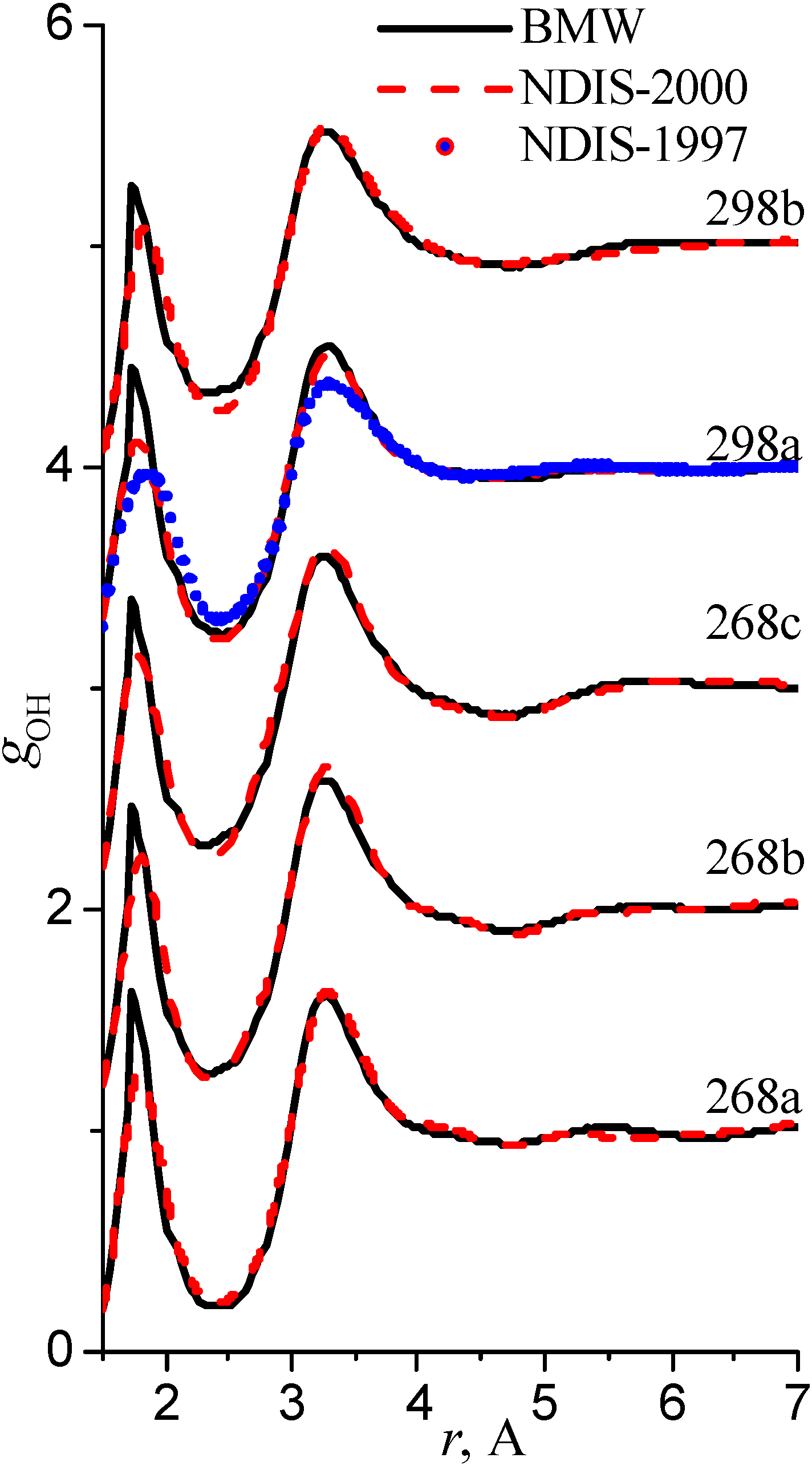
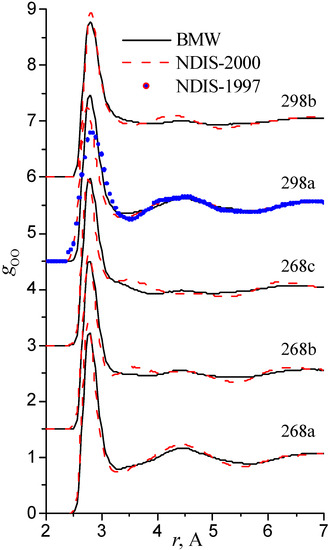
Figure 7.
Radial distribution functions gOO
Figure 7.
