Introduction
Furocoumarins, such as psoralene and the angelicine derivatives are naturally occurring compounds. They are known to possess a high photobiological activity [
1]. Psoralene derivatives have been used for many years in the treatment of skin diseases [
2]. Furocoumarin/ultraviolet therapy, known as photopheresis, has recently become an effective treatment of cutaneous T cell lymphoma, Sezary syndrome and related diseases [
3,
4]. The photochemotherapeutic effects of furocoumarins are based on intercalation of the molecules between the pyrimidine bases of the microorganism’s DNA. The intercalation is then followed by the UV light activated cycloaddition reactions of furocoumarins with the pyrimidine bases. These [2+2] photocycloaddition reactions result in a cross-linking of DNA and prevent a microorganism’s reproduction.
Psoralens, which are linear furocoumarins, have the highest photosensitivity. Their molecules have two active sites in the [2+2] photocycloaddition reactions: the pyrone ring and furan ring double bonds. This kind of difunctionality of the psoralens (and of the angelicines in to a lesser extent) has been suggested to cause undesirable side effects in their medical use. Mutagenicity and carcinogenicity should be mentioned among these side effects of some psoralens [
5,
6,
7,
8,
9,
10,
11].
Even though some methods of furocoumarin synthesis have been known for a long time, the successful use of furocoumarins as effective medicines requires access to as many new derivatives as possible. For example, it has been found that furocoumarin analogs that contain other heteroatoms besides the oxygen atom in the lactone ring are free of some side effects [
4]. Better intercalation properties and higher hydrophilicity should be also specific for new furocoumarins recommended for pharmaceutical testing. Therefore, the search for new synthetic approaches to the fucoumarins and their analogous heterocyclic compounds is a promising trend in photochemotherapy [
12,
13,
14,
15,
16,
17,
18,
19].
Fries rearrangement of acetoxyheteroarenes
Acylhydroxyheteroarenes are convenient intermediates in furoheteroarene synthesis. The key reaction for preparation of these intermediates is the Fries rearrangement of acyloxyheteroarenes. Nevertheless, this reaction with heteroarene derivatives has not been studied much when compared with that of benzene derivatives.
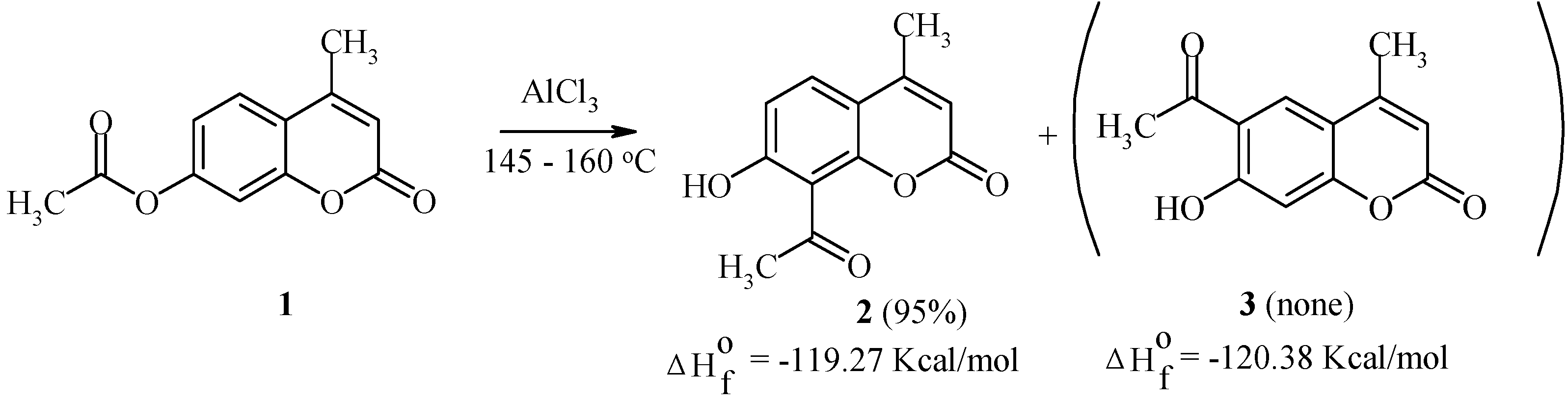
We have studied the Fries rearrangement of 7-acyloxycoumarins in detail [
20,
21,
22]. Some of the results are given below. For example, 7-acetoxy-4-methylcoumarin (
1) undergoes a smooth transformation into 8-acetyl-7-hydroxy-4-methylcoumarin (
2) as the exclusive product of the rearrangement (
Scheme 1). This rearrangement is largely temperature independent.
Kinetic factors seem to explain this result, since both 8-acetyl- and 6-acetyl-isomers (compounds 2 and 3, respectively) possess similar thermodynamic stability.
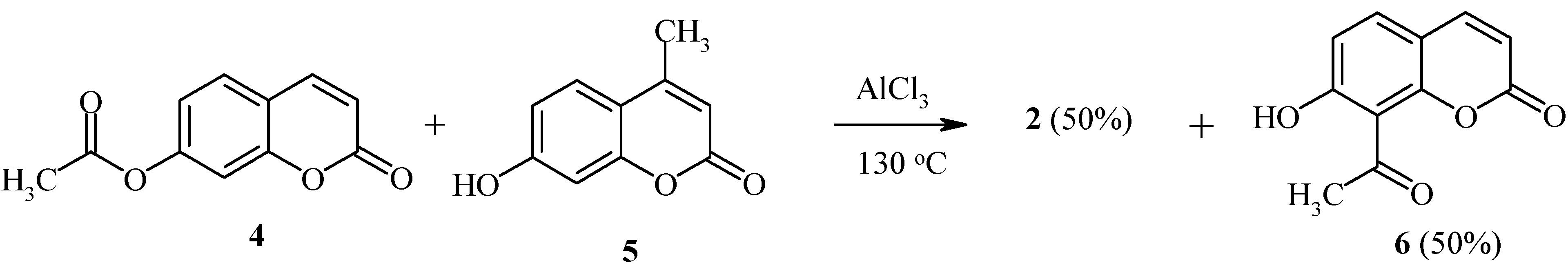
We have found that the Fries rearrangement of 7-acyloxycoumarins takes place by an intermolecular mechanism [
20]. Thus, rearrangement of 7-acetoxycoumarin (
4) in the presence of 7-hydroxy-4-methylcoumarin (
5) results in the formation of equimolar amounts of 8-acetyl-7-hydroxycoumarin (
6) and 8-acetyl-7-hydroxy-4-methylcoumarin (
2) (
Scheme 2).
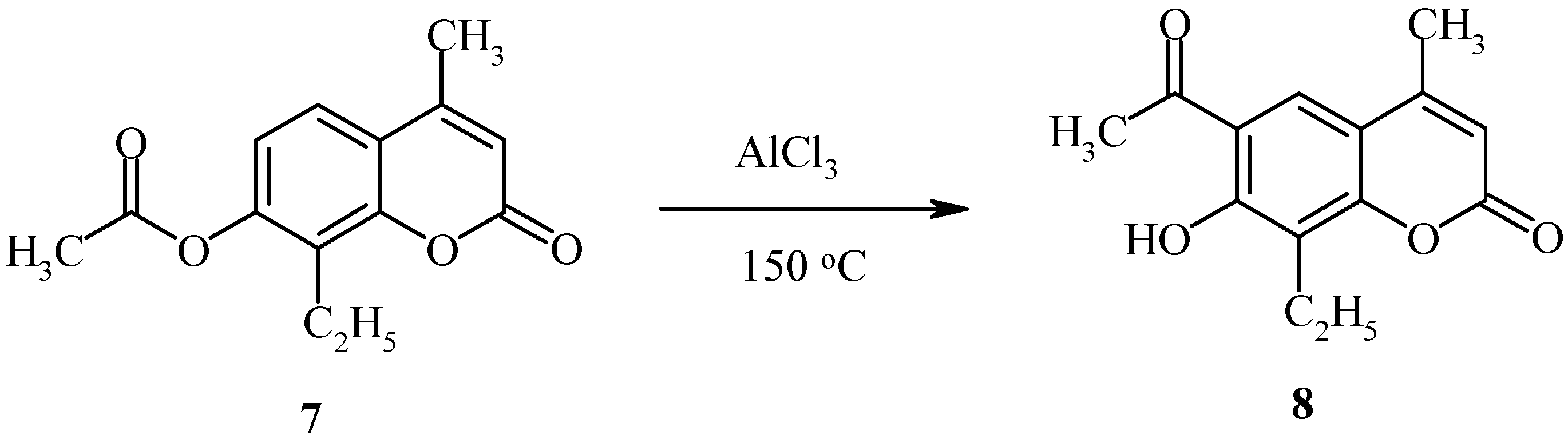
The deacylated product, 7-hydroxycoumarin, has also been found in the final reaction mixture. When the position 8 of the starting coumarin derivative is occupied (as in compound
7,
Scheme 3), the acyl group rearrangement involves the 6 position.
