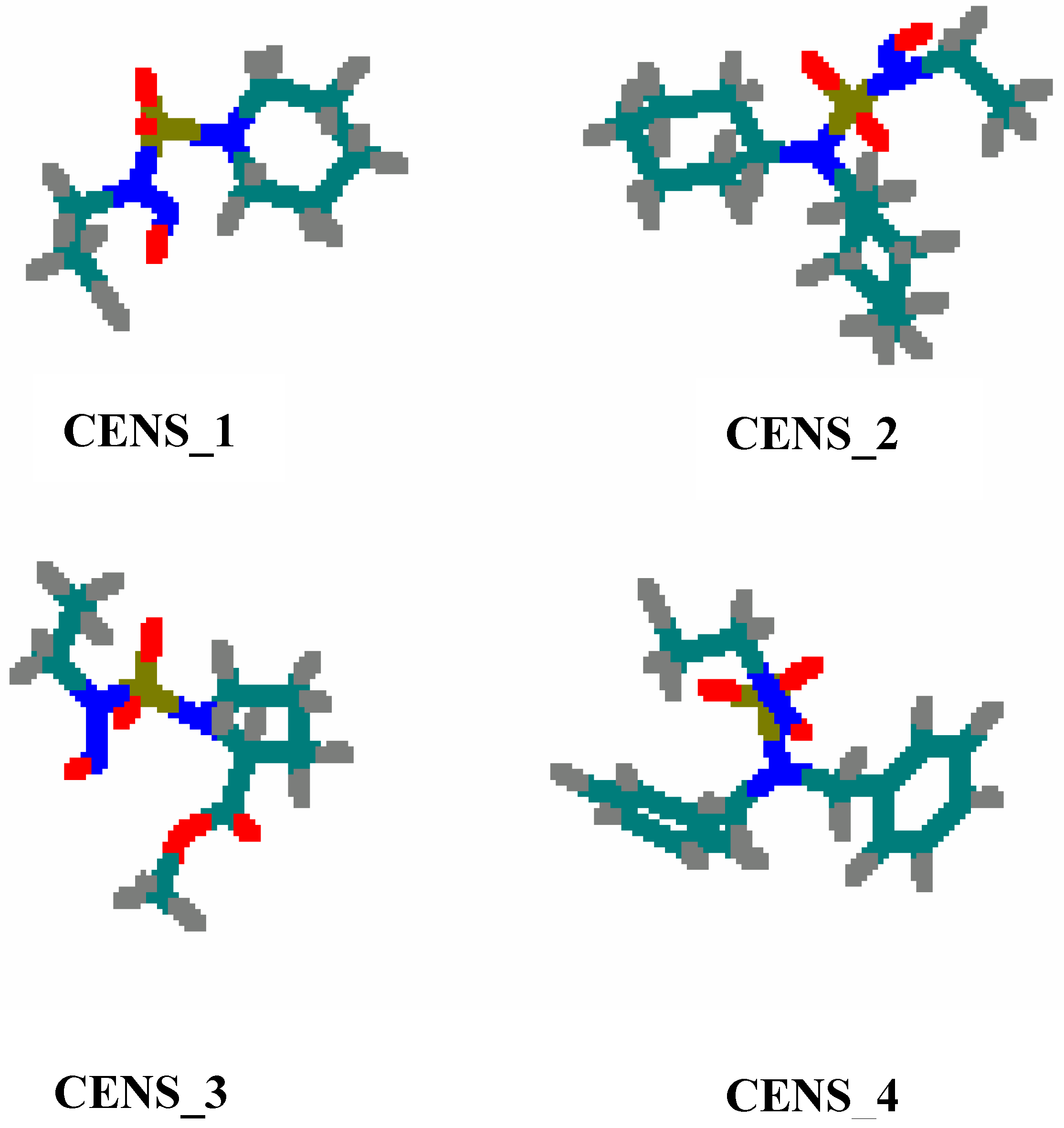
Results and Discussion
The 2-chloroethylnitrososulfamide molecules used in our theoretical study have been prepared in our laboratory by Abdaoui
et al. [
2,
3]. They are shown in
Figure 1.
