A Comparative Study of O2, CO and CN Binding to Heme IX Protein Models

Institut Universitari de Ciència Molecular, Universitat de València, Dr. Moliner 50, E 46100 Burjassot (València), Spain
Molecules 2004, 9(8), 632-649; https://doi.org/10.3390/90800632
Submission received: 26 April 2004
/
Accepted: 7 June 2004
/
Published: 31 July 2004
Abstract
:Parametrization of a molecular-mechanics program to include terms specific for five‑ and six-coordinate transition metal complexes results in computer-simulated structures of heme complexes. The principal new feature peculiar to five and six coordination is a term that measures the effect of electron-pair repulsion modified by the ligand electronegativity and takes into account the different structural possibilities. The model system takes into account the structural differences of the fixing centre in the haemoglobin subunits. The customary proximal histidine is added. The prosthetic group heme IX is wholly considered in our model. The calculations show clearly that certain conformations are much more favourable that others for fixing O2. From the O2 binding in haemoglobin, myoglobin and simple Fe porphyrin models it is concluded that the bent O2 ligand is best viewed as bound superoxide O2–. Axial ligands are practically free- rotating. A small modification of the model in both crystal and protein matrix affects the orientation of the ligands in experimental systems.
Introduction
The heme prosthetic group is in the active centre of a number of relevant proteins as the oxygen (O2) transport proteins haemoglobin (Hb) and myoglobin (Mb) [1,2], as well as enzymes involved in catabolism as peroxidases [3], catalases, oxidases [4] and cytochromes [5]. The replacement of Fe by Mg in heme produces chlorophyll [6], and the replacement of Fe by other transition metals coupled with modifications in the aromatic ring produces species as vitamin B12 [7] and cofactor F‑430 [8,9].
Rohmer’s group characterized the electronic state of Fe(P) (P = porphyrin) complexes [10,11,12], and predicted an electronic structure, which was later proven by experiment [13]. Other theoretical studies are the real position of the CO group in Fe(P)(Im)(CO) (Im = imidazole) [14,15], the position of the CN group in Fe(mdi)2(py)(CN) (mdi = malondialdiminate) complexes [16], the role of distal and proximal histidines (His) on the binding of O2 in Hb [17,18,19,20], and structural aspects of the binding of O2 and other ligands to heme [21,22,23,24,25,26,27]. The amount of information obtained from the calculations is seriously limited by the size of the heme group, which has only recently allowed the appearance of theoretical studies on reactivity [28,29,30,31,32,33]. The coordination of O2 to Fe(P)(Im) 5‑coordinate species leads to 6‑coordinate species with octahedral geometry: the biomimetic forms of Mb–O2 and Hb–O2. X-ray data have been reported only for two complexes: Fe(TpivPP)[1‑(Me)Im](O2) [34] and Fe(TpivPP)[2‑(Me)Im](O2) [35]. Both are quite similar, sharing the same porphyrin TpivPP = meso‑tetrakis(α,α,α,α‑o‑pivalamidophenyl)porphyrin. Maseras’ group optimized the geometry of Fe(TpivPP)[1‑(Me)Im](O2) with hybrid quantum mechanics/molecular mechanics (QM/MM) IMOMM(DFT-B3LYP:MM3) [36] and pure QM DFT-B3LYP [37]. Ghosh and Bocian optimized the geometry of Fe(P)(Im)(CO) with density functional theory (DFT) [19]. Salzmann et al. optimized under constraint the geometry of a Fe(TpivPP)[1-(Me)Im](CO) model with DFT-B3LYP [38]. Han et al. calculated a heme model–CO system employing the ab initio pseudopotential method with local density approximation (LDA) exchange correlation [39].