Radial distribution functions gOO
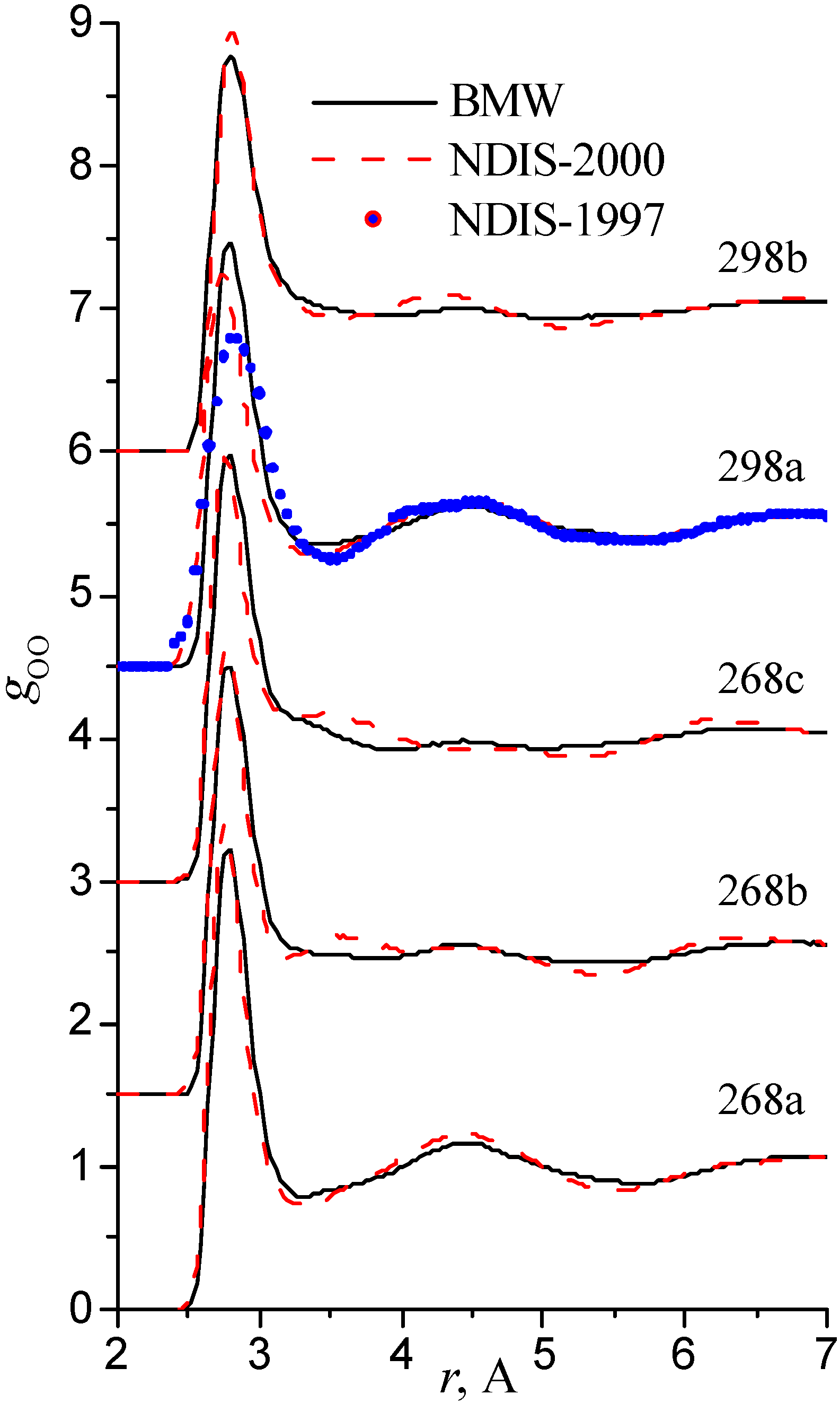
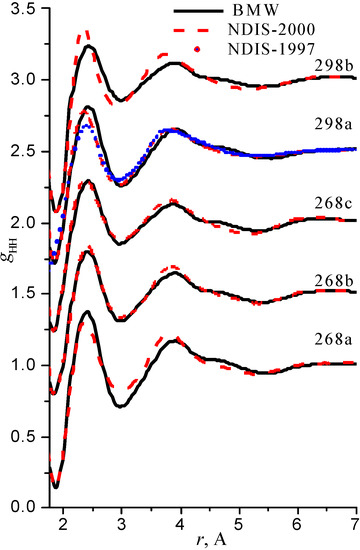
Figure 8.
Radial distribution functions gHH
Figure 8.
Radial distribution functions gHH
RDFs within the high temperature/ high pressure regions are illustrated in figure 9, figure 10 and figure 11.