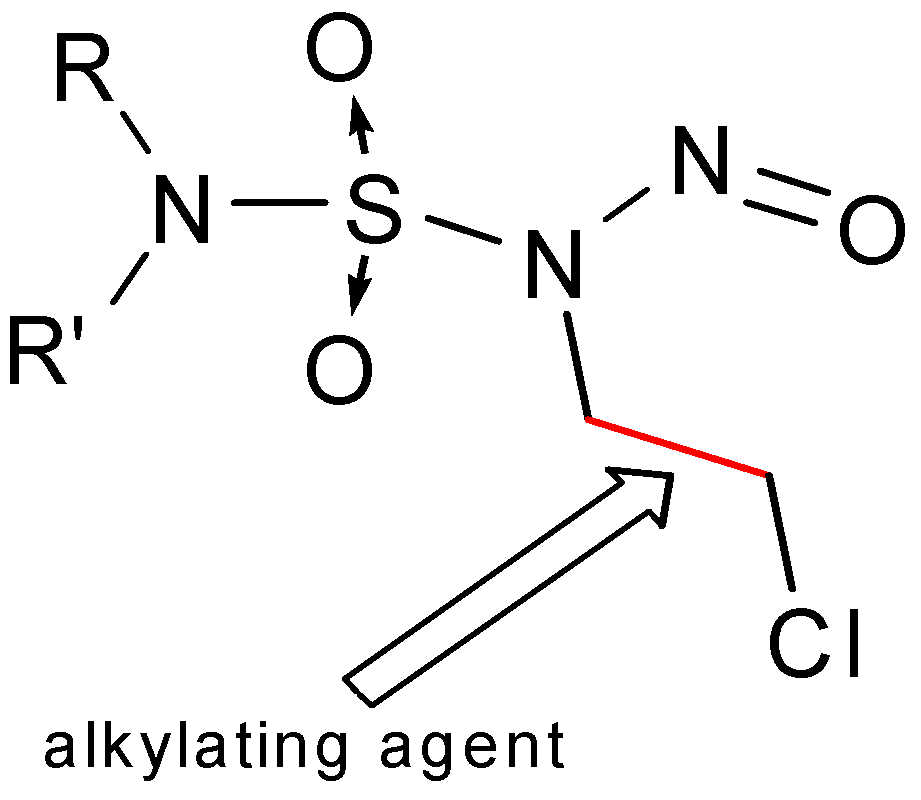
Figure 2 shows the common alkylating moiety of these molecules.
Figure 1.
The structures of the four CENS.
Figure 1.
The structures of the four CENS.
CENS_1: N-Nitroso-N-(2-chloroethyl)-N’-sulfamoylpiperidyne
CENS_2: N-Nitroso-N-(2-chloroethyl)-N’-dicyclohexylsulfamide
CENS_3: N-Nitroso-N-(2-chloroethyl)-N’-sulfamoylprolinate
CENS_4: N-Nitroso-N-(2-chloroethyl)-N’-dibenzylsulfamide
Figure 2.
Alkylating moiety in CENS
Figure 2.
Alkylating moiety in CENS
All the molecular mechanics calculations were carried on a Pentium 133 MHz with the MM+ force field as implemented in Hyperchem 6.0. The different 2-chloroethylnitrososulfamide compounds were built using Hyperchem. The search of the lowest energy conformation was performed with Allinger’s software including a Metropolis Monte Carlo (MMC) approach for generation of the new conformations using random variations of the randomly selected torsional angles [
5]. This search was performed with a range for ring acyclic or ring torsion variation of ±10-120°. The Random Walk, and Metropolis Criterion use T= 300 K, switching to 400 K. The structures which were found by MMC were reminimized by Polack-Ribiere optimization using MM+.
The neutral form of compounds has been used, with a dielectric constant of 1.5 applied in the calculations of the electrostatic interaction [
6]. Energy minimization was terminated when the gradient root mean square was below 0.01 kcal/mol. After the energy minimization, acceptance was determined by the following two criteria: 1) execution of a conformational search by the simulated annealing method [heat time: 0.1ps, run time: 0.5 ps, starting temperature: 100 K, simulation temperature: 300K, temperature step: 30K] which is described by Choe
et al. [
7]; 2) the structure obtained was minimized with a semi-empirical method (PM3) and we verified that there were no negative frequencies in the vibration spectrum.
Table 1 gives the detailed energy data for the most favored conformations of the four CENS, which are shown in
Figure 3.
Table 1.
The detailed energies of the most favored conformations of the four CENS
Table 1.