The reversible binding of O2 and carbon monoxide (CO) plays a central role in studies of heme-protein structure and function. The affinity of Hb for CO is 200 times that for O2, which accounts for the risk for CO poisoning. Numerous encumbered FeII porphyrin models were synthesized in an effort to elucidate the structural details of small ligand binding. The steric bulk of certain axial ligands bonded to synthetic FeII porphyrins provided model compounds of reduced O2 and CO affinity, and models of the tense (T) state of hemoproteins. There is only one single-crystal X‑ray structure determination on such a complex [40]. There was much discussion on the mechanistic basis of the variation of affinity values in heme proteins and model compounds. This focused on the nature of the axial ligand, distal steric effects, distal polar effects, and enforced doming and ruffling of the porphyrin skeleton. Johansson et al. showed by QM calculations on a heme a model that, upon reduction, the spin pairing at Fe is accompanied by effective delocalization of electrons from the Fe towards the periphery of the porphyrin ring, including its substituents [41,42]. In previous works, both non‑interacting (NID) and interacting (ID) induced-dipole polarization models were implemented in the program molecular mechanics (MM2) [43]. The polarizing force field (MMID2) [44] was applied to For‑Gly‑NH2 [45]. In the present report, terms specific for five‑ and six-coordination have been included in MMID2. The new program is called MMIDX. The next section presents the computational method. Following that the results are discussed. Finally, the conclusions are summarized.
Computational method
The molecular polarizability, αabmol, is defined as the linear response to an external electric field,
where μaind is the induced molecular dipole moment. Considering a set of N interacting atomic polarizabilities, the atomic μind has a contribution also from the other atoms,
where T(2)pq,bc is the interaction tensor as modified by Thole [46]
where vpq = rpq/spq if rpq < spq; otherwise vpq = 1, with s = 1.662(αpαq)1/6; δ represents the Kronecker delta function: δab = 1 if a = b, and δab = 0 if a ≠ b. Molecular polarizability can be written as
where B is the relay matrix defined as (in a supermatrix notation)
1,3‑Interactions between atoms bonded to a common atom are not specifically included in Equation (6) in the MM2+polarization approach, because they are effectively already included in the bond-length and bond-angle strainless parameters.
For 5‑coordinate structures, there is a need to consider the effect of 1,3‑interactions because an energy term is needed for ligand–ligand repulsion to account for (1) the stability difference of various possible geometries and (2) structural effects due to the variation of ligand electronegativity. The geometries of the methylfluorophosphoranes (CH3)nPF5‑n (n = 0→3) were qualitatively correlated with Gillespie’s VSEPR theory, which was adopted as a model for the present approach. Bond electron-pair repulsion (EPR) terms were introduced via the non‑bonded term Enb of Equation (6) modified to express EPR for atoms bonded to 5‑ or 6‑coordinated atoms. The unmodified term is
where P = [rVDW(A)+rVDW(B)]/rAB, rVDW is the van der Waals radius of the specific atom and rAB is the non‑bonded distance between A and B; ε = (εAεB)1/2, where εA and εB are parameters specific to atoms A and B, and are related to the hardness of the atoms. Hill evaluated the constants in Equation (7), which give the energy Enb(AB) in units of kilocalories per mole [47]. The modification of Equation (7) to express EPR terms is [48]
The addition of a scaling factor D, to obtain a suitable balance between this term and the other terms in Equation (6), and the replacement of P with P* (where P* = [rVDW(A)+rVDW(B)]/r*AB and r*AB is the distance between atoms A and B calculated from modified bond lengths, d*CA and d*CB, between the central atom C and either atom A or B) provide the necessary adjustments to quantitatively reproduce the (CH3)nPF5‑n structures [49]. The variation in ligand electronegativity is introduced by a distance factor RA in the relation d*CA = dCARA. The magnitude of R is inversely related to the electronegativity difference between atoms C and A. The R factors are the means of including EPR between atoms A and B. If the electronegativity difference ΔXCA is large, the bonding electron pair can be considered to move away from atom C, thus decreasing EPR between C–A and C–B bonds. When ΔXCA > ΔXCA’, EPR term E(1,3)AB < E(1,3)A’B even when the actual bond lengths are equal. A set of distance factors R may be obtained from the bond ionic character I,
and using the relation
where rA and rC are covalent radii of atoms A and C.
