Synthesis of Plasmepsin II Inhibitors – Potential Antimalarial Agents

Abstract
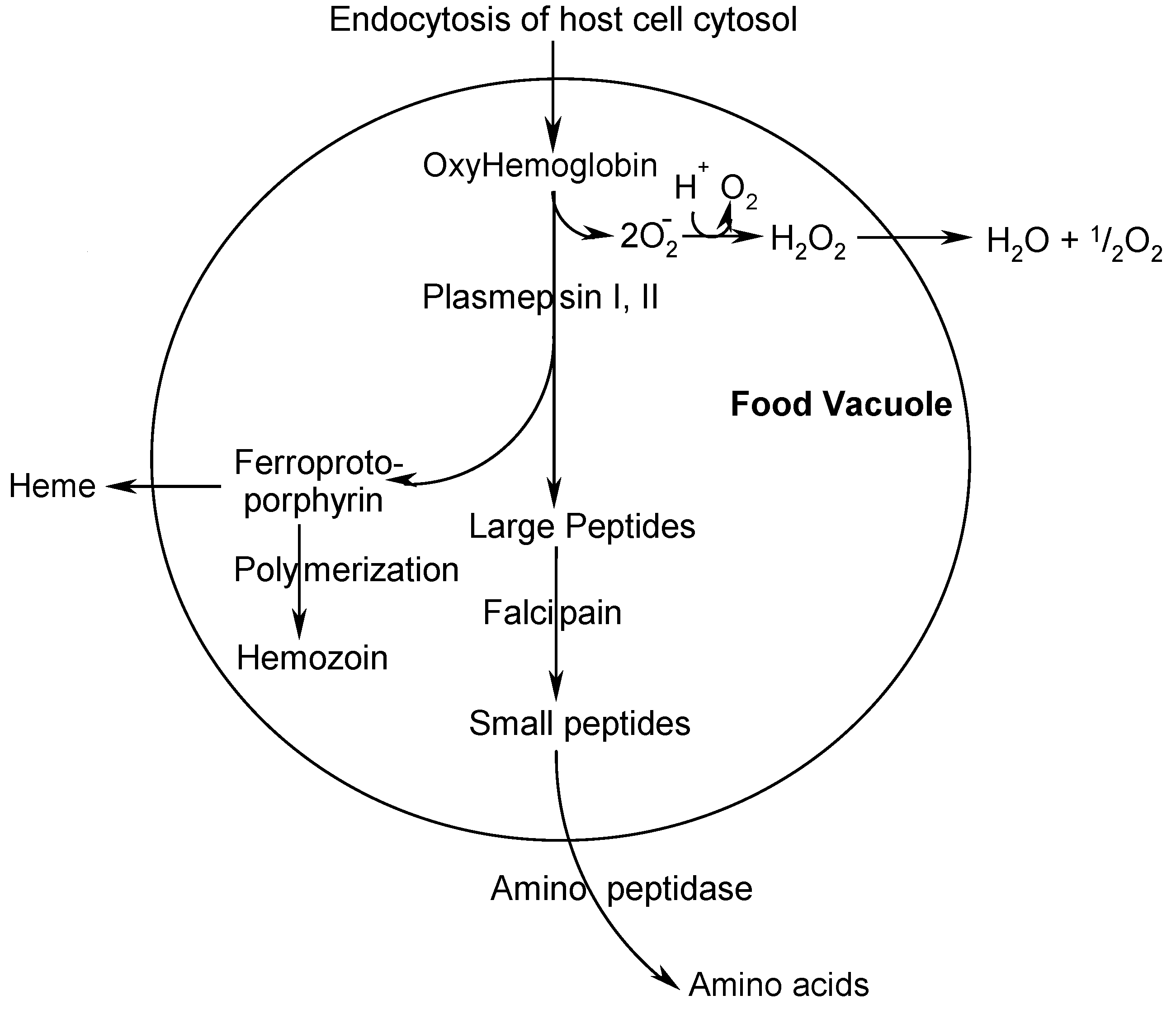
:Introduction




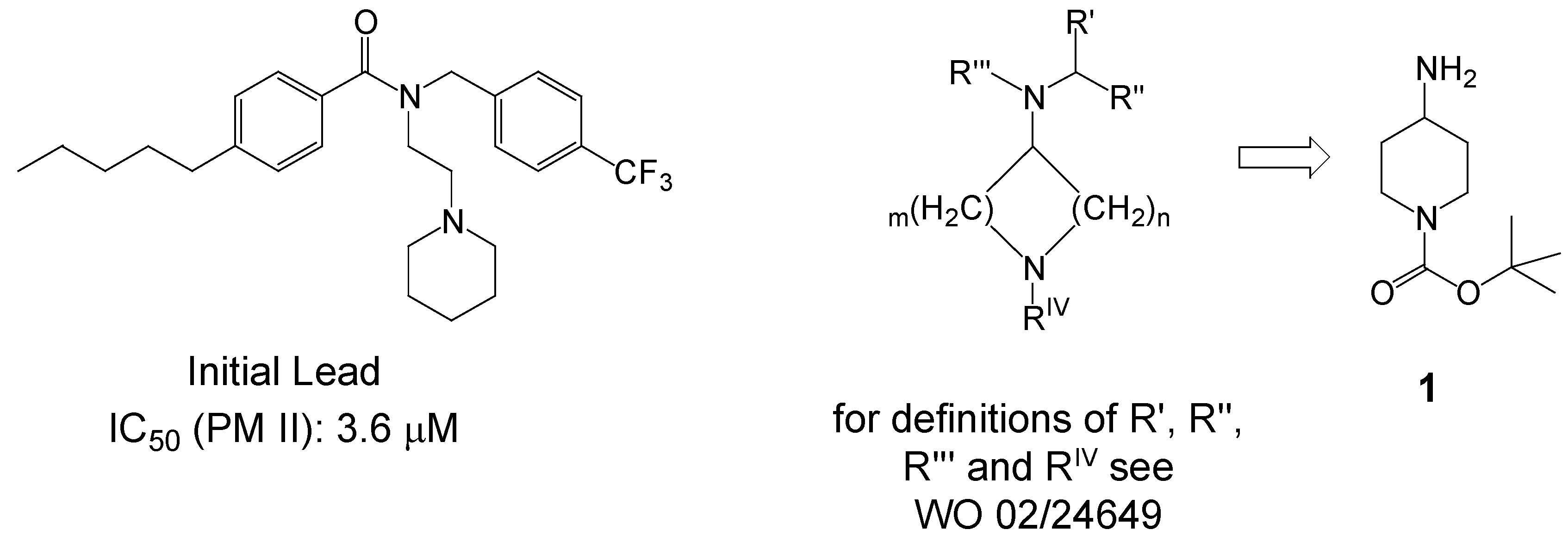
Results and Discussion





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Experimental
General
Selected Procedures [11]
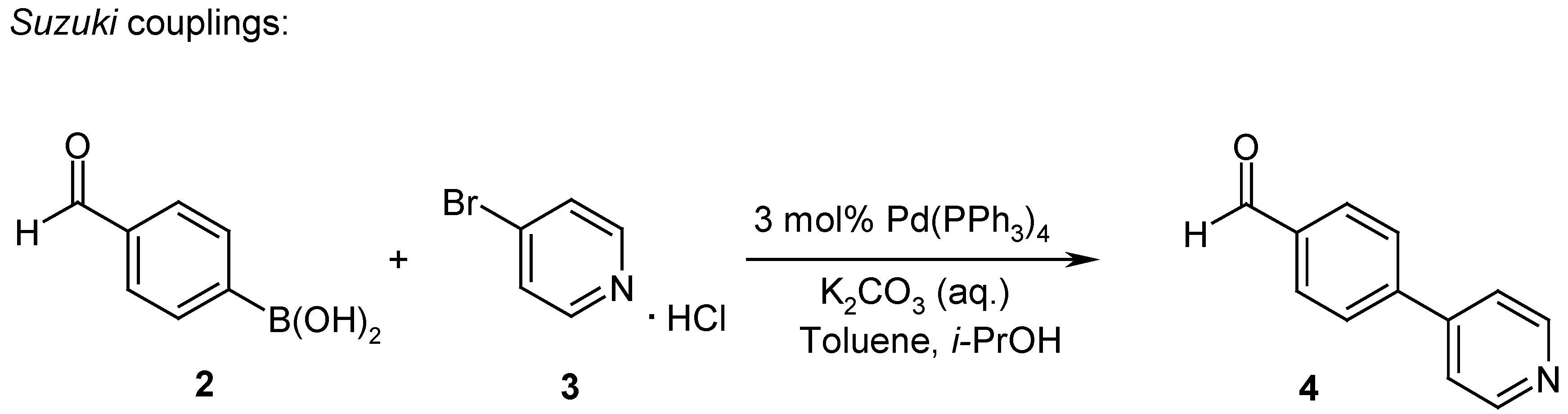
4-Pyridin-4-yl-benzaldehyde (4)
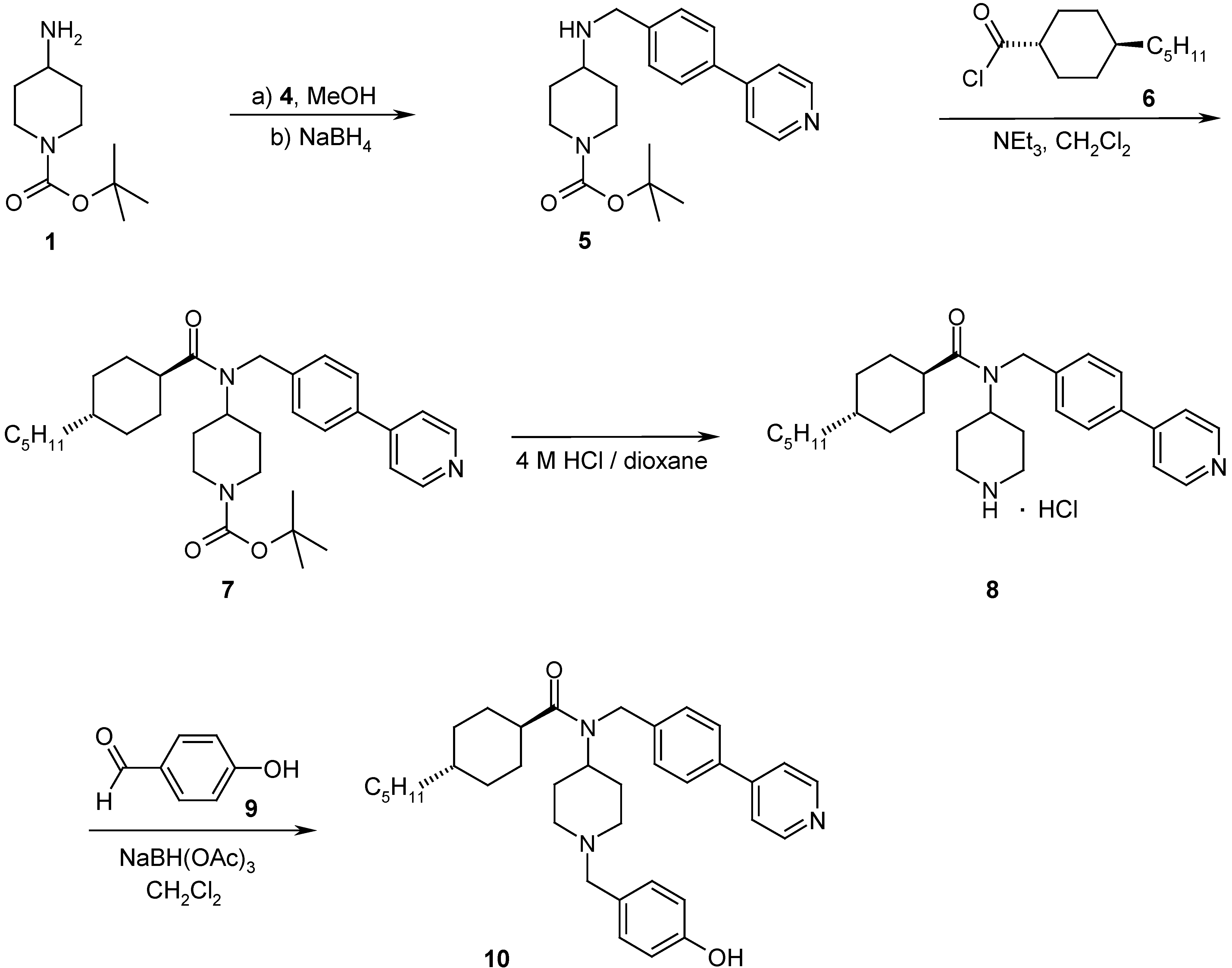
4-(4-Pyridin-4-yl-benzylamino)-piperidine-1-tert-butyl carbamate (5)
4-[(4-trans-Pentyl-cyclohexanecarbonyl)-(4-pyridin-4-yl-benzyl)-amino]-piperidine-1-tert-butyl carbamate (7)
4-trans-Pentyl-cyclohexanecarboxylic acid piperidin-4-yl-(4-pyridin-4-yl-benzyl)-amide hydrochloride (8)