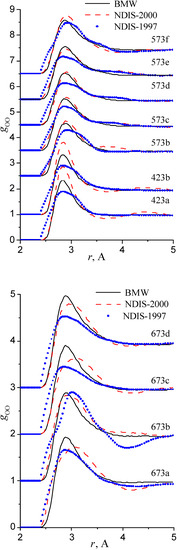
Figure 9.
Radial distribution functions gOO. Graphics are shifted along the y-axis for clarity.
Figure 9.
Radial distribution functions gOO. Graphics are shifted along the y-axis for clarity.
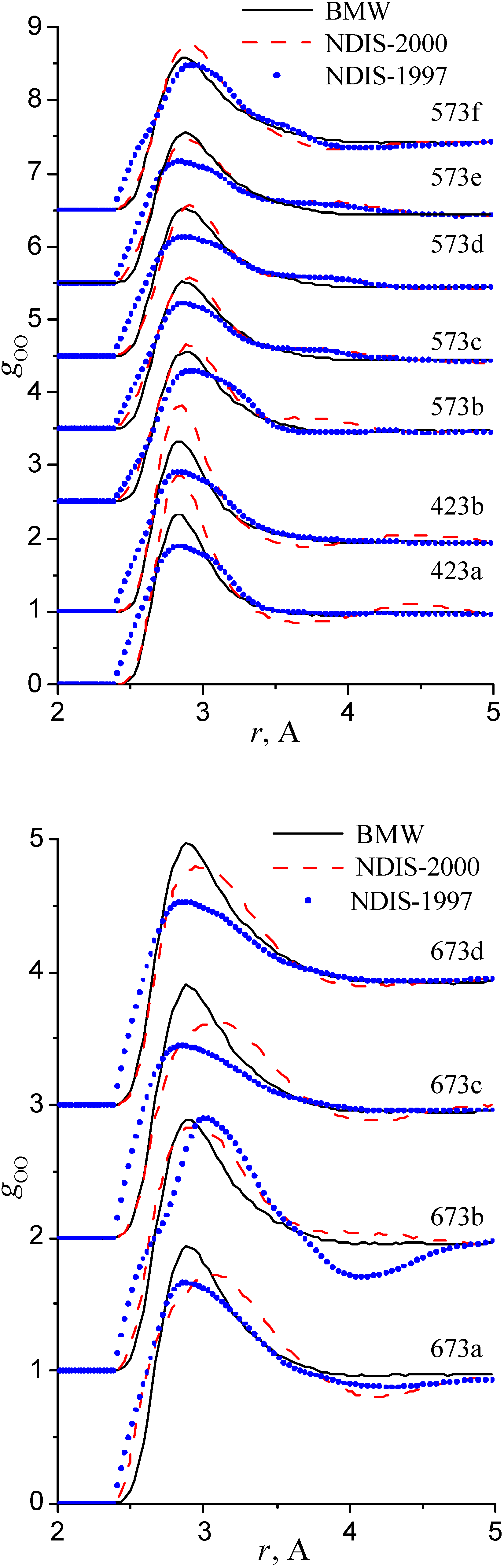
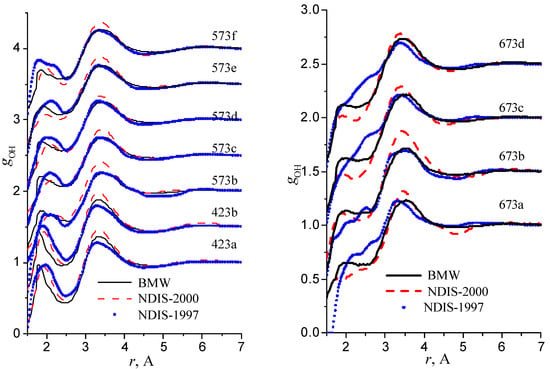
Figure 10.
Radial distribution functions gOH
Figure 10.