Calculation Results and Discussion
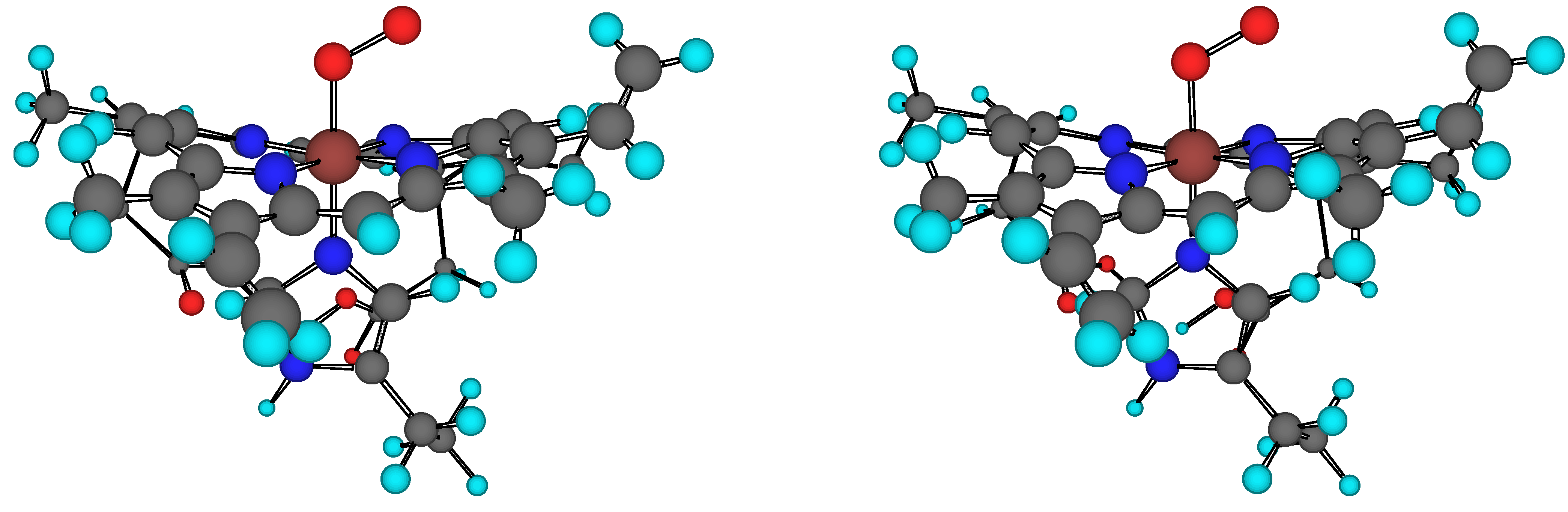
The structure of the heme(–His)–O2 model is shown in Figure 1. Heme is the prosthetic group of oxy–Hb. The molecular mechanics calculations use the MM2/MMX+polarization force fields. Van der Waals parameters for the Fe atom have been taken from the force field UFF [50]. Torsional contributions involving dihedral angles with the metal atom and the bending terms involving Fe in central position have been set to zero.
Molecular mechanics dipole moments μ are given in Table 1. In general, μ increases with the oxidation state of Fe. In particular, μheme(FeIII)(–His)–CN results the greatest due to the highly polar Feδ+–C–Nδ‑ complex. The μheme(FeIII)(–His)–O2 is relatively large due to the extremely polar Feδ+–O–Oδ‑. The inclusion of polarization in MM2 corrects, in general, μ in the correct direction when compared with MMX+ID. In particular, μheme(–His) remains almost constant (MM2+polarization). The binding of His in heme increases μ by a factor of 5. Moreover, the subsequent binding of CN doubles μ and the binding of O2 or CO increases μ 7–16%.
Figure 1.
Stereo view of the heme(–His)–O2 model. Heme is the prosthetic group of oxyhaemoglobin.