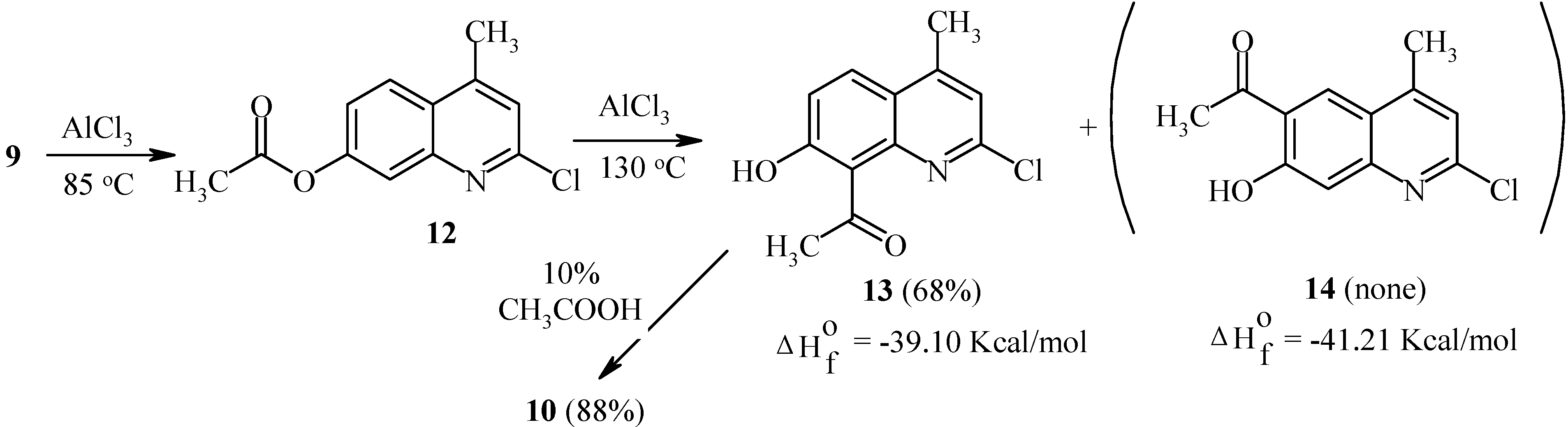
Rearrangement of 7-acetoxycoumarins thus provides intermediates for both angular and linear furocoumarins. We have found the regioselectivity of the Fries rearrangement of acyloxyheteroarenes to be very dependent on the structure of the heteroarene [
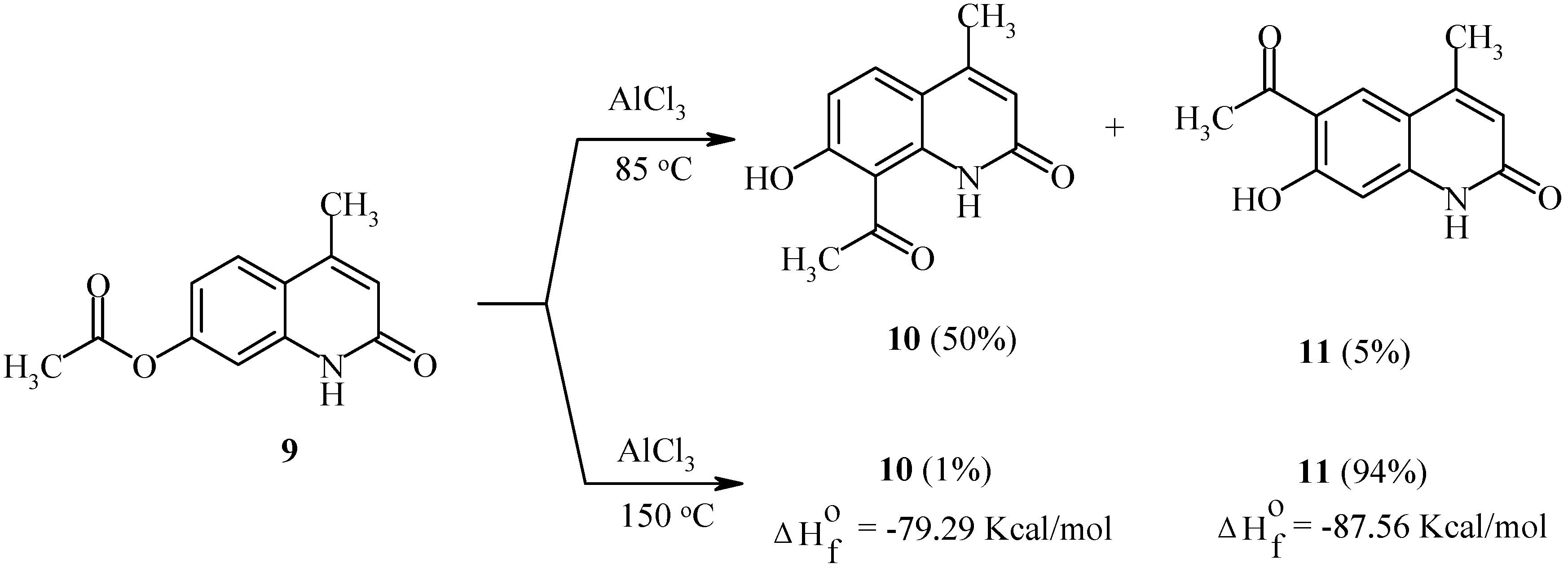
23]. Regioselectivity data of rearrangements of acyloxycoumarins, bis(acyloxy)coumarins, acyloxyquinolin-2-ones, acyloxy-2-chloroquinolines and their analogs are shown below. In contrast to the reactions of acyloxycoumarins, the regioselectivity of Fries rearrangement of 7-acyloxyquinolin-2-one
9 is very dependent on the temperature.
At relatively low temperature (85°C), 8-acetyl-7-hydroxy-4-methylquinolin-2-one (
10) is the predominant product of the rearrangement: 50% of 8-isomer
10 and 5% of 6-isomer
11 have been found in the final reaction mixture. However, an increase in temperature to 150°C results in the formation of the 6-isomer
11 as the major product (
Scheme 4).
It should be noted that isomers
10 and
11 possess different calculated thermodynamic stabilities. As can be seen, a high rearrangement temperature results in predominant formation of the thermodynamically more stable isomer
11. By contrast, isomer distributions of products at lower temperatures seem to follow a kinetic trend. Substitution of a chlorine atom for the oxo-function in quinolinone
9 greatly affects the regioselectivity of the Fries rearrangement. Thus, 7-acetoxy-2-chloro-4-methylquinoline (
12) undergoes the rearrangement with exclusive formation of the 8-acetyl-isomer
13 independent of the temperature (
Scheme 5). A possible isomer
14 was not found. Heating of
13 in aqueous acetic acid furnished quinolone
10.
As can be seen from
Scheme 5, isomers
13 and
14 exhibit similar calculated thermodynamic stabilities. Therefore, the exclusive formation of
13 appears to be a result of a kinetically controlled rearrangement. As already noted, a similar selectivity has been observed for acetoxycoumarin
1.
The following conclusions concerning regioselectivity of the acyloxyheteroarenes Fries rearrangement agree with the given results. If two isomers, the products of the rearrangement, have different thermodynamic stabilities, one can see a definite effect of the reaction temperature on the regioselectivity. If two isomeric products of the rearrangement possess similar thermodynamic stabilities, then the regioselectivity seems to be independent of the reaction temperature.
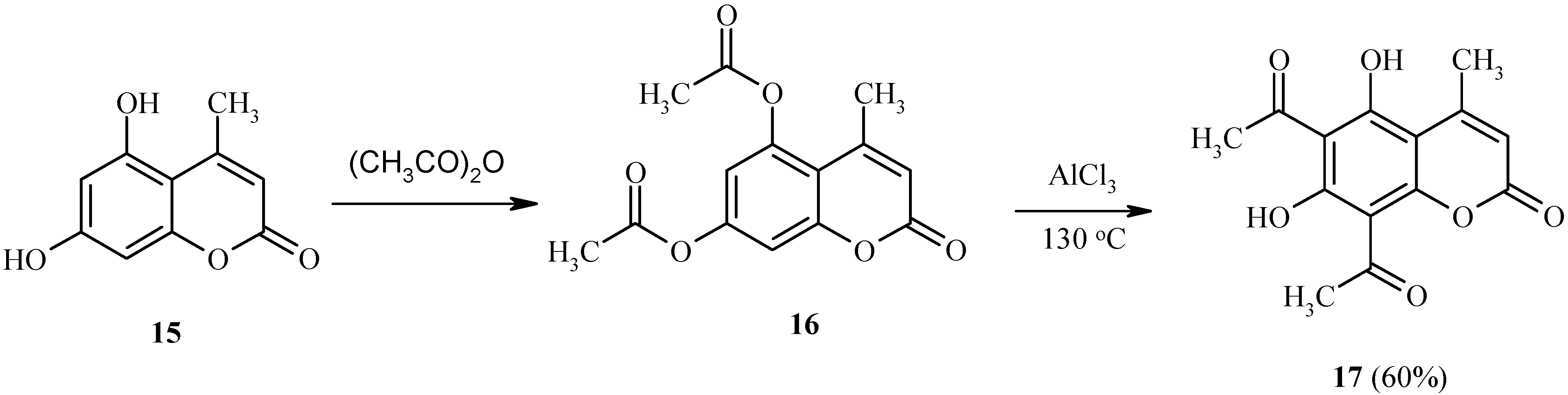
An interesting example of the Fries rearrangement is provided by the transformation of 5,7-bis(acetoxy)-4-methylcoumarin (
16), derived from dihydroxycoumarin
15 (
Scheme 6). In the rearrangement of
16, two acetoxy functions are simultaneously rearranged in the same molecule to give
17. In summary, the Fries reaction seems to be a convenient method for the synthesis of heteroarenes with adjacent acyl and hydroxyl functions.