Radial distribution functions gOH
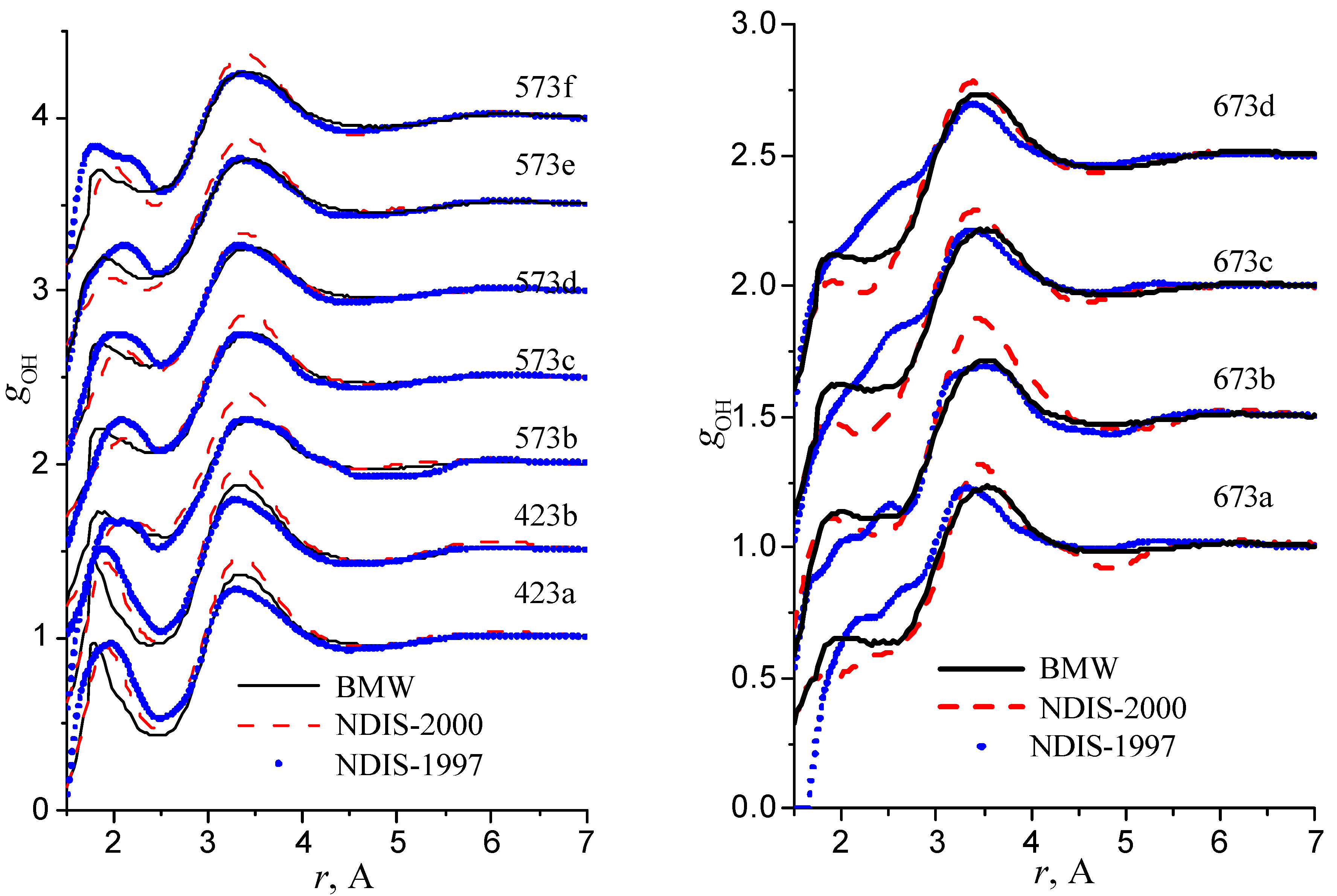
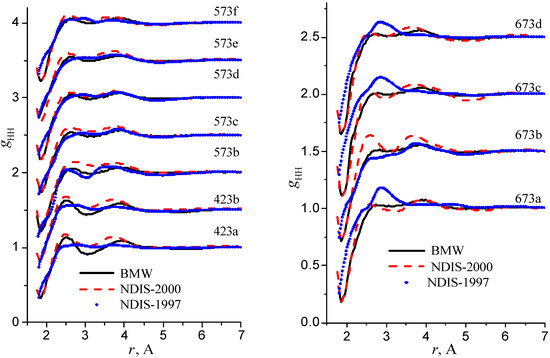
Figure 11.
Radial distribution functions gHH
Figure 11.