{kind=link}
| Adduct | Fe oxid. state | MM2 | MM2+NIDa | MM2+IDb | MMX | MMX+NIDa | MMX+IDb |
|---|---|---|---|---|---|---|---|
| heme | 2 | 0.467 | 0.483 | 0.539 | 0.467 | 0.483 | 0.539 |
| heme(–His) | 2 | 2.840 | 2.725 | 2.819 | 2.136 | 2.969 | 2.304 |
| heme(–His)–O2 | 3 | 2.512 | 2.372 | 3.091 | 3.348 | 2.978 | 2.342 |
| heme(–His)–CO | 2 | 2.723 | 2.725 | 2.905 | 3.185 | 3.054 | 3.388 |
| heme(–His)–CN | 3 | 5.090 | 5.538 | 5.470 | 6.111 | 6.824 | 6.553 |
a NID: polarization by non‑interacting induced dipoles.b ID: polarization by interacting induced dipoles.
The 4-coordinate Fe(P) system
The heme group is a natural starting point for both experimental and theoretical studies. Crystal structures were reported for a number of heme derivatives with different substituents in the ring. In particular, Fe(TPP) (TPP = meso‑tetraphenylporphyrin) has been chosen. Its electronic state is well known experimentally to correspond to a low spin triplet (S = 1). Selected structural parameters are listed in Table 2. The agreement in bond angles between both MM2/MMX+polarization geometries and the X‑ray structure is good, with discrepancies smaller than 5°. Differences in bond distances are larger in a number of cases as for the C–Cbridge, C–C’ and C’–C’’ distances. These have values of 1.395Å, 1.439Å and 1.365Å, respectively, in X‑ray, and ≈1.336Å, 1.340Å and 1.332Å, correspondingly, in MM2/MMX+polarization.
Table 2.
Selected geometric parameters (Å and degrees) from the geometry optimization of Fe(P) with the pure B3LYP and with the IMOMM(B3LYP:MM3) methods [37].a
| Parameter | MM2 | MM2+NIDb | MM2+IDc | MMXd | MMXd+NIDb | MMXd+IDc |
|---|---|---|---|---|---|---|
| Fe–Ne | 1.881 | 1.878 | 1.877 | 1.881 | 1.878 | 1.877 |
| N–C | 1.353 | 1.352 | 1.350 | 1.353 | 1.352 | 1.350 |
| C–Cbridge | 1.337 | 1.336 | 1.334 | 1.337 | 1.336 | 1.334 |
| C–C’ | 1.341 | 1.340 | 1.339 | 1.341 | 1.340 | 1.339 |
| C’–C’’ | 1.332 | 1.332 | 1.332 | 1.332 | 1.332 | 1.332 |
| Fe–N–C | 128.6 | 128.6 | 128.6 | 128.6 | 128.6 | 128.6 |
| N–Fe–N | 90.2 | 90.2 | 90.2 | 90.2 | 90.2 | 90.2 |
| N–C–Cbridge | 120.8 | 120.8 | 120.9 | 120.8 | 120.8 | 120.9 |
| N–C–C’ | 112.0 | 112.1 | 112.1 | 112.0 | 112.1 | 112.1 |
| Fe-planef | 0.104 | 0.102 | 0.105 | 0.104 | 0.102 | 0.105 |
| Parameter | Experiment | Pure QM | QM/MM | |||
| Fe–N | 1.966 | 2.016 | 1.940 | |||
| N–C | 1.378e | 1.397 | 1.362 | |||
| C–Cbridge | 1.395e | 1.402 | 1.369 | |||
| C–C’ | 1.439e | 1.459 | 1.345 | |||
| C’–-C’’ | 1.365e | 1.367 | 1.333 | |||
| Fe–N–C | 127.2e | 127.4 | 127.7 | |||
| N–Fe–N | 90.0e | 90.0 | 90.0 | |||
| N–C–Cbridge | 125.3e | 125.5 | 126.2 | |||
| N–C–C’ | 110.6e | 110.4 | 110.3 | |||
| Fe-plane | 0.000 | 0.000 | 0.000 | |||
a Experimental data on the Fe(TPP) system are also provided for comparison [13].b NID: polarization by non‑interacting induced dipoles.c ID: polarization by interacting induced dipoles.d Scaling factor D = 0.05.e Average values.f Distance of the Fe atom to the mean plane of the porphyrin N atoms.