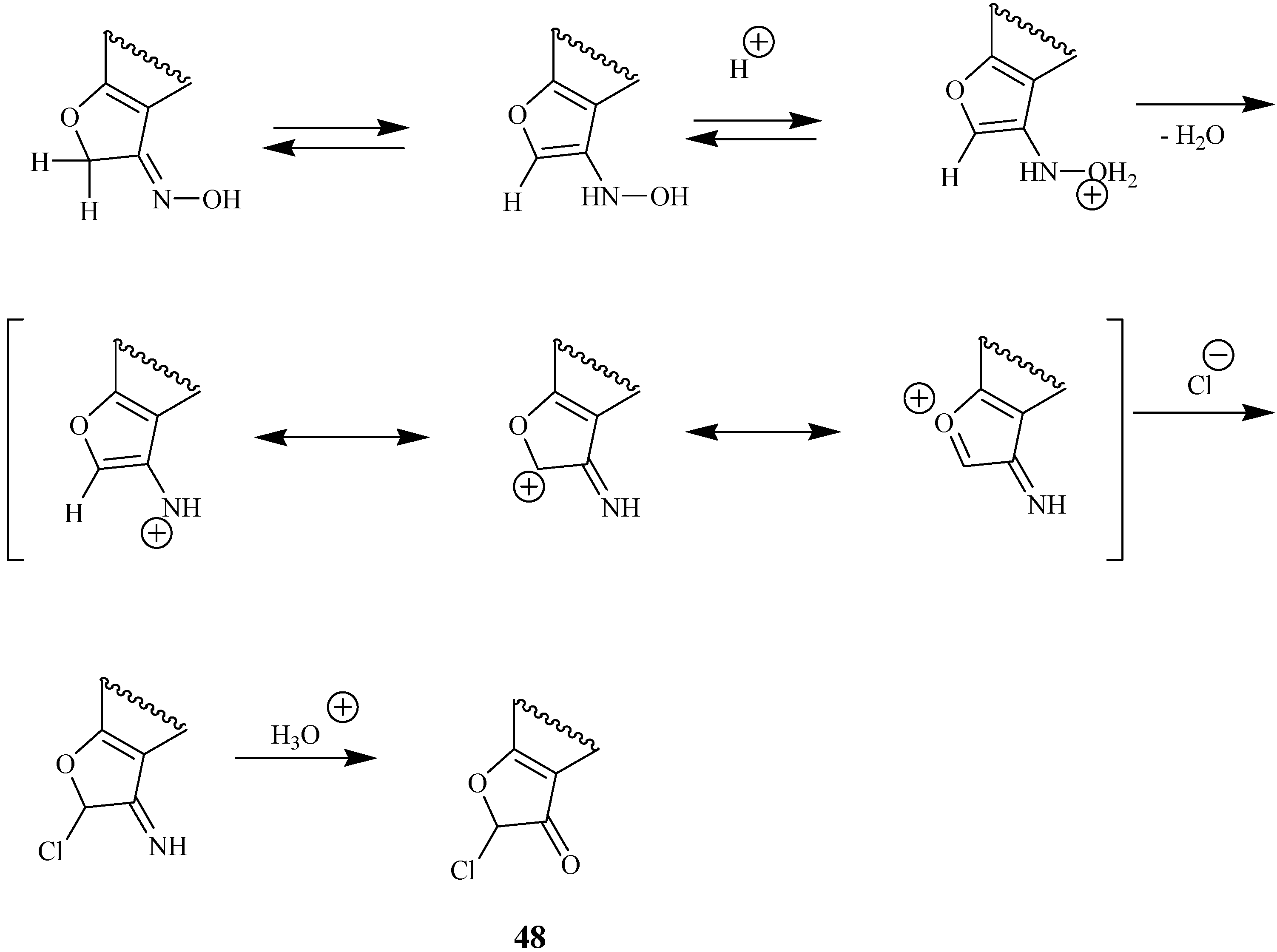
Unusual Fries rearrangements of chloroacetoxyheteroarenes
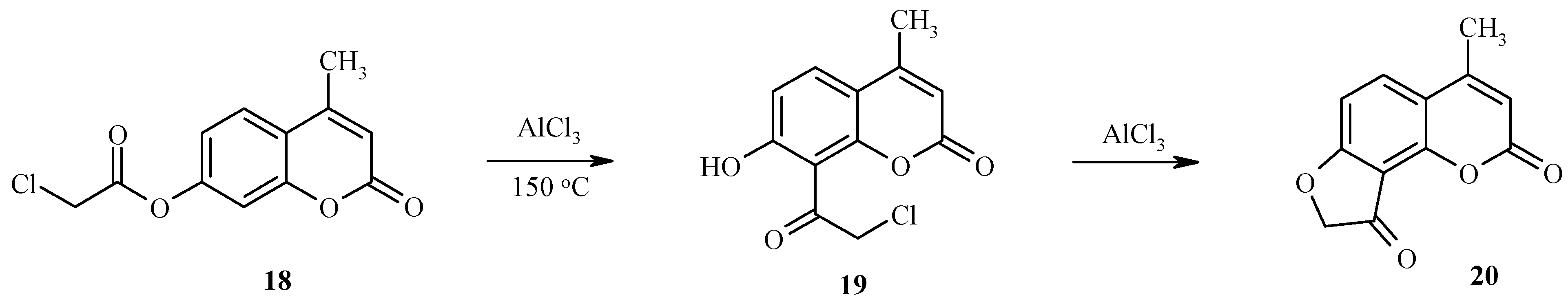
Unusual results have been found for the treatment of 7-chloroacetoxyheteroarenes with excess aluminum trichloride [
22]. Unexpectedly, the classical rearrangement is followed by an intramolecular cyclization of an intermediate chloroacetyl hydroxy derivative with formation of a condensed dihydrofuranone. As an example, the rearrangement of 7-chloroacetoxy-4-methylcoumarin (
18) to give the final product
20 via the intermediate
19 is shown in
Scheme 7.
Thus, compound 18 and similar 7-chloroacetoxy substituted coumarins are important substrates for the synthesis of angular furocoumarins.
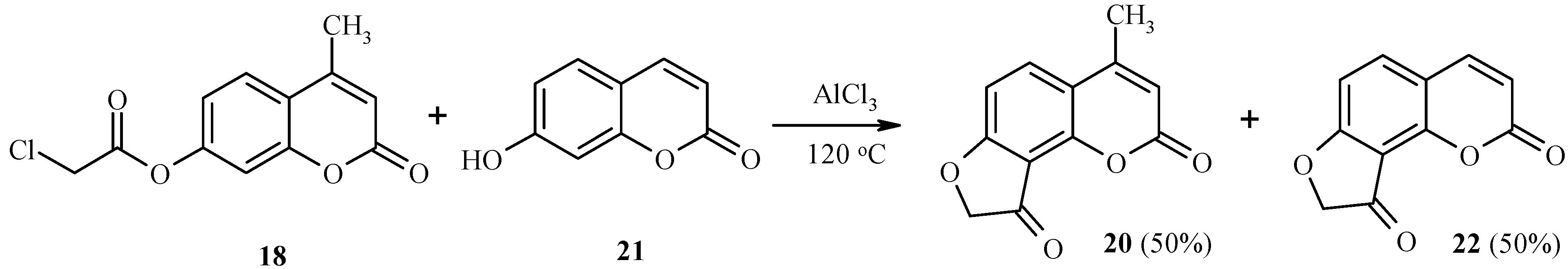
As with 7-acetoxycoumarins, 7-chloroacetoxycoumarins undergo this unusual Fries rearrangement by an intermolecular mechanism as well. Thus, rearrangement of
18 in the presence of 7-hydroxy-4-methylcoumarin (
21) gives equimolar amounts of a 4-methyl derivative
20 and its analog
22, lacking the methyl group (
Scheme 8) [
20].
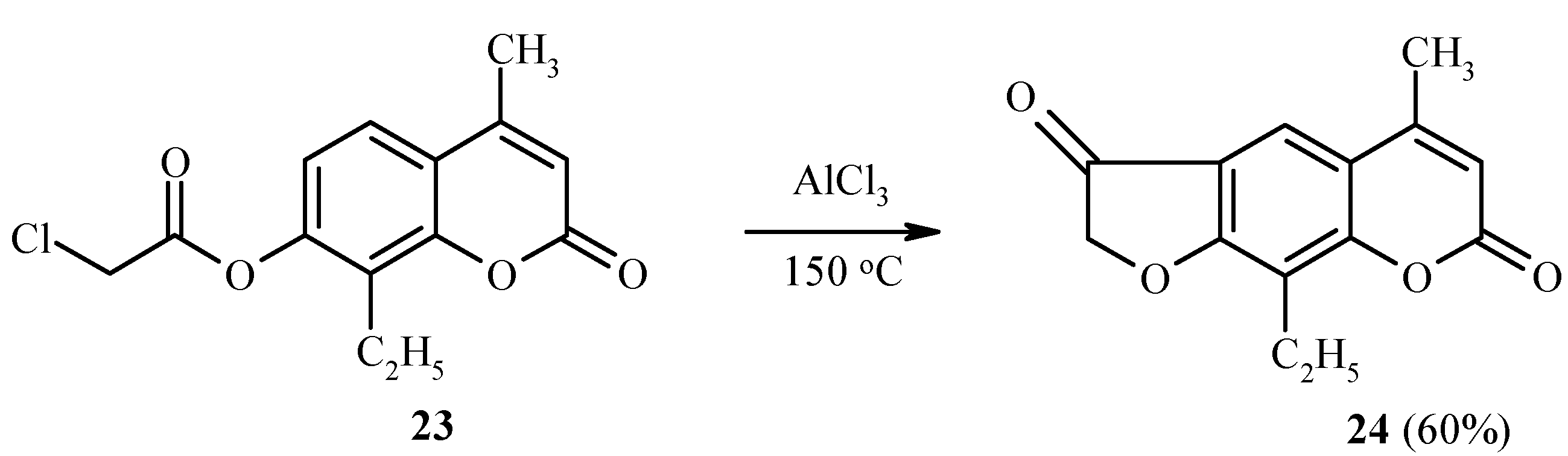
Substrates which have a substituent at position 8 of the coumarin ring system, undergo rearrangement into position 6 [
22]. This is illustrated in
Scheme 9 by the synthesis of
24 from
23.