Radial distribution functions gHH
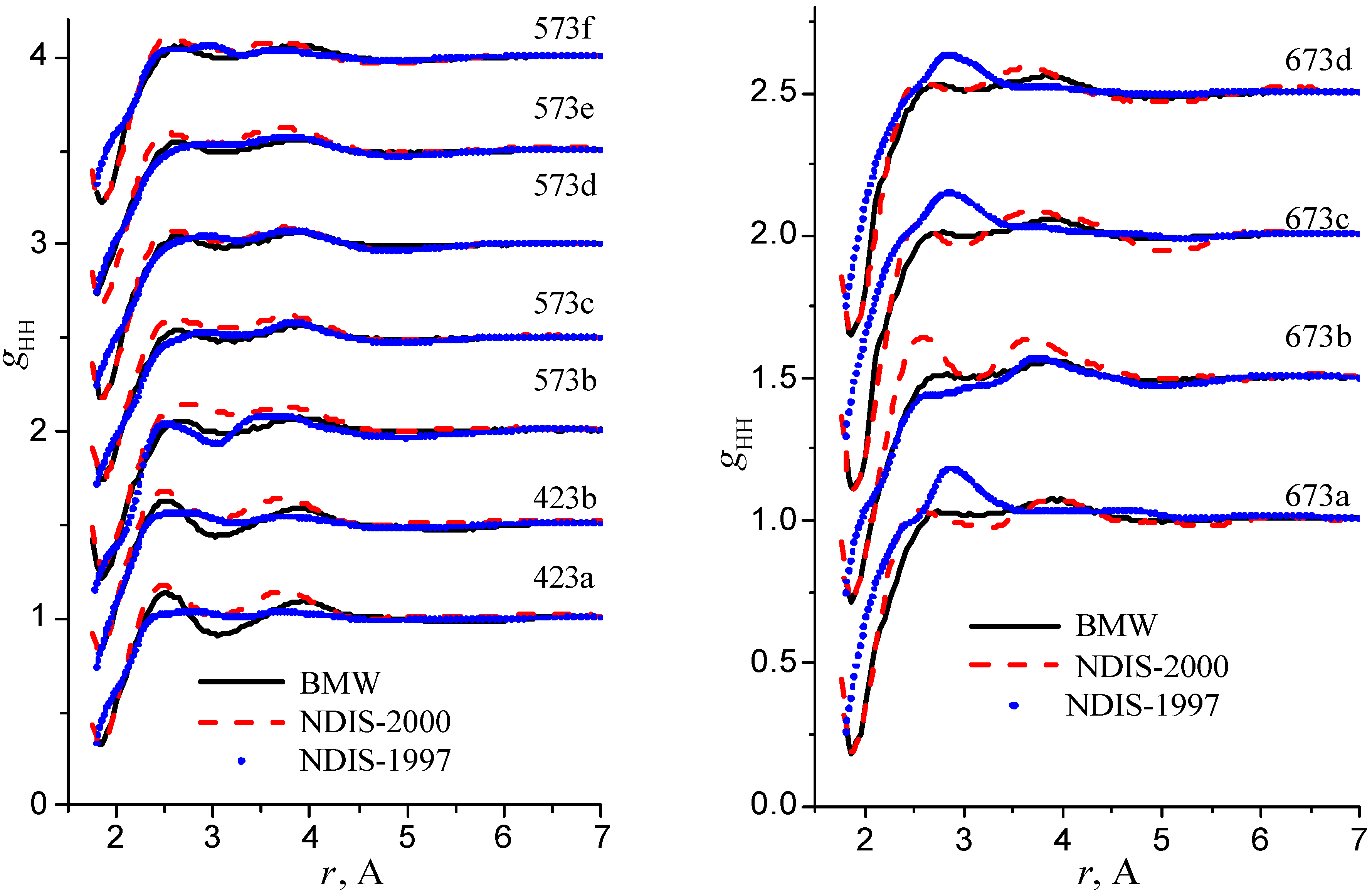
Our simulated results are in much better agreement with the latest RDFs (NDIS-2000) than with older 1997 RDFs. They seems quite satisfactory, but honestly, one should not give too much credit to this good agreement, since often differences between the two “experimental” curves exceed their respective differences with our BMW computed curves. As stressed before, the “experimental” RDFs are not raw experimental data. Neutron Scattering experiments are unusually complex and costly “big science” experiments. The raw experimental are obtained in the momentum space. A first series of possible uncertainties affect the collection of the data: the reactor initial beam coherence quality, the temperature distribution of the moderator, subtraction of the scattering due to the container vessel and many other technical issues. A second source of errors lies within the extraction of the partial structure factors. It relies on the implicit assumption that the structure of light and heavy water are absolutely identical. A third source of errors lies within the mathematical procedure itself when transforming data from momentum space to spatial coordinates. Fourier transformations are not accurate enough due to the truncation of the momentum space and they are quite sensitive to the data noise. A more detailed analysis of those delicate problems as well as a more trustful direct comparison between BMW computed partial and total structure factors with momentum experimental data will be presented elsewhere.