The detailed energies of the most favored conformations of the four CENS
| Parameter | Compound |
|---|
| CENS_1 | CENS_2 | CENS_3 | CENS_4 |
|---|
| Bond | 0.7717 | 1.8569 | 0.8473 | 0.8245 |
| Angle | 4.1127 | 6.0393 | 10.0222 | 4.2251 |
| Dihedral | -2.9549 | 2.5646 | -0.8142 | -14.4350 |
| Stretch-bend | 0.1267 | 0.3744 | -0.1083 | 0.1374 |
| Vdw | 6.2340 | 7.7230 | 4.6044 | 5.3712 |
| Electrostatic | -14.3593 | -13.9257 | -13.2188 | -14.4369 |
| E steric | -6.0740 | 6.2890 | 1.3825 | -18.3134 |
Table 1 contains some significant information: 1) the primary contribution to the energy is the electrostatic energy for CENS_1, CENS_2 and CENS_3, but for the CENS_4 compound the strongest contribution to the energy are the dihedral and electrostatic energies; 2) the bond and stretch-bend energies are relatively unimportant in the total energy; 3) the angle energy and Vdw interaction have a big influence of the energy.
Figure 3.
The most favored conformations of the four CENS
Figure 3.
The most favored conformations of the four CENS
During the global minimum conformational research for CENS_4, two methods were used for energy optimization: MM+ and a semi empirical method (PM3) [
8]. The CENS_4 geometry was optimized with MM+, and the obtained structure was used as initial structure for the PM3 calculation. Once the minimal energy of the structure was given using the semi empirical method, we used it as the basic structure for a new calculation with molecular mechanics.