Agreement between experiment and MM2/MMX+polarization is good (≈0.06Å). All these atoms are, in part, purely described with MM2. The optimized MM2/MMX+polarization values are close to the optimal bond distance for these types of atoms in the applied force field, which is 1.337Å. Another discrepancy in the geometries appears in the Fe–N distance. This is more puzzling, because the calculated distance 1.877–1.881Å is smaller than the experimental value of 1.966Å. The calculated displacement of the Fe atom to the mean plane of the porphyrin N‑atoms ring (0.1Å) is not observed experimentally.
The 5-coordinate Fe(P)(Im) system
Table 3.
Selected geometric parameters (Å and degrees) from the geometry optimization of Fe(P)(NH=CH2) with the pure B3LYP and of Fe(P)[1‑(Me)Im] with the IMOMM(B3LYP:MM3) methods [37].a
| Parameter | MM2 | MM2+NIDb | MM2+IDc | MMXd | MMXd+NIDb | MMXd+IDc |
|---|---|---|---|---|---|---|
| Fe–Nporphe | 1.897 | 1.894 | 1.890 | 1.895 | 1.898 | 1.895 |
| Fe–NIm | 1.866 | 1.863 | 1.864 | 1.873 | 1.885 | 1.865 |
| NIm–Cε | 1.327 | 1.327 | 1.326 | 1.398 | 1.388 | 1.389 |
| NIm–Cδ | 1.344 | 1.343 | 1.338 | 1.392 | 1.393 | 1.387 |
| Fe–NIm–Cε | 126.3 | 126.3 | 126.1 | 125.1 | 117.7 | 106.7 |
| Fe–NIm–Cδ | 126.7 | 126.6 | 126.3 | 121.7 | 121.0 | 131.9 |
| Nporph–Fe–NIm–Cε | 115.8 | 115.7 | 114.9 | 135.2 | 100.2 | 99.9 |
| Fe-planef | 0.097 | 0.088 | 0.095 | 0.130 | 0.274 | 0.191 |
| Parameter | Experiment | Pure QM | QM/MM | |||
| Fe–Nporph | 2.075 | 2.101 | 2.029 | |||
| Fe–NIm | 2.134 | 2.252 | 2.233 | |||
| NIm–Cε | 1.350 | 1.279 | 1.299 | |||
| NIm–Cδ | 1.250 | –g | 1.414h | |||
| Fe–NIm–Cε | 126.5 | 126.1 | 136.8 | |||
| Fe–NIm–Cδ | 120.4 | 120.6 | 122.6 | |||
| Nporph–Fe–NIm–Cε | 80.8 | 90.0 | 136.8 | |||
| Fe-plane | 0.310 | 0.284 | 0.379 | |||
a Experimental data on the Fe(Piv2C8)[1‑(Me)Im] system [59] are provided for comparison. b NID: polarization by non‑interacting induced dipoles.c ID: polarization by interacting induced dipoles.d Scaling factor D = 0.05.e Average values.f Distance of the Fe atom to the mean plane of the porphyrin N atoms.g Frozen in calculation.h Corresponds to N‑H in this calculation.
Coordination of an Im ligand to the heme group leads to a 5‑coordinate species with a square pyramidal geometry. These compounds are good biomimetic models of Mb and Hb, with Im replacing the proximal His of the biological systems. The need to avoid dimerization and formation of 6‑coordinate species with two axial ligands poses serious restrictions on the nature of the porphyrins able to give this kind of complexes. For this study, the species Fe(Piv2C8)[1‑(Me)Im] {Piv2C8 = α,α,5,15‑[2,2’‑(octanediamido)diphenyl]‑α,α,10,20‑bis(o‑pivalamidophenyl)porphyrin} has been chosen. This species has the advantage of having 1‑methylimidazole as axial ligand, in constrast with the more common 2‑methylimidazole, which is more sterically demanding. Neither for Fe(Piv2C8)[1‑(Me)Im] nor for other 5‑coordinate derivatives of heme is the electronic state experimentally known. Electronic spectroscopy, magnetic susceptibility and Mössbauer measurements identify it as high spin (S = 2). Selected parameters are collected in Table 3. The Fe–Nporph distances are longer than those in the 4‑coordinate system by approximately 0.02Å (MMX+ID). This trend is in agreement with the reference values. The result is fully consistent with the shift from low spin to high spin in the metal. Overall agreement in the geometric parameters is correct. Moreover, one has to view with suspicion the X‑ray parameters of Im, which would make the N=C double bond NIm–Cε in Im longer than the N–C single bond NIm–Cδ. Furthermore, the MM calculations are, in general, in agreement with the QM/MM reference, which provides the expected result.