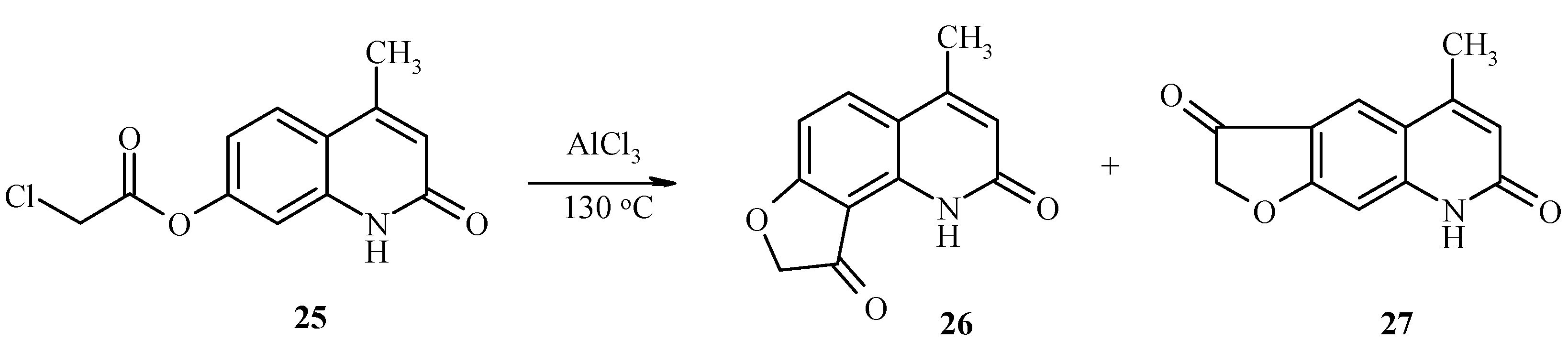
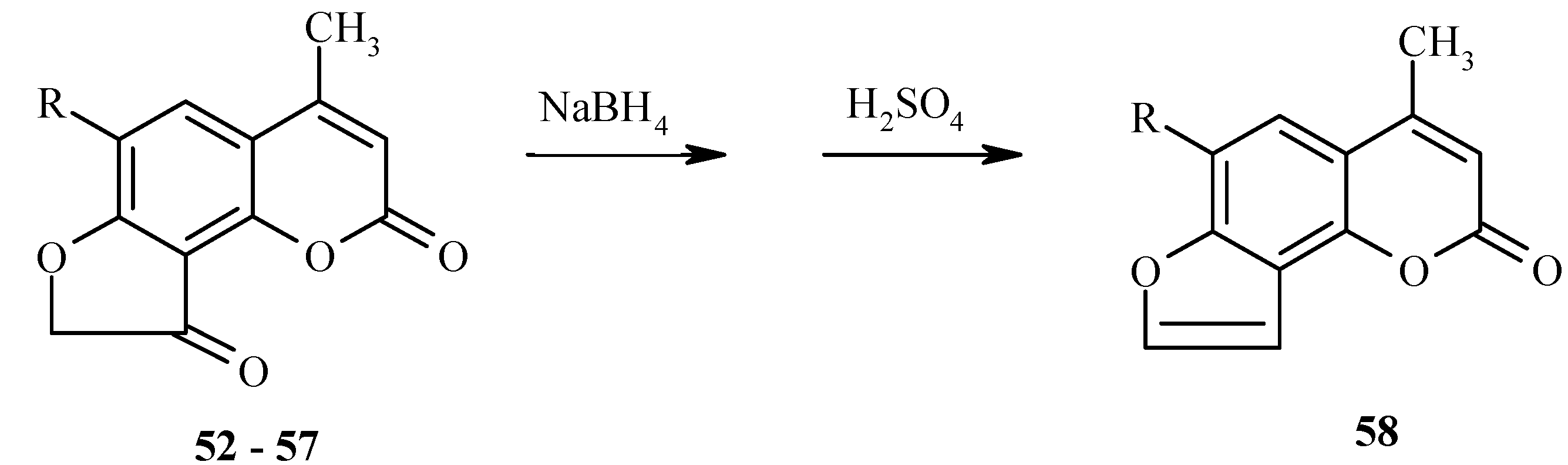
7-Chloroacetoxy-4-methylquinolin-2-one (
25) also undergoes the a Fries rearrangement that is followed by an intramolecular cyclization to give a mixture of furoquinolines
26 and
27 (
Scheme 10). Due to low regioselectivity, this reaction does not have much synthetic value.
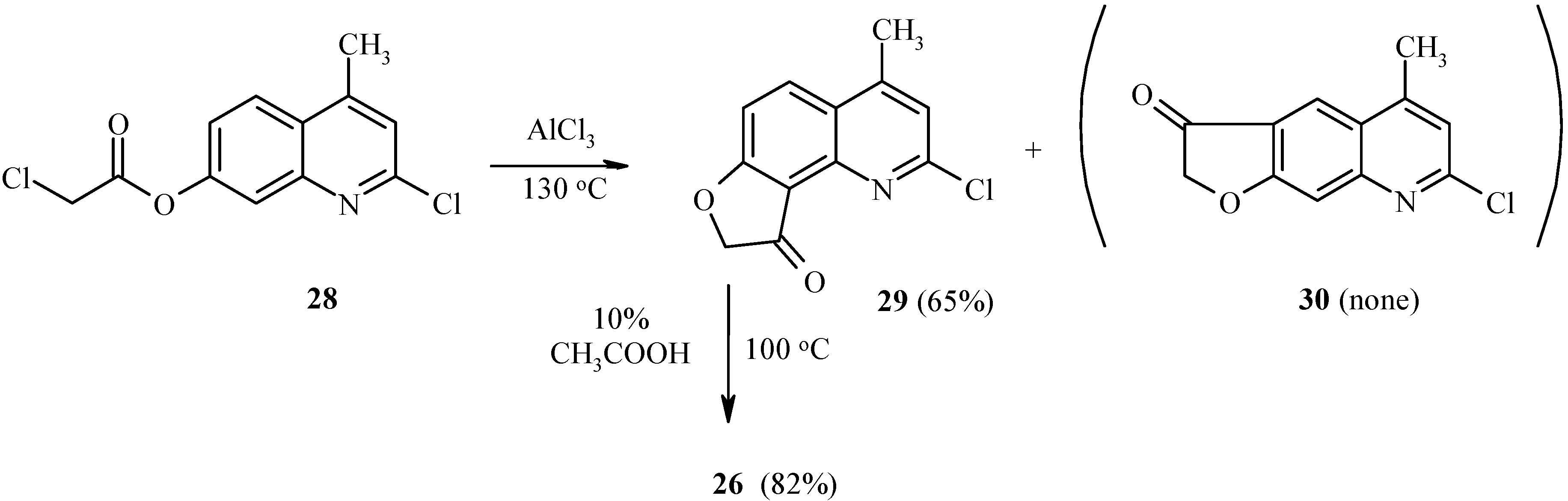
As with 7-acetoxy derivatives, regioselectivity of the Fries rearrangement of 7-chloroacetoxy-substituted substrates is much affected by substitution of the chlorine atom for the 2-oxo function. Exclusive formation of
29 without traces of isomer
30 by rearrangement of
28 can be noted (
Scheme 11). The chlorine atom in
29 can be replaced by an oxo or amino function, as exemplified by hydrolysis of
29 to furoquinolinone
26.
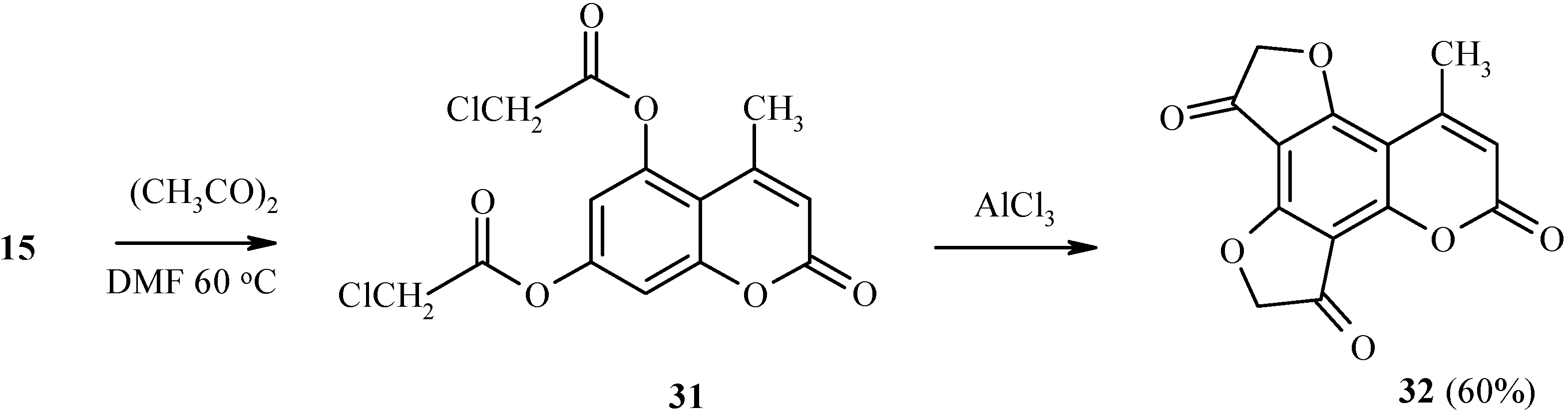
Two dihydrofuranone rings in the same heteroarene molecule can be constructed under Fries rearrangement as well (
Scheme 12). For example, 5,7-bis(chloroacetoxy)coumarin
31 has been prepared by chloroacetylation of compound
15. Heating of
31 with a large excess of aluminum trichloride gave
32 in good yield.