From PM3 method, as implemented in Hyperchem 6.0, a large volume of data can be produced. The values for heat of formation, electric dipole and binding energy, for the lowest-energy conformation of the four CENS are summarized in
Table 2.
Table 2.
Bonding energy, heat of formation (kcal/mol) and electric dipoles (Debye) for the four CENS as calculated by the PM3 semi-empirical method.
Table 2.
Bonding energy, heat of formation (kcal/mol) and electric dipoles (Debye) for the four CENS as calculated by the PM3 semi-empirical method.
| Parameter | CENS_1 (kcal/mol) | CENS_2 (kcal/mol) | CENS_3 (kcal/mol) | CENS_4 (kcal/mol) |
|---|
| Binding energy | -2610.314 | -4455.257 | -2972.178 | -4291.6479 |
| Heat of formation | -71.58632 | -96.26391 | -143.470 | -6.504729 |
| Dipole (D) | 3.013 | 3.232 | 4.273 | 3.712 |
According to the bonding energy, quantitatively the order of the stability of the four CENS can be established as: CENS_2> CENS_4> CENS_3> CENS_1.
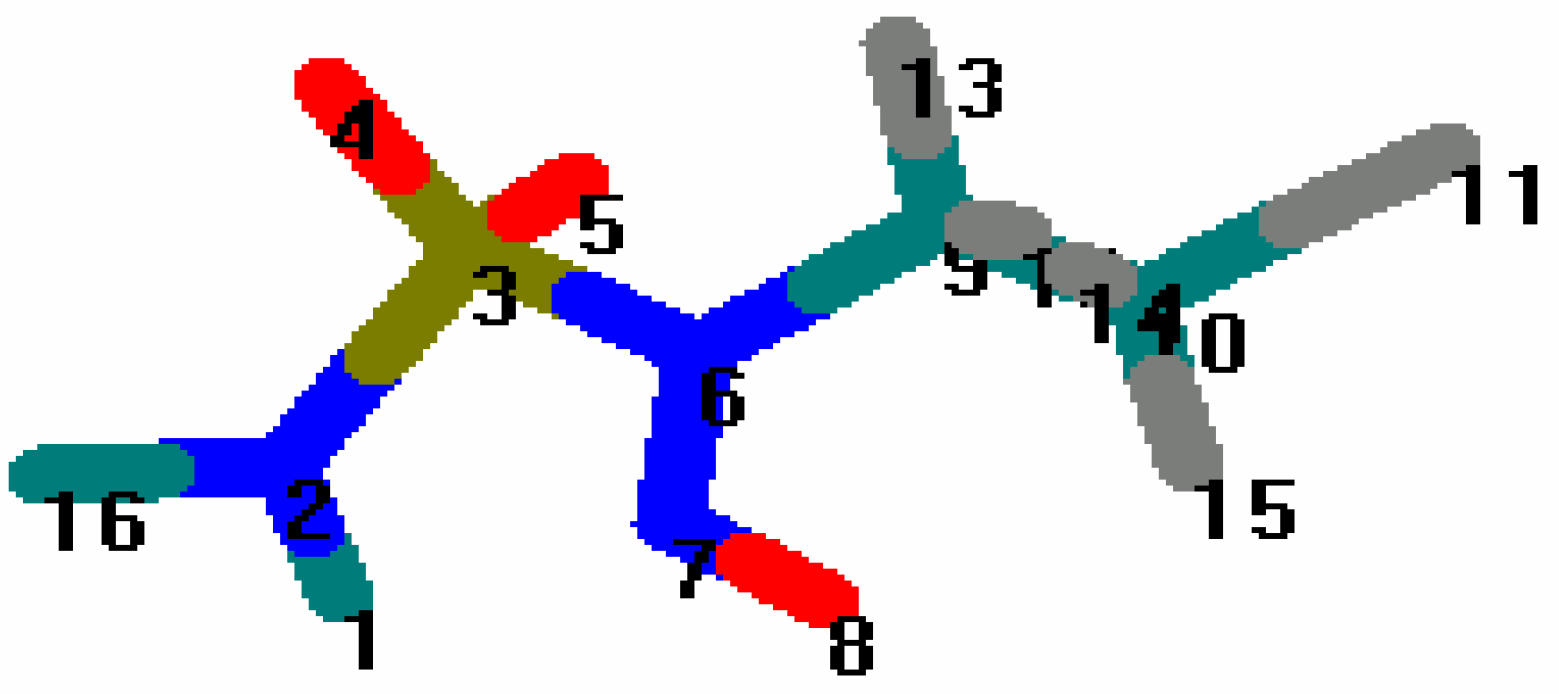
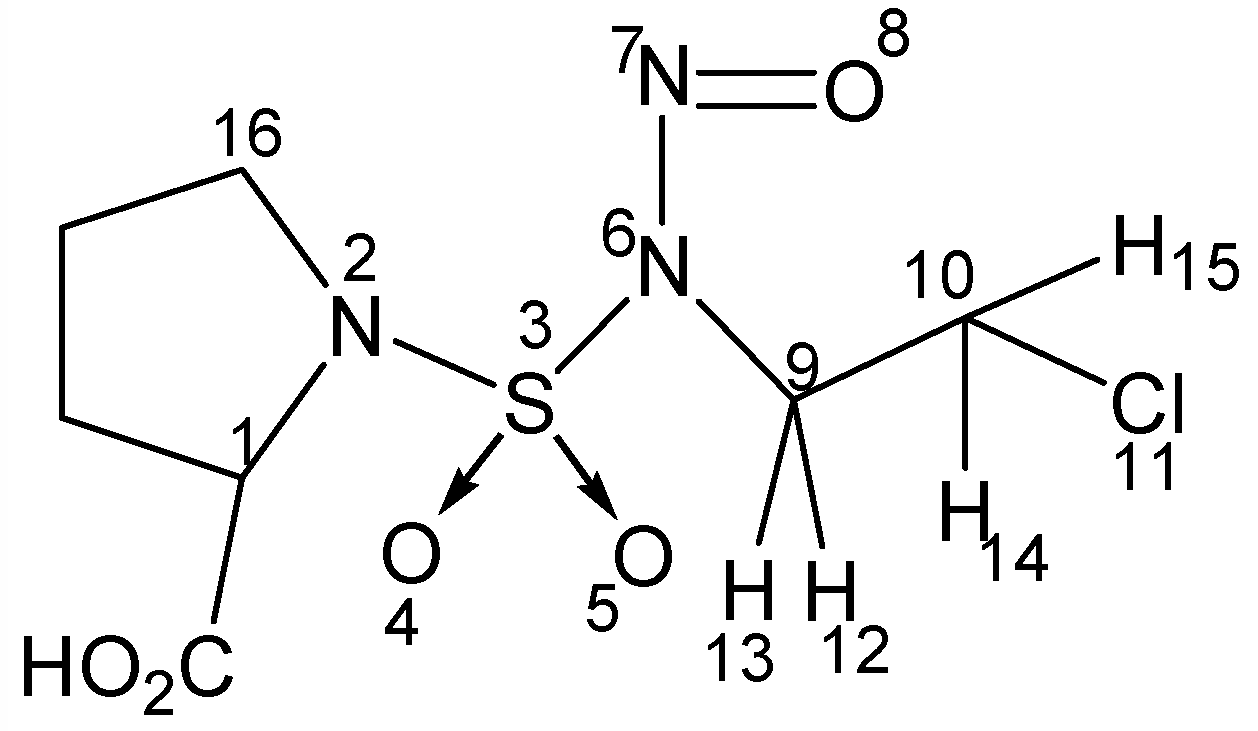
The bond lengths and bond angles of the common fragment of the four CENS (
Figure 4) calculated by molecular mechanics (
Table 3 and
Table 4) was compared with those obtained using X-ray crystallography for N-nitroso-N-(2-chloroethyl)-N’-sulfamoylproline [2b] derived from CENS_3 (
Figure 5).