The sharper discrepancy concerns the Nporph–Fe–NIm–Cε dihedral angle. This angle measures the rotation around the Fe–NIm single bond, and rules the placement of the Im plane with respect to the porphyrin ring. Its sign is arbitrary, because the x and y directions are equivalent in absence of an axial ligand. In this work, a positive sign has been chosen for consistency with data on the 6‑coordinate complexes presented below. An angle of 90° (as in the pure QM reference) means that the Im plane is eclipsing one of the Fe–Nporph bonds, while an angle of 135° (≈135.2° in MMX) indicates a staggered orientation of Im with respect to the Fe–Nporph bonds. Therefore both pure QM and MMX values are just opposite with the experimental value, 126.0°, lying in between, although closer to MMX results. The structural minima lead to structures where the Im ligand is located about the bisector of an angle Nporph–Fe–Nporph. The MMX+polarization results lie, in general, in the range 90–133° of the references. The importance of the large discrepancy between different values is, however, arguable, because there is also a large dispersion in different experimental 5‑coordinate derivatives of heme, as well as in experimental reports of Mb and Hb. The corresponding interpretation is that the rotation around this single bond has a rather low barrier.
A final number to mention is the displacement of the Fe atom out of the porphyrin N‑atoms plane. This is one of the most important parameters, because it seems to play a critical role in the cooperativity of Hb involved in the allosteric transition. In this case, MMX+NID calculation of 0.274Å lies near the experimental and reference values ca. 0.3Å. The positive sign corresponds to the fact that the displacement is towards Im, and away from the empty site.
The 6-coordinate Fe(P)(Im)(ligand) systems
Coordination of O2 to the 5‑coordinate heme–His species leads to 6‑coordinate species with octahedral geometry. These are the biomimetic forms of Mb–O2 and Hb–O2. X‑ray data are reported only on two complexes: Fe(TpivPP)[1‑(Me)Im](O2) and Fe(TpivPP)[2‑(Me)Im](O2). Both are quite similar, sharing the same porphyrin TpivPP, which is meso‑tetrakis(α,α,α,α‑o‑pivalamidophenyl)-porphyrin. Fe(TpivPP)[1‑(Me)Im](O2), containing the less sterically demanding 1‑methylimidazole ligand, has been chosen for comparison.
| Parameter | MM2 | MM2+NIDb | MM2+IDc | MMXd | MMXd+NIDb | MMXd+IDc | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Fe–Nporphe | 1.880 | 1.876 | 1.870 | 1.899 | 1.891 | 1.899 | ||||
| Fe–NIm | 1.871 | 1.862 | 1.861 | 1.871 | 1.908 | 1.875 | ||||
| Fe–O | 1.847 | 1.841 | 1.845 | 1.866 | 1.903 | 1.866 | ||||
| O–O | 1.211 | 1.210 | 1.210 | 1.256 | 1.261 | 1.244 | ||||
| Fe–O–O | 123.5 | 121.5 | 122.8 | 121.3 | 121.7 | 121.3 | ||||
| O–Fe–NIm | 171.7 | 169.5 | 168.6 | 146.2 | 131.6 | 157.3 | ||||
| Nporph–Fe–NIm–Cε | 179.0 | 178.2 | 176.8 | 177.2 | 160.7 | 173.5 | ||||
| Nporph–Fe–O–O | -3.3 | -5.0 | -16.5 | -25.9 | -11.0 | -24.1 | ||||
| Fe-planef | -0.181 | -0.150 | -0.159 | -0.074 | -0.114 | -0.044 | ||||
| Parameter | Experiment | Pure QM | QM/MM | LSD | ||||||
| Fe–Nporph | 1.979 | 2.035 | 1.949 | 2.010 | ||||||
| Fe–NIm | 2.068 | 2.050 | 2.167 | 2.070g | ||||||
| Fe–O | 1.745 | 1.757 | 1.759 | 1.770 | ||||||
| O–O | 1.163 | 1.268 | 1.286 | 1.300 | ||||||
| Fe–O–O | 129.4 | 121.1 | 117.0 | 121.0 | ||||||
| O–Fe–NIm | 180.0 | 175.8 | 179.4 | – | ||||||
| Nporph–Fe–NIm–Cε | 159.5 | 177.9 | 137.0 | – | ||||||
| Nporph–Fe–O–O | -42.4 | -44.6 | -44.1 | – | ||||||
| Fe-plane | -0.030 | -0.034 | -0.076 | – | ||||||
a Experimental data on the Fe(TpivPP)[1‑(Me)Im](O2) system are provided for comparison [34].b NID: polarization by non‑interacting induced dipoles.c ID: polarization by interacting induced dipoles.d Scaling factor D = 0.05.e Average values.f Distance of the Fe atom to the mean plane of the porphyrin N atoms.g Frozen in calculation.