Within the framework of the 3A-model, some unconventional metastable clusters or chains of ID and BD are expected. Not surprisingly, our BMW potential MC simulations are producing unusual molecular configurations that may be metastable in water and aqueous solutions. These configurations may play a significant role in triggering chemical and physical processes. Needless to say, those clusters might account for native microstructural defects or micro heterogeneities of liquid water [Bushuev 97], [Bushuev 99].
A rather striking example of one of those transient clusters is shown in Figure 12. It consists of six molecules and has the form of chain. The sequence of intermolecular interactions is :
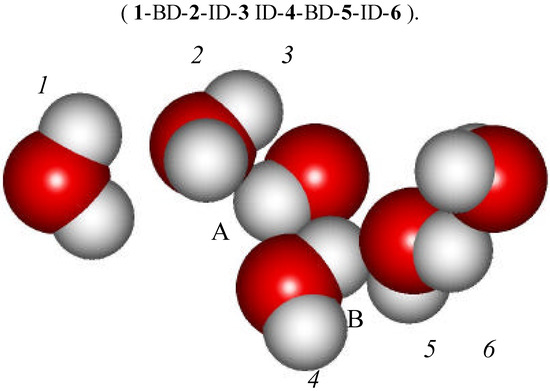
Figure 12.
A selected water cluster formed by BD and ID interactions.
Figure 12.
A selected water cluster formed by BD and ID interactions.
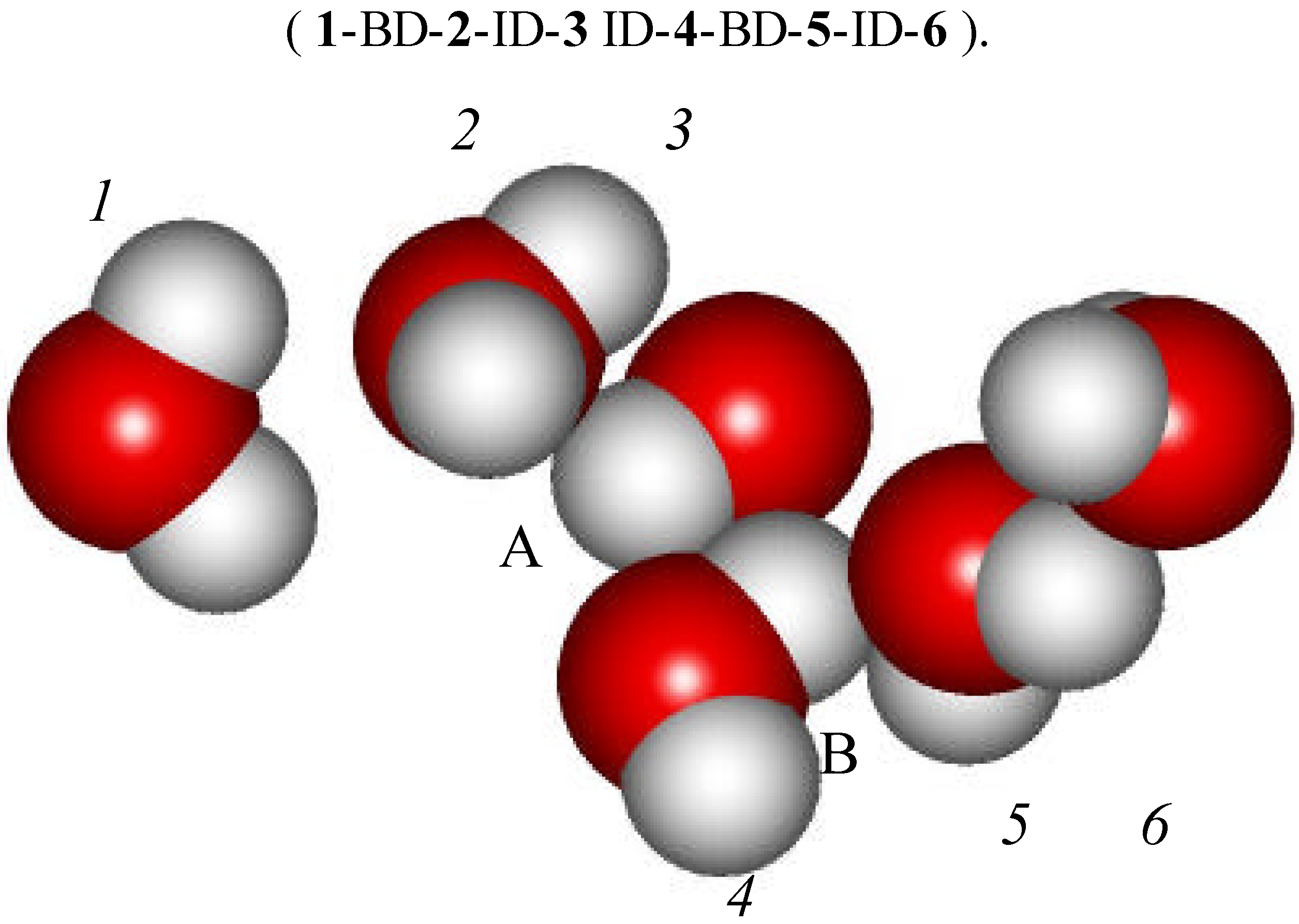
It is interesting to notice that H atoms belonging to water molecules 3 and 4 (labeled respectively A and B) may also be viewed as forming twofold H-bonds. Proton A is located between the oxygen atoms of water molecules 2 and 4. Similarly proton B lies between the O atoms of water molecules 3 and 5. It is therefore quite possible that twofold H-bonds are providing yet another stabilization interaction to this cluster. In fact, in constrained crystalline structures, occurrence of the 2-fold H-bond is quite common. In the present case, it is the ID/BD structure that provides the constraint. This cluster is likely going to be the future topic of our ab initio investigations, but only when we are feeling confident enough that proper theoretical methods are available for correcting the BSSE and taking into account the ZPE. Within the context of the 3-A model, two-fold bonds may play a complementary and totally new role by further stabilizing BD/ID clusters. The BMW MC simulation has brought this hindsight. One should also expect that these types of weakly bonded clusters are going to be entropy favored. The non-tetrahedral BD / ID clusters may lead to the formation of a significant population of two-fold 3-body H-bonds.
In connection with high temperature / high pressure liquid water structure, where a significant number of ID are expected, it has been proposed [Bassez 99] that it is the specific structure of supercritical liquid water or high pressure water that triggered prebiotic synthesis reactions. The 3-A model brings therefore some support to the idea that the chemistry in ocean hydrothermal vent systems might constitute the cradle of life.