Figure 4.
Common fragment of the four CENS.
Figure 4.
Common fragment of the four CENS.
Figure 5.
N-nitroso-N-(2-chloroethyl)-N’- sulfamoylproline
Figure 5.
N-nitroso-N-(2-chloroethyl)-N’- sulfamoylproline
Table 3.
Comparison of bond length (Å) parameters obtained by X-ray crystallography for the common fragment of the N-nitroso-N-(2-chloroethyl)-N’-sulfamoylproline and that with those obtained by molecular mechanics calculations.
Table 3.
Comparison of bond length (Å) parameters obtained by X-ray crystallography for the common fragment of the N-nitroso-N-(2-chloroethyl)-N’-sulfamoylproline and that with those obtained by molecular mechanics calculations.
| Bond | Crystallographic data drawn from N-nitroso-N-(2-chloroethyl)-N’-sulfamoylproline | MM+ calculations for |
|---|
| CENS_1 | CENS_2 | CENS_3 | CENS_4 |
|---|
| C(1)-N(2) | 1.48 | 1.45 | 1.47 | 1.44 | 1.45 |
| C(16)-N(2) | 1.45 | 1.45 | 1.46 | 1.45 | 1.45 |
| N(2)-S(3) | 1.57 | 1.65 | 1.65 | 1.64 | 1.65 |
| S(3)-O(4) | 1.44 | 1.44 | 1.44 | 1.44 | 1.44 |
| S(3)-O(5) | 1.40 | 1.44 | 1.44 | 1.44 | 1.44 |
| S(3)-N(6) | 1.68 | 1.65 | 1.65 | 1.65 | 1.65 |
| N(6)-N(7) | 1.37 | 1.35 | 1.36 | 1.35 | 1.35 |
| N(7)-O(8) | 1.20 | 1.17 | 1.17 | 1.17 | 1.17 |
| N(6)-C(9) | 1.47 | 1.45 | 1.46 | 1.45 | 1.45 |
| C(9)-C(10) | 1.50 | 1.54 | 1.53 | 1.54 | 1.53 |
| C(10)-Cl(11) | 1.73 | 1.79 | 1.80 | 1.79 | 1.79 |
Table 4.
Comparison of bond angle (°) parameters obtained by X-ray crystallography for the common fragment of the N-nitroso-N-(2-chloroethyl)-N’-sulfamoylproline and that with those obtained by molecular mechanics calculations.
Table 4.
Comparison of bond angle (°) parameters obtained by X-ray crystallography for the common fragment of the N-nitroso-N-(2-chloroethyl)-N’-sulfamoylproline and that with those obtained by molecular mechanics calculations.
| Bond | Crystallographic data drawn from N-nitroso-N-(2-chloroethyl)-N’-sulfamoylproline | MM+ calculations for |
|---|
| CENS_1 | CENS_2 | CENS_3 | CENS_4 |
|---|
| N(6)N(7)O(8) | 113.5 | 123.2 | 124.1 | 123.3 | 123.1 |
| N(2)S(3)O(4) | 109.7 | 110.3 | 110.4 | 109.2 | 108.5 |
| N(6)S(3)O(5) | 103.0 | 109.7 | 108.9 | 108.7 | 110.0 |
| N(6)S(3)N(2) | 105.1 | 98.4 | 97.4 | 97.2 | 100.7 |
| O(4)S(3)O(5) | 121.7 | 118.8 | 120.3 | 120.3 | 117.2 |
According the results of
Table 3 and
Table 4 it should be noted that the differences between the calculated and experimental values for the bond lengths are all less than 0.05 Å except for the N2-S3 bond length; the difference then reaches 0.08 Å. In the same way, the differences between the calculated and experimental values for the bond angles does not exceed 8° except in the case of the N6-N7-O8 bond angle; the difference in this case being 10.8°. By taking into account the difference between the various structures of the CENS and the structure of the carboxylic acid of the CENS_3 (
Figure 5) with which we made the comparison, concerning the common part of the CENS we can conclude that the calculated parameters show good agreement with experimental results and this opens the possibility of using detailed molecular information obtained by molecular mechanics (MM+) for analysis of the characteristics of CENS.