The state of this system is a low spin open-shell singlet (S = 1) resulting in a FeIII––O2– charge distribution. Selected parameters are reported in Table 4. The parameters concerning the coordination of O2, which are probably the most critical for the biochemical activity of Hb, are well reproduced. The computed values for the Fe–O distance, 1.8–1.9Å, are close to the experimental value of 1.746Å. The calculated values for the O–O distance, 1.21–1.26Å, on the other hand, are far from the experimental value of 1.163Å, but the experimental value, even shorter than the 1.21Å for free O2, is suspect, because of the disorder on the placement of the ending O atom within the crystal, as admitted by the authors of the X‑ray experiment [34]. The O–O interatomic distance increases from 1.21Å in free O2 to 1.261Å (MMX+NID), suggesting that electronic charge is transferred from FeP to O2, in agreement with the experimental observation that the Fe–O2 bond can be formally described as FeIII–O2– [9].
The most significant feature of the present structure is the bent Fe-O-O bond. The Fe-O-O bond angles are indicative of a bent η1 coordination mode, where only one O atom is directly attached to the metal. These calculations are in agreement with the experiment and calculation references. Atomic net charges have been calculated with our program POLAR [51]. The results, qFe = 3.078, qO binding = –0.412 and qO end = –0.531e are in agreement with Weiss’ model for the Fe–O2 bond in which the bond might be ionic between a Fe3+ and a superoxide ion, net charge being transferred from Fe to O2 (Fe3+O2–) [52]. Experimental support for Weiss’ model was advanced by Misra and Fridovich [53]. The geometry agrees with Pauling’s prediction of a bent FeO2 bond, and the O–O distance is close to that of 1.27Å, predicted by him [54]. It is slightly shorter than that of 1.34Å in O2– close to the electron spin resonance results, which show that no more than 2/3 of the density of one electron is transferred from the metal to the antibonding π* orbitals of O2.
The greatest discrepancy concerns the Nporph–Fe–O–O dihedral angle. This angle measures the rotation around the Fe–O single bond, and rules the placement of the Fe–O–O plane with respect to the porphyrin ring. An angle of 0° (approximately the –3.3° in MM2) means that the Fe–O–O plane is eclipsing one of the Fe–Nporph bonds, while an angle of –45° (approximately the –44.6° in pure QM) indicates a staggered orientation of the O2 with respect to the Fe–Nporph bonds. The structural minima lead to structures where the O2 ligand is located about the bisector of an angle Nporph–Fe–Nporph. The MMX/MMX+NID results lie near the reference results. The structural minimum correspond to a trans isomer, where the ending O is placed above the opposite quadrant where the Nδ is located. Finally, the Fe atom is computed by MMX+ID to lie in the energy minimum rather close to the porphyrin N‑atoms plane, with a deviation of 0.044Å, always towards the O atom. This is again in agreement with the experiment and reference calculations, and rather different from the behaviour of the 5‑coordinate system.