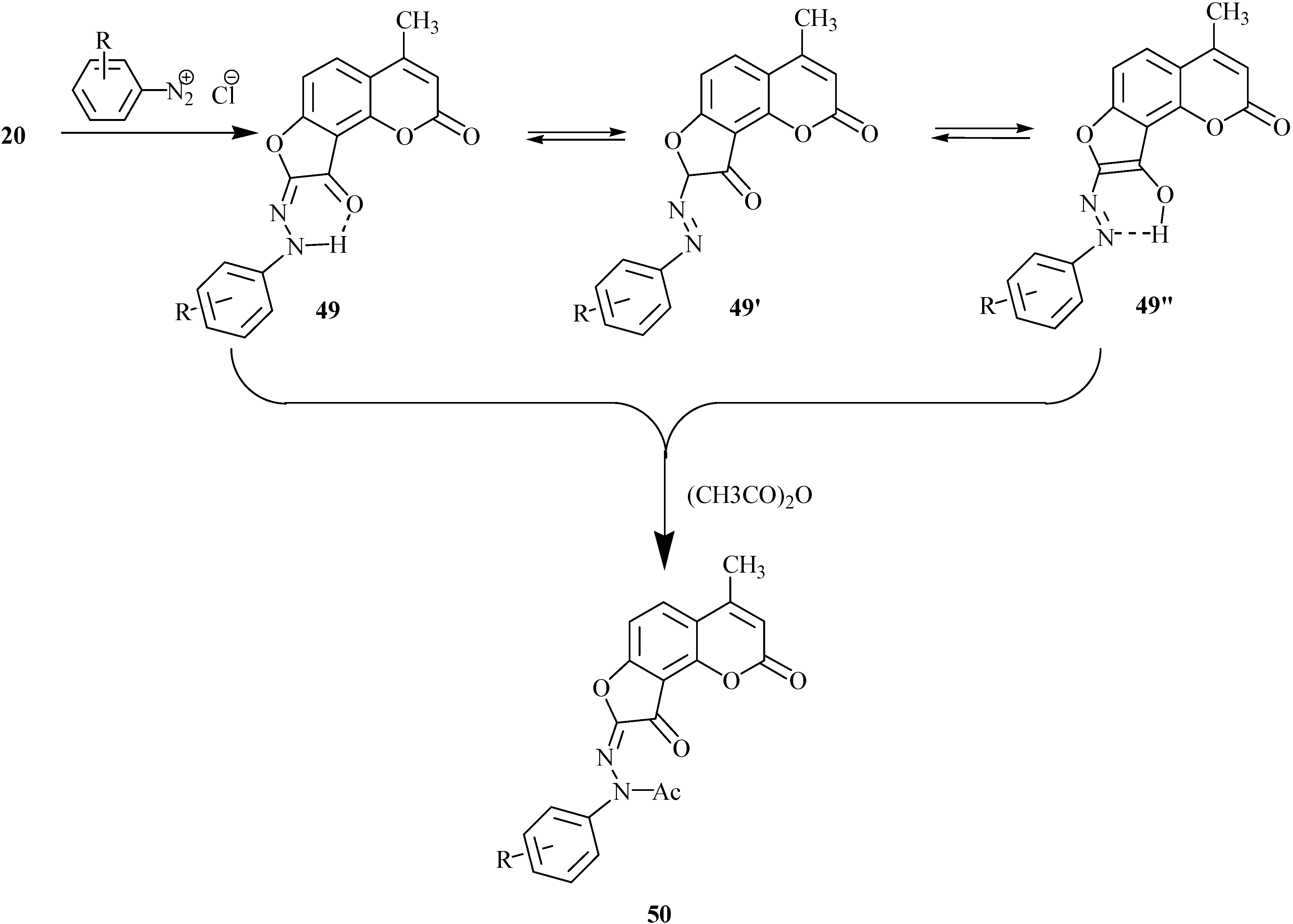
Base-catalyzed condensation of acetylhydroxyheteroarenes with α-halocarbonyl compounds
Base-catalyzed condensation of acetylhydroxyheteroarenes with α-halocarbonyl compounds is a preparative method for the construction of a furan ring in condensed heteroarenes [
23,
24]. This approach is shown in
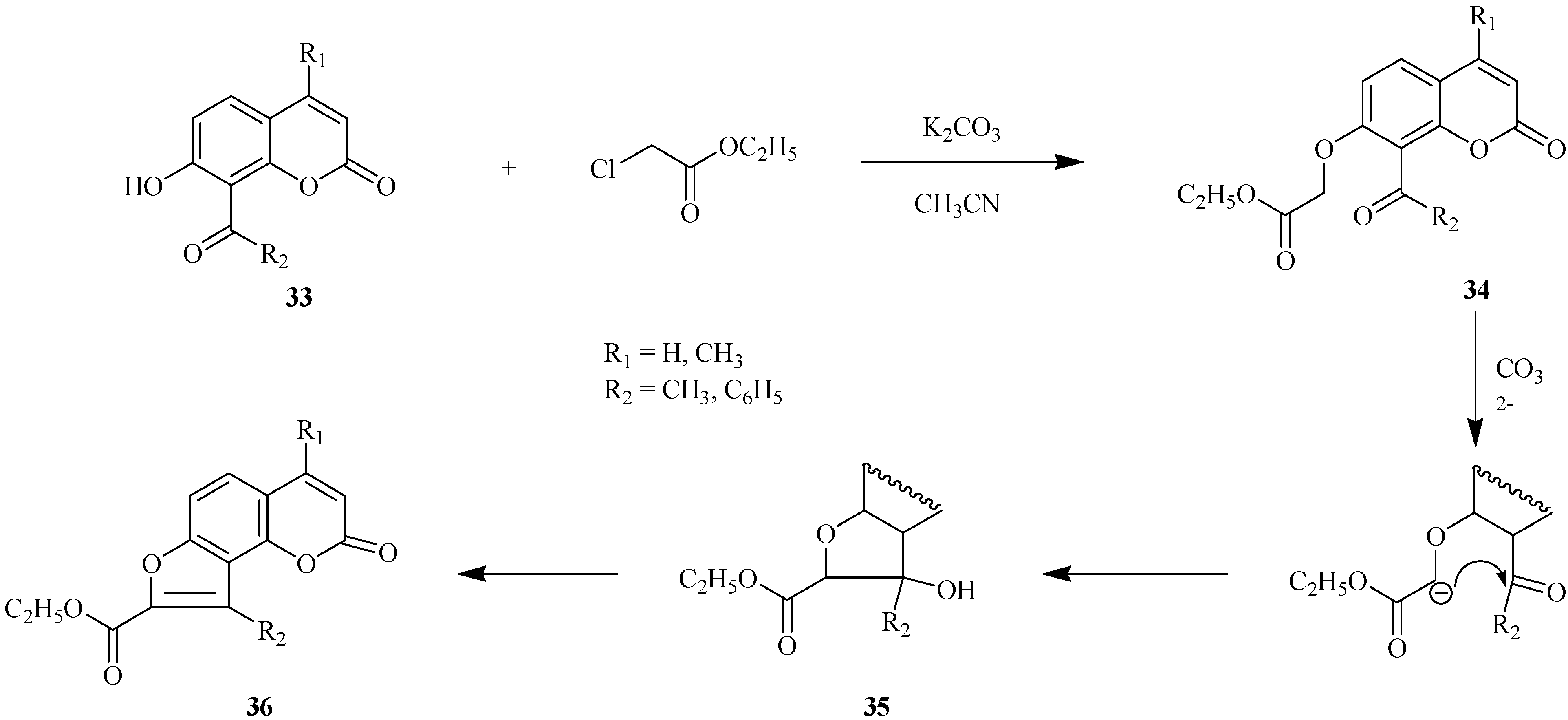
Scheme 13 for condensation of 8-acyl-7-hydroxycoumarins
33 with ethyl chloroacetate.
Ethyl coumarinyloxyacetate 34 is formed in the first step of the reaction. This step is followed by ionization of the oxymethylene group and intramolecular cyclization with successive formation of intermediate 35. Compound 35 undergoes dehydration to 8-(ethoxycarbonyl)furo[2,3-h]coumarin 36 which is an angelicine analog. These transformations provide a convenient way for preparation of furocoumarins with hydrophilic functional groups at the furan ring of the final furoheteroarene.