A detailed (ID, LD, BD) population analysis of the configurations produced in our BMW MC simulations, in comparison to standard SPC/E MC simulations will be presented elsewhere.
Our future studies should concern two fronts: A first issue is to a get a detailed understanding of the water dimer and n-mer PES with ab initio methods. This line of study might require a significant amount of time since new theoretical developments are needed. Without waiting, yet an another assault will be attempted by designing BMW potentials and performing Monte-Carlo and Molecular Dynamics simulations.
Conclusions
According to the precepts of the 3-attractors water model, a new class (BMW of effective 2-body potentials for classical simulation has been proposed. A first version BMW v. 0.1 has been designed starting from the SPC/E potential to be used in Monte-Carlo simulations. Our (N,p,T) Monte-Carlo simulations show that thermodynamical predictions by the BMW potential seem to be the best in our comparisons. Concerning structural predictions in relationship with Neutron Scattering experiments, the BMW potential RDFs predictions appear very satisfactory, but until a comparison can be made between computed and measured structure factors, extreme caution is warranted.
There is no positive proofs in natural sciences, contrary to mathematics. A model may be considered as valid until falsified. Our Monte-Carlo simulations have not highlighted any element allowing one to falsify the 3-attractor water model. On the contrary, considering the comparative performance of the BMW potential in regards to the SPC/E potential, the 3-attractor water model appears as a most plausible model. Furthermore, Monte-Carlo simulations have uncovered a totally new role for 2-fold H-bonds.
Despite the preliminary nature of our simulations, results are obviously very encouraging. At the same time, because of the preliminary nature of our simulations, there is much room for improvement, while carrying out a cautious assessment of both theoretical and experimental data
Acknowledgments
We are extremely grateful to Alan K. Soper for providing us with experimental RDFs in a table form and for stimulating discussions. We acknowledge useful and lively discussions with Marie-Claire Belissent-Funel.