Structural parameters of Fe(P)(Im)(CO) are summarized in Table 5. The geometric parameters concerning the coordination of CO, which are probably the most critical for the biochemical activity of Hb, are well reproduced. The computed values for the Fe–C distance, 1.97–2.01Å, are close to the experimental value of 1.770Å. The calculated values for the C–O distance, 1.11–1.14Å, are also close to the experimental report of 1.120Å. The C–O distance is similar in the free molecule, 1.171Å, calculated with AM1 [55] and in Fe(P)(Im)(CO), 1.143Å (MMX). The interpretation is that electronic charge is not transferred from FeP to CO. This is in agreement with the experimental observation that the Fe–CO bond can be formally described as FeII–CO [9]. The most significant feature of the present structure is the linear Fe–C–O bond. While such a linear bond is to be expected based on the extensive literature on transition metal CO complexes, the result is nonetheless highly significant since bent Fe–C–O bonds with bond angles of 135–145º appear to be the rule in various CO complexes of hemoproteins [56,57,58].
Table 5.
Selected geometric parameters (Å and degrees) from the complete geometry optimization of Fe(P)(Im)(CO) with LDA [39], partial geometry optimization of Fe(TpivPP)[1‑(Me)Im](CO) with DFT (B3LYP and BPW91) [38], and complete geometry optimization of Fe(mdi)2(py)(CO) with NLDFT [16], of Fe(P)(Im)(CO) with LDFT [19] and of Fe(TpivPP)(Im)(CO) with LSD [21].a
| Parameter | MM2 | MM2+NIDb | MM2+IDc | MMXd | MMXd+NIDb | MMXd+IDc | |
|---|---|---|---|---|---|---|---|
| Fe–Nporphe | 1.878 | 1.873 | 1.871 | 1.900 | 1.893 | 1.896 | |
| Fe–NIm | 1.868 | 1.863 | 1.862 | 1.876 | 1.877 | 1.886 | |
| Fe–C | 1.971 | 1.970 | 1.970 | 1.998 | 1.989 | 2.008 | |
| C–O | 1.110 | 1.109 | 1.110 | 1.143 | 1.137 | 1.138 | |
| Fe–C–O | 180.0 | 180.0 | 180.0 | 179.6 | 179.8 | 177.4 | |
| C–Fe–NIm | 179.7 | 174.6 | 174.3 | 158.3 | 172.7 | 149.3 | |
| Nporph–Fe–NIm–Cε | 136.0 | 178.9 | 176.0 | 135.7 | 161.4 | 169.0 | |
| Fe-planef | -0.182 | -0.180 | -0.159 | -0.037 | -0.052 | -0.064 | |
| Parameter | Experiment | LDA | B3LYP | BPW91 | NLDFT | LDFT | LSD |
| Fe–Nporph | 2.015 | 1.990 | – | – | 1.961 | 1.983 | 2.020 |
| Fe–NIm | 2.100 | 1.960 | – | – | 2.139 | 1.966 | 2.070g |
| Fe–C | 1.770 | 1.790 | 1.801 | 1.743 | 1.739 | 1.733 | 1.720 |
| C–O | 1.120 | 1.160 | 1.147 | 1.167 | 1.166 | 1.165 | 1.170 |
| Fe–C–O | 179.0 | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 |
| C–Fe–NIm | 177.5 | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 | – |
| Nporph–Fe–NIm–Cε | – | 174.2 | – | – | – | – | – |
| Fe-plane | -0.020 | – | – | – | – | – | – |
a Experimental data on Fe(TpivPP)[1‑(Me)Im](CO) system are provided for comparison [38].b NID: polarization by non‑interacting induced dipoles.c ID: polarization by interacting induced dipoles.d Scaling factor D = 0.05.e Average values.f Distance of the Fe atom to the mean plane of the porphyrin N atoms.g Frozen in calculation.
The global energy minimum is linear for Fe–C–O (MM2/MMX+polarization). These calculations are in agreement with the experiment and calculation references. The Im ring is rotated so that the N atom is directed toward the Fe atom, and the rotation angle Nporph–Fe–NIm–Cε