4-trans-Pentyl-cyclohexanecarboxylic acid [1-(4-hydroxy-benzyl)-piperidin-4-yl]-(4-pyridin-4-yl-benzyl)-amide (10)
Acknowledgements
References
- (a) Bell, A. I. Drugs 2000, 3, 310–317. (b) Bell, A. Curr. Opin. Anti-infect. Invest. Drugs 2000, 2, 63–70.
- Rosenthal, P. J.; Sijwali, P.S.; Singh, A.; Shenai, B. R. Curr. Pharm. Des. 2002, 8, 99–110.
- http://sites.huji.ac.il/malaria/hemoglobindigest.html
- Rosenthal, P. J. Antimalarial Chemotherapy: Mechanism of Action, Resistance, and New Directions in Drug Discovery; Humana Press: New Jersey, 2001. [Google Scholar]
- Berry, C. Curr. Opin. Drug Disc. Dev. 2000, 3, 624–629.
- Boss, C.; Richard-Bildstein, S.; Weller, T.; Fischli, W.; Meyer, S.; Binkert, C. Curr. Med. Chem. 2003, 10, 883–907.
- Actelion Pharmaceuticals Ltd (Boss, C.; Fischli, W.; Meyer, S.; Richard-Bildstein, S.; Weller, T.), 2002, WO 02/24649
- (a) Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. (b) Watanabe, T.; Miyaura, N.; Suzuki, A. Synlett 1992, 207–210. (c) Gong, Y.; Pauls, H.W. Synlett 2000, 829–831.
- Abdel-Magid, A. F.; Carson, K. G.; Harris, B. D.; Maryanoff, C. A.; Shah, R. D. J. Org. Chem. 1996, 61, 3849–3862.
- Matayoshi, E. D.; Wang, G. T.; Krafft, G.A.; Erickson, J. Science 1990, 247, 954–958.
- Mueller, R. Diploma Thesis, University of Applied Sciences Basel, Muttenz, 2002.
- Taber, D. F.; Deker, P. B.; Gaul, M. D. J. Am. Chem. Soc. 1987, 109, 7488–7494.
- Sample Availability: Not available.
© 2003 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Mueller, R.; Huerzeler, M.; Boss, C. Synthesis of Plasmepsin II Inhibitors – Potential Antimalarial Agents. Molecules 2003, 8, 556-564. https://doi.org/10.3390/80700556
Mueller R, Huerzeler M, Boss C. Synthesis of Plasmepsin II Inhibitors – Potential Antimalarial Agents. Molecules. 2003; 8(7):556-564. https://doi.org/10.3390/80700556
Chicago/Turabian StyleMueller, Reto, Marianne Huerzeler, and Christoph Boss. 2003. "Synthesis of Plasmepsin II Inhibitors – Potential Antimalarial Agents" Molecules 8, no. 7: 556-564. https://doi.org/10.3390/80700556
APA StyleMueller, R., Huerzeler, M., & Boss, C. (2003). Synthesis of Plasmepsin II Inhibitors – Potential Antimalarial Agents. Molecules, 8(7), 556-564. https://doi.org/10.3390/80700556