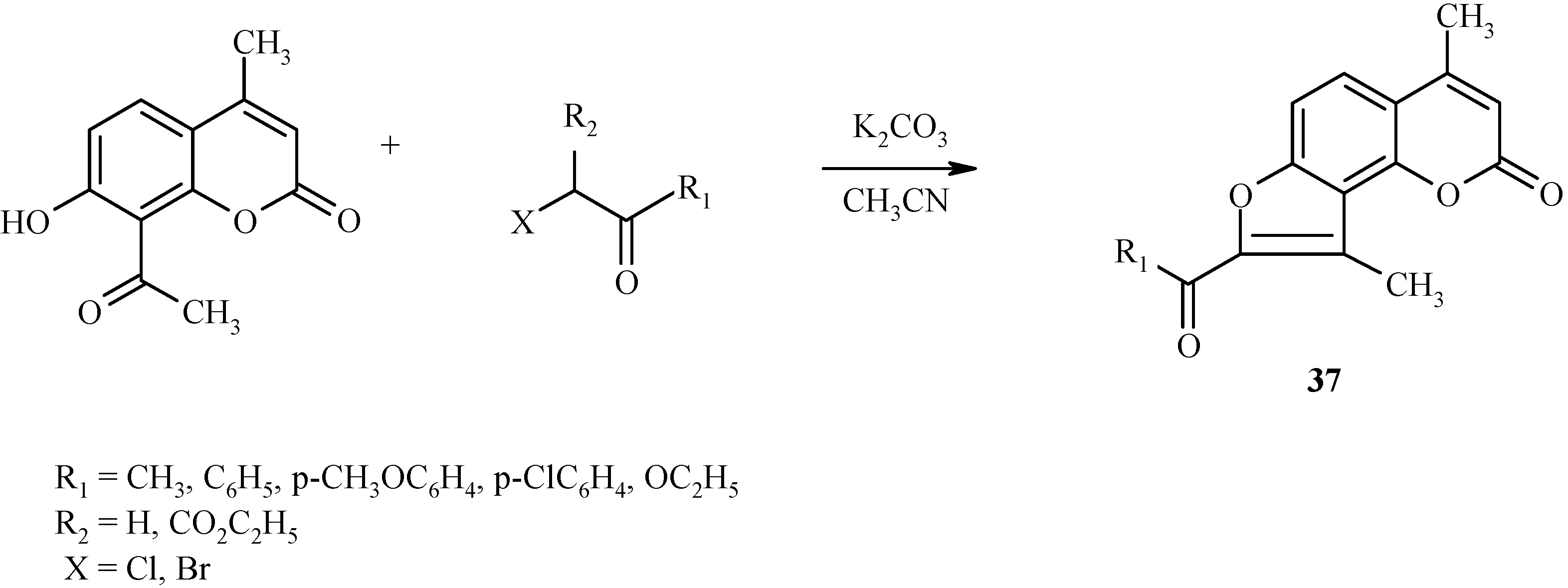
A similar condensation takes place with α-haloketones, as shown in
Scheme 14 for the preparation of angelicine derivative
37.
Base-catalyzed condensation of acylhydroxycoumarins with ethyl chloroacetate is also a useful reaction for preparation of psoralen derivatives, as exemplified in
Scheme 15 by the synthesis of a substituted psoralen
38.
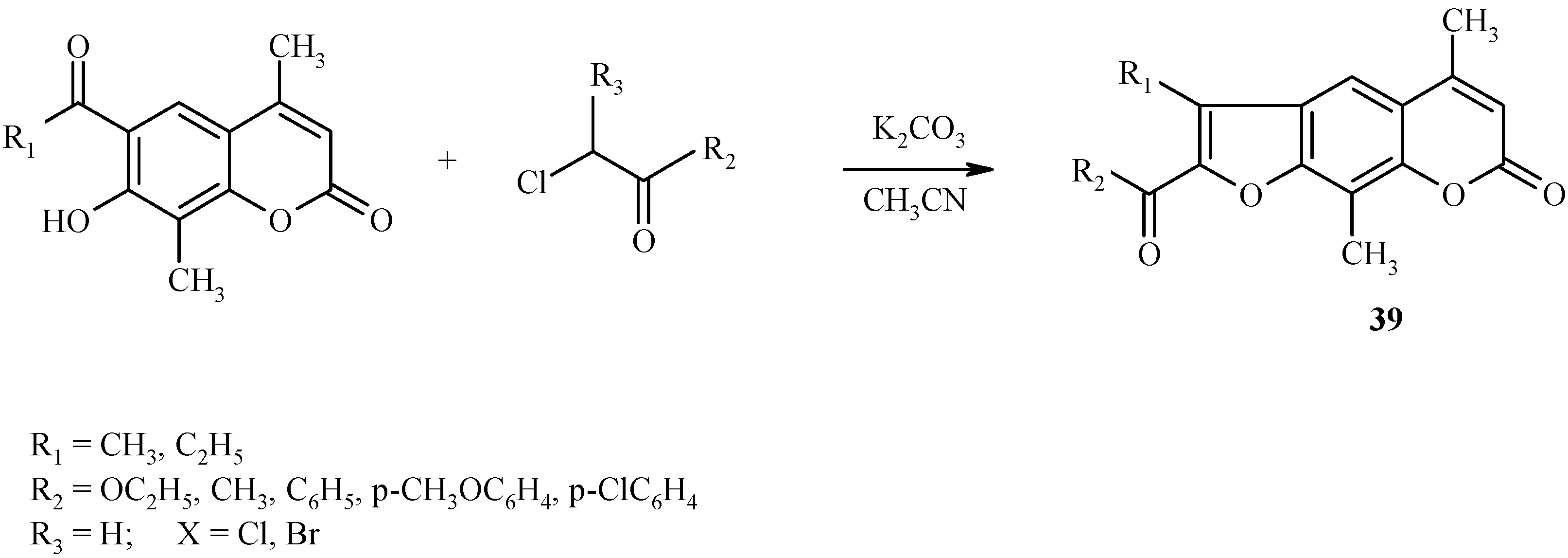
Similar condensations leading to other psoralen derivatives
39 are shown in
Scheme 16.
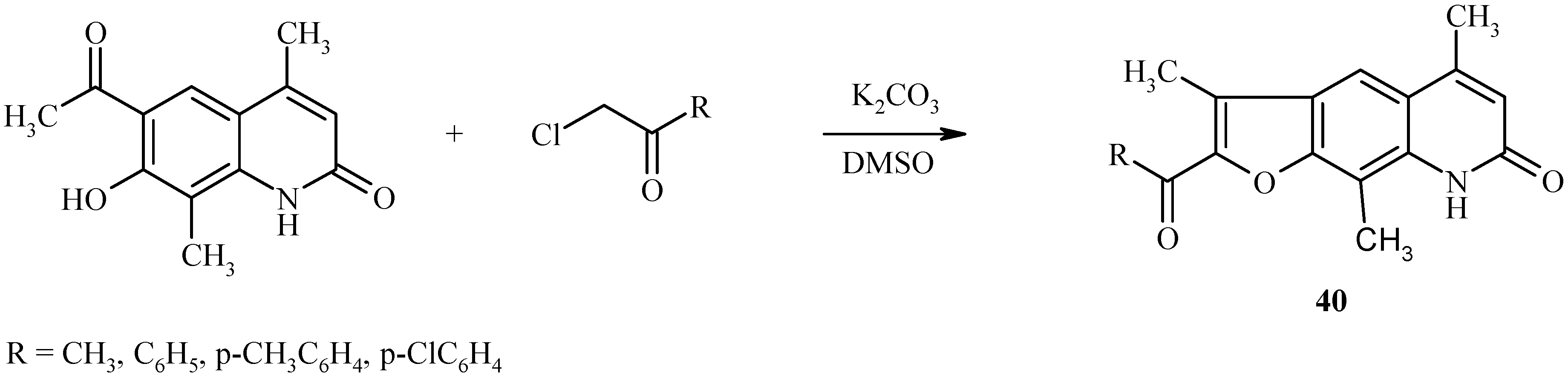
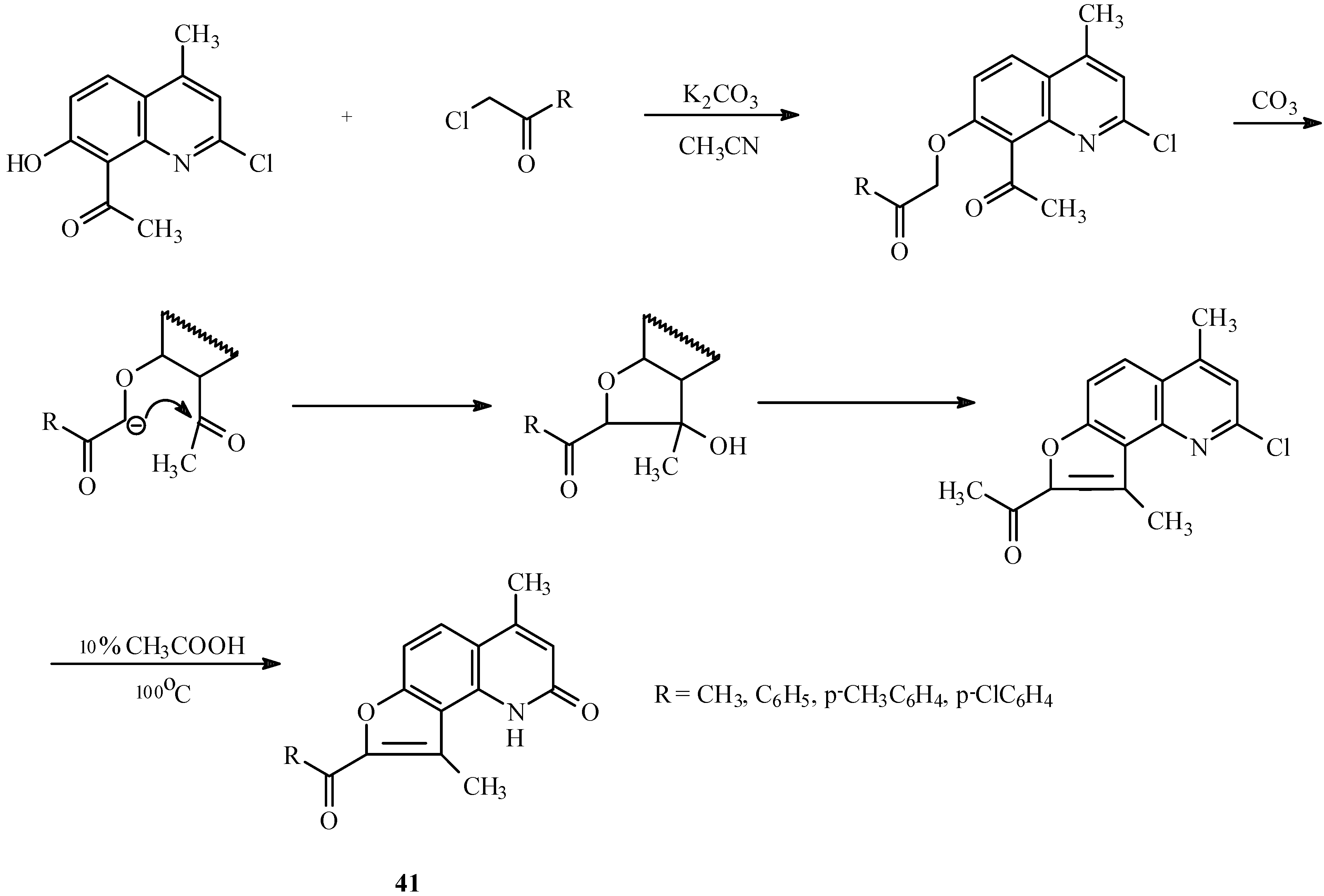
The base-catalyzed condensation of the acyl/hydroxyl functionality with α-halocarbonyl compounds can also be used for the synthesis of other fused furoheteroarenes as well (23, 24). For example, both linear (
40,
Scheme 17) and angular furoquinolines (
41,
Scheme 18) have been prepared by this method.
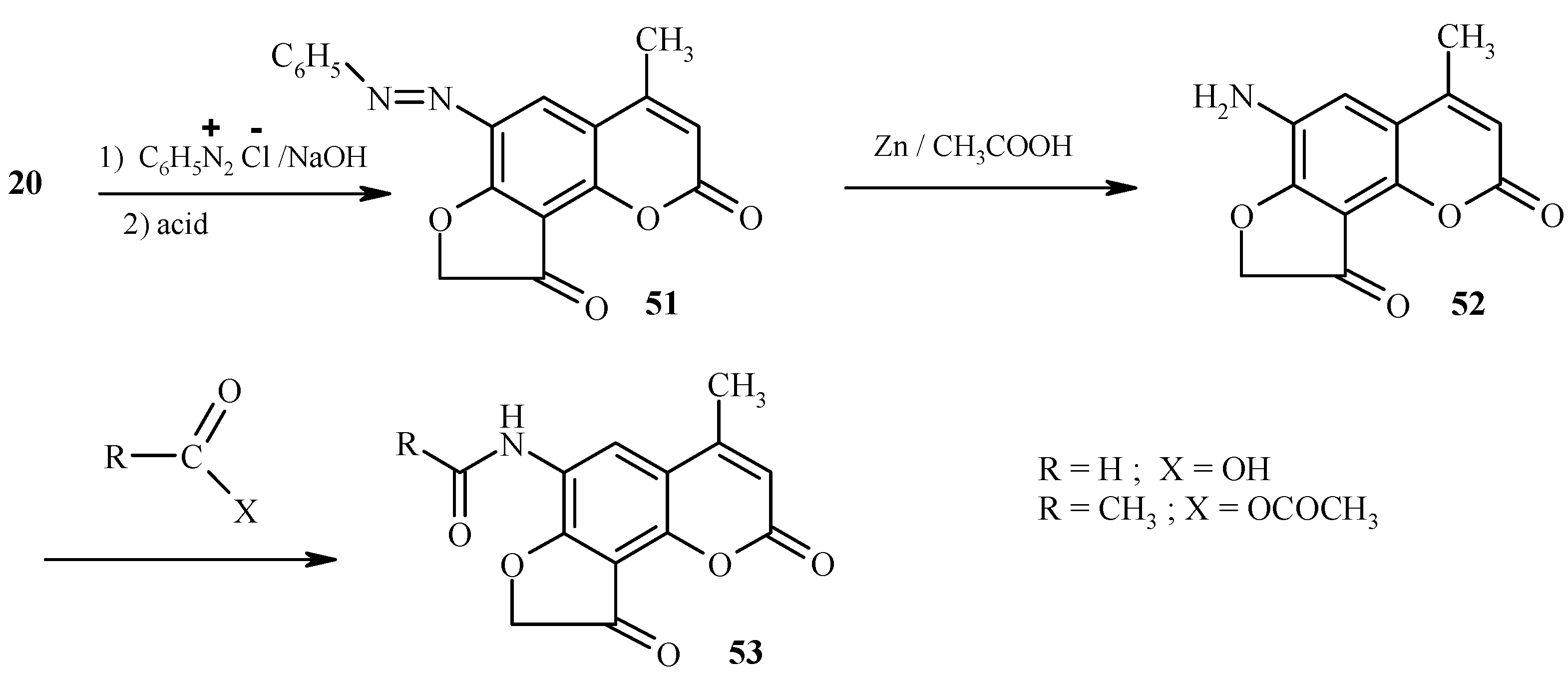
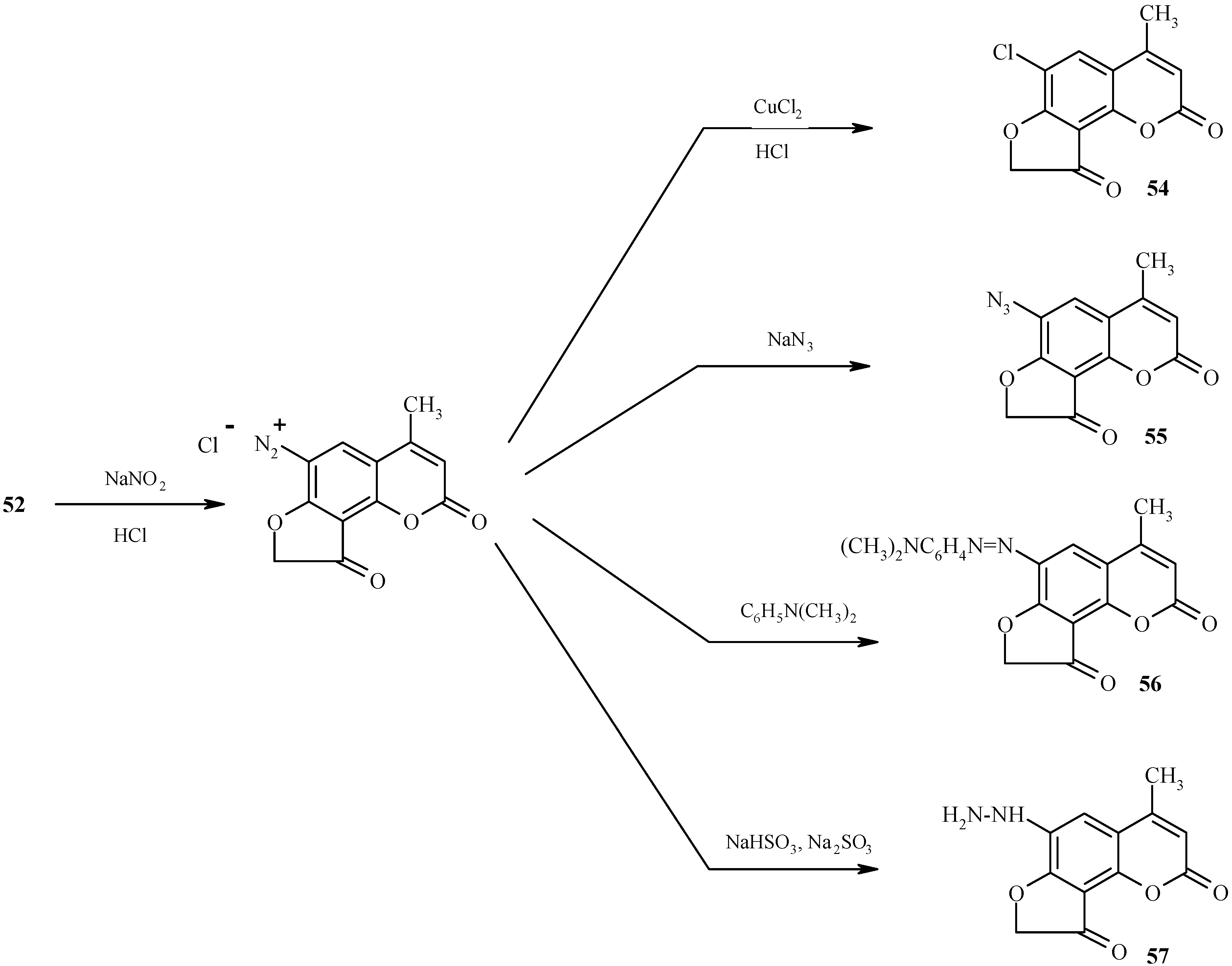
Pyrrolofurocoumarins
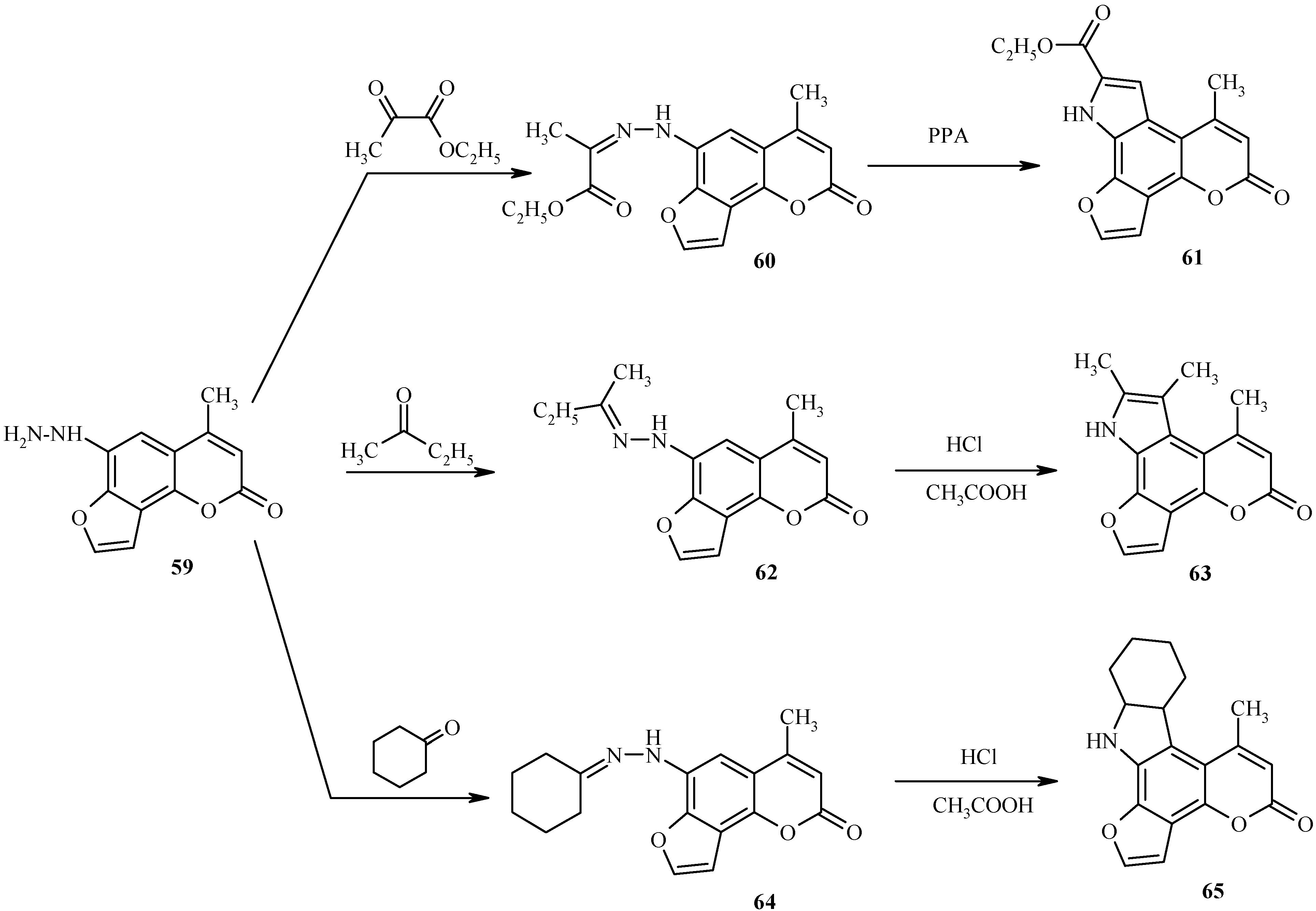
6-Hydrazino substituted furocoumarin
59 (
Scheme 29) is obtained in good yield upon diazotization of the amine
58 (R = NH
2) followed by reduction of the resultant diazonium salt with tin dichloride in concentrated hydrochloric acid [
34]. As shown in this scheme, the hydrazine
59 is a practical starting material for high-yield syntheses of pyrrole-fused furocoumarins
61, 63 and
65 through the intermediacy of the respective hydrazones
60, 62 and
64. Since these hydrazones are quite unstable compounds, their Fisher cyclization should be performed immediately after their preparation to ensure high yields of the cyclized products.
A success of the Fisher cyclization of hydrazones depends on the structure of hydrazone, acid catalyst, temperature and other factors. As a rule, strong mineral acids are required for the Fisher reaction. We have successfully used several different acid media such as sulfuric acid – acetic acid, hydrogen chloride – acetic acid, thionyl chloride – ethanol, and polyphosphoric acid.
Under optimized conditions, polyphosphoric acid is the best solvent for cyclization of 60 to 61. On the other hand, compounds 63 and 65 are obtained in yields greater than 85% when the Fisher cyclization of the respective hydrazones 62 and 64 are conducted in a mixture of hydrochloric and acetic acids.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
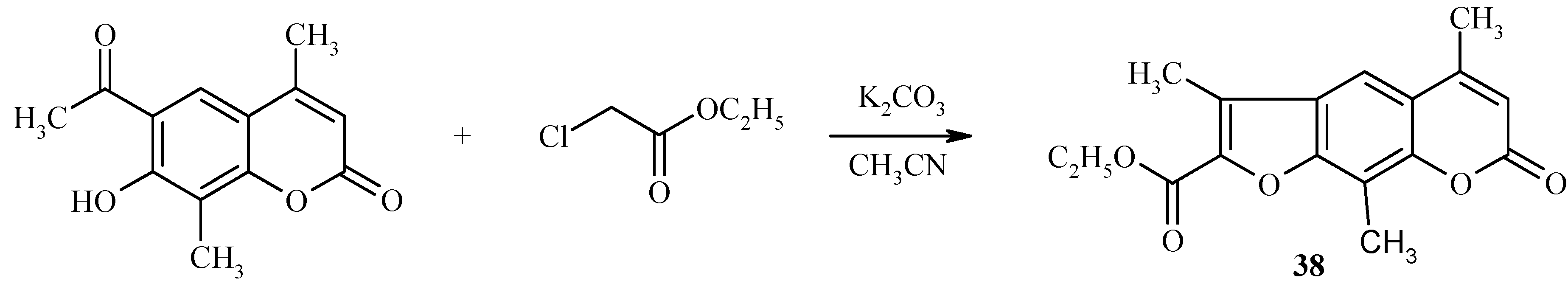{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
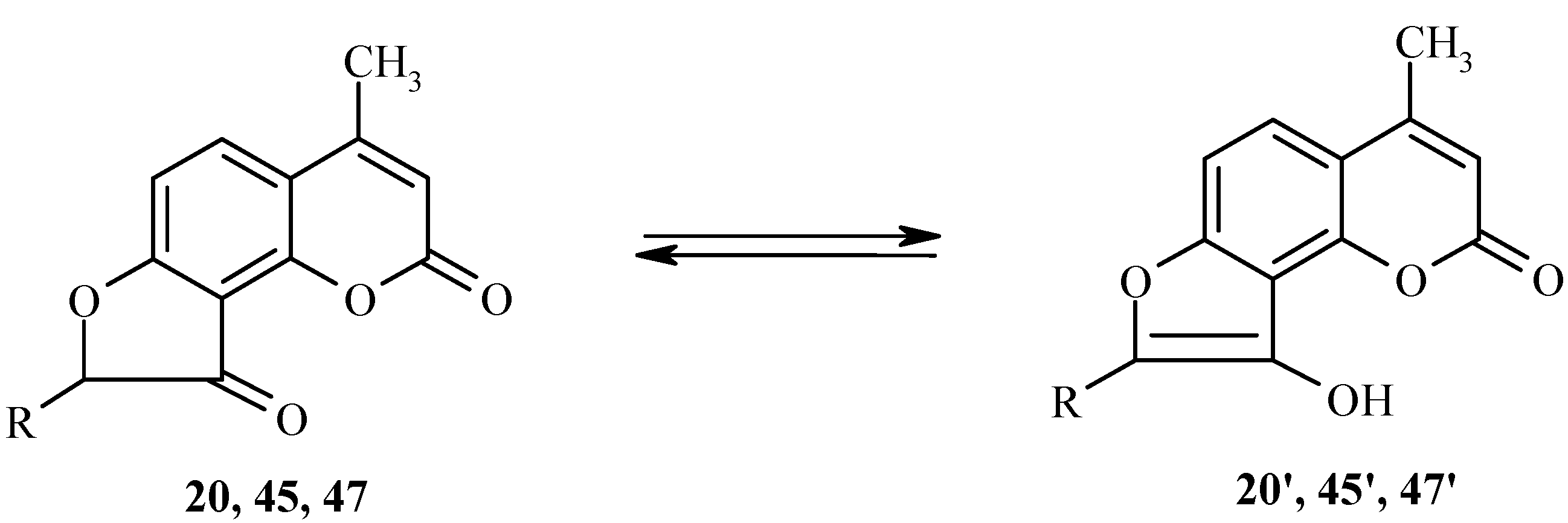{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




