Anti and syn calculations
The relative stabilities of the
anti and
syn conformers and the interconversion between them are very important to understand reactivity [
9]. We have calculated the lowest-energy of the
anti and
syn conformers of the four CENS and the transition state (TS) for the
anti-
syn conformer using MM+, AM1, PM3 and
ab initio methods.
Figure 6.
Anti, syn and transition state structures
Figure 6.
Anti, syn and transition state structures
The syn conformation was built starting from the most stable conformation obtained previously, by making a 180° rotation of the N=O bond around the N-N bond. The obtained structure is optimized with MM+ by using the Polack-Ribiere algorithm to a maximum energy gradient of 0.01 kcal/ (Å.mole).
Table 5.
The values of the steric energy (kcal/mol) of the anti and syn structures and the difference between these two energies as calculated by MM+.
Table 5.
The values of the steric energy (kcal/mol) of the anti and syn structures and the difference between these two energies as calculated by MM+.
| | CENS_1 | CENS_2 | CENS_3 | CENS_4 |
|---|
| anti | -6.074 | 1.382 | 6.289 | -18.313 |
| syn | -5.435 | 2.338 | 6.436 | -14.553 |
| ΔE (syn-anti) | 0.638 | 0.955 | 0.147 | 3.760 |
The MM+ calculations (
Table 5) show that
anti conformation is the most stable one for the four CENS molecules studied. This difference varies from 0.147 (CENS_3) to 3.760 kcal/mole (CENS_4). Using the molecular mechanics results we can say that quantitatively the
anti and
syn conformations should exist in similar amounts (ΔE< 1) for CENS_1, CENS_2 and CENS_3, whereas for CENS_4, only the
anti conformation is possible because ΔE>2 [
10,
11,
12].
Semi – empirical methods
The
anti and
syn conformations obtained for the four CENS with MM+ were re-optimized with PM3 and AM1 by using the Polack-Ribiere algorithm to a maximum energy gradient of 0.01 kcal/(A. mole). The structure of the transition state was obtained by imposing a geometrical constraint making the N-N=O bond angle equal to 180°. The search for the transition state (TS) and vibrational calculations are done using the Eigenvector following Algorithm. The confirmation of its localization was done by checking in the vibration spectrum for the presence of only one imaginary frequency [
13]. The results obtained are given in
Table 6:
Table 6.
The relative energy (kcal/mol) of the anti, syn and TS structures of the four CENS calculated by PM3 and AM1 semi empirical methods.
Table 6.
The relative energy (kcal/mol) of the anti, syn and TS structures of the four CENS calculated by PM3 and AM1 semi empirical methods.
| | | CENS_1 | CENS_2 | CENS_3 | CENS_4 |
|---|
| PM3 | anti | -2611.166 | -2972.178 | -4455.257 | -4291.156 |
| syn | -2609.039 | -2971.424 | -4453.520 | -4288.289 |
| TS | -2551.511 | -2912.350 | -4396.771 | -4231.334 |
| |∆E (anti-syn)| | 2.127 | 0.754 | 1.737 | 2.867 |
| |ΔE (TS-anti)| | 59.655 | 59.828 | 58.486 | 59.822 |
| AM1 | anti | -2626.291 | -2983.727 | -4475.774 | -4300.583 |
| syn | -2620.866 | -2977.450 | -4471.331 | -4295.431 |
| TS | -2544.697 | -2900.914 | -------- | -4231.334 |
| |∆E (anti-syn)| | 5.425 | 6.277 | 4.443 | 5.152 |
| |ΔE (TS-anti)| | 81.594 | 82.813 | -------- | 69.249 |
According to the results (
Table 6) we can note:
The two methods AM1 and PM3 give the same most stable conformation (anti).
The energy difference between the syn and anti conformations is higher in the PM3 than with the AM1 method.
In the PM3 method, the energy difference between the anti conformation and the transition state is almost constant for the four CENS (around 59.kcal/mole). But in the AM1 method it varies from 69.249 to 82.813kal/mole. According to the magnitude of this difference the transition between the anti to syn conformation is difficult.