References
- Alimi, R.; Garcia-Vela, A.; Gerber, R.B. A remedy for zero-point energy problems in classical trajectories: A combined semiclassical/classical molecular dynamics algorithm. Journal of Chemical Physics 1992, 96, 2034–2038. [Google Scholar]
- Bassez, M-P. La Structure de l'Eau Supercritique et l'Origine de la Vie, Science et Technologie Regards Croisés. In CNRIUT'99; Editions L'Harmattan: Paris, 1999; pp. 583–591. [Google Scholar]
- Bellissent-Funel, M.-C.; Tassaing, T.; Zhao, H.; Beysens, D.; Guillot, B.; Guissani, Y. The structure of supercritical heavy water as studied by neutron diffraction. Journal of Chemical Physics 1997, 107, 2942–2949. [Google Scholar]
- Bellissent-Funel, M.-C. Is there a liquid-liquid phase transition in supercooled water? Europhys. Lett 1998, 42, 161–166. [Google Scholar]
- Berendsen, H.J.; Grigera, J.C.; Straatsma, T.P. The missing term in effective pair potentials. Journal of Chemical Physics 1987, 91, 6269–6271. [Google Scholar]
- Botti, A.; Bruni, F.; Ricci, M-A.; Soper, A. K. Neutron diffraction study of high density supercritical water. Journal of Chemical Physics 1998, 109, 3180–? [Google Scholar]
- Bushuev, Y.G. Properties of the network of hydrogen bonds in water. Russian Chemical Bulletin 1997, 46, 888–891. [Google Scholar]
- Bushuev, Y.G.; Davletbaeva, S.V.; Korolev, V.P. Structural properties of liquid water. Russian Chemical Bulletin 1999, 48, 831–841. [Google Scholar]
- Clementi, E.; Habitz, P. A new two-body Water-Water potential. Journal of Chemical Physics 1983, 87, 2815–2820. [Google Scholar]
- Chatzidimitriou-Dreismann, C.A. Proton nonlocality and decoherence in -condensed matter predictions and experimental results. In Advances in Chemical Physics; Prigogine, I., Rice, S.A., Eds.; John Wiley: New York, 1997; Volume 109, pp. 393–430. [Google Scholar]
- Jedlovszky, P.; Richardi, J. Comparison of different water models from ambient to supercritical conditions : A Monte Carlo simulation and molecular Ornstein-Zernike study. Journal of Chemical Physics 1999, 110, 8019–8031. [Google Scholar]
- Matsuoka, O.; Clementi, E.; Yoshimine, M. CI study of the water dimer potential surface. Journal of Chemical Physics 1976, 64, 1351–1361. [Google Scholar]
- Muguet, F.F. Investigation of diffuse intermolecular electronic systems. Ph.D. Dissertation, UMI, Ann Arbor (MI, USA), 1992. [Google Scholar]
- Muguet, F. F.; Robinson, G.W. Towards a new correction method for the BSSE: Application to the ammonia dimer. Journal of Chemical Physics 1995, 102, 3648–3654. [Google Scholar]
- Muguet, F. F. Towards a multi-attractor model of liquid water. Electronic Computational Chemistry Conference n°4 ECCC 4 1997, paper 66. [Google Scholar]
- Muguet, F. F. Ab initio vibrational study of the bifurcated, inverted and linear water dimers. Water in the Gas Phase (June 21-24, 1998).
- Muguet, F. F. The Water Dimer: Symmetries and ZPE Corrections According to Intermolecular Distance. QSCP4 (April 22-27, 1999).
- Muguet, F. F. The Water Dimer: Symmetries and ZPE Corrections According to Intermolecular Distance. Internet J. Chem 2000, 3, 6–? [Google Scholar]
- Robinson, G.W.; Zhu, S-B.; Singh, S.; Evans, M.W. Water in Biology, Chemistry and Physics: Experimental Overviews and Computational Methodolgies; World Scientific: Singapore, 1996. [Google Scholar]
- Singh, S.; Chul Hee Cho; Robinson, G.W. Understanding all of water's anomalies with a non local potential. Journal of Chemical Physics 1997, 107, 7979–7998. [Google Scholar]
- Robinson, G.W.; Chul Hee Cho; Urquidi, J. Isosbestic points in liquid water : Further strong evidence for the two-state mixture model. Journal of Chemical Physics 1999, 111, 698–702. [Google Scholar]
- Soper, A. K; Bruni, F.; Ricci, M-A. Site-site pair correlation functions of water from 25 to 400°C: Revised analysis of new and old diffraction data. Journal of Chemical Physics 1997, 106, 247–254. [Google Scholar]
- Soper, A. K. The radial distribution functions of water and ice from 220 to 673K and at pressures upto 400 mPa. Chemical Physics 2000, 258, 121–137. [Google Scholar]
- Starr, F. W.; Bellissent-Funel, M.-C.; Stanley, H. E. Structure of supercooled and glassy water under pressure. Phys. Rev. E 1999, 60, 1084–1087. [Google Scholar]
- Tassaing, T.; Bellissent-Funel, M.-C.; Guillot, B.; Guissani, Y. The partial pair correlation functions of dense supercritical water. Europhys. Lett 1998, 42, 265–270. [Google Scholar]
- Tassaing, T.; Bellissent-Funel, M.-C. The dynamics of supercritical water: A quasielastic incoherent neutron scattering study. Journal of Chemical Physics 2000, 113, 3332–3337. [Google Scholar]
© 2003 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.