One notices in our search for the transition state of CENS_3 that the imposed constraint N-N=0 equal to 180° is not respected because at the beginning of each optimization this parameter changes considerably. Consequently, we could not locate a transition state for this structure.
Ab Initio method
In order to confirm this result we used an
ab initio method. For this purpose we optimized the
syn and
anti conformations by the same procedure used in PM3 and AM1 calculations using the RHF method with a minimal basic function STO-3G. In the simplest case of the CENS_1 molecule the system is formed with 97 basis functions and 291 Gaussian primitives and this represents a great challenge for calculation with an IBM Pentium III 133 MHz personal computer (in terms of computational time). The transition state was obtained with the same procedure as that used in AM1 and PM3 methods. The calculation of the transition state was given with the same method RHF/ STO-3G). The results obtained are shown in
Table 7:
Table 7.
The relative energy (kcal/mol) of the anti, syn and TS structures of the four CENS calculated by the RHF method with a minimal STO- 3G basis function.
Table 7.
The relative energy (kcal/mol) of the anti, syn and TS structures of the four CENS calculated by the RHF method with a minimal STO- 3G basis function.
| | CENS_1 | CENS_2 | CENS_3 | CENS_4 |
|---|
| E anti | -941384.75 | -1057519.50 | -1110126.50 | -1154040.75 |
| E syn | -941386.78 | -1057521.00 | -1110126.67 | -1154045.37 |
| E (TS) | -941300.25 | -1057434.37 | -1110041.50 | -1153959.62 |
| |ΔE (anti-syn)| | 2.03 | 1.50 | 0.17 | 4.62 |
| |ΔE (TS-syn)| | 84.53 | 86.63 | 85.17 | 85.74 |
These results allow the following observations:
We obtain a completely contrary order of stability to that obtained by the other methods (molecular mechanics and semi empirical). Thus, the syn conformation is now slightly more stable than the anti conformation.
The energy barrier between the syn conformation and the transition state is around 85 kcal/mole.
Since the energetic barrier between the syn conformation and the transition state is very important (85 kcal/mol) we can say that the transition between the syn to anti conformations is very difficult.
From the results of
Table 8 some important information can be observed. On the one hand, the N center atoms of CENS_1, CENS_2 and CENS_3 undergo a reduction of 2/3 of their electronic density (obtained by the PM3 method) in the various transition states but for CENS_4 it loses only a half of its electronic density. On the other hand, the electronic density in the SO
2 group in CENS_1, CENS_2 and CENS_3 varies considerably, but in CENS_4 it remains almost constant. We thus note that in the CENS_1, CENS_2 and CENS_3 the SO
2 group plays a significant role in the formation of transition state but in CENS_4 it does not appear to participate in the structure of the transition state.
Table 8.
The charge of selected atoms obtained by PM3 calculations (four CENS in syn, anti and transition form).
Table 8.
The charge of selected atoms obtained by PM3 calculations (four CENS in syn, anti and transition form).
| | N (center) | N (nitroso) | O (nitroso) | S (nitroso) | O4, O5 |
|---|
| CENS_1 |
| anti | -0.584 | 0.338 | -0.279 | 2.264 | -0.813, -0.796 |
| syn | -0.561 | 0.360 | -0.286 | 2.232 | -0.798, -0.825 |
| TS | -0.186 | -0.044 | -0.103 | 0.774 | -0.322, -0.340 |
| CENS_2 |
| anti | -0.572 | 0.332 | -0.280 | 2.279 | -0.826, -0.800 |
| syn | -0.560 | 0.359 | -0.295 | 2.211 | -0.811, -0.797 |
| TS | -0.167 | -0.044 | -0.091 | 0.786 | -0.341, -0.349 |
| CENS_3 |
| anti | - 0.562 | 0.349 | -0.269 | 2.273 | -0.832, -0.793 |
| syn | - 0.564 | 0.363 | -0.270 | 2.231 | -0.791, -0.828 |
| TS | -0.173 | -0.024 | -0.092 | 0.788 | -0.352, -0.341 |
| CENS_4 |
| anti | -0.545 | 0.335 | -0.290 | 2.253 | -0.822, -0.810 |
| syn | -0.548 | 0.348 | -0.276 | 20204 | -0.805, -0.821 |
| TS | -0.257 | 0.191 | -0.330 | 2.140 | -0.810, -0.797 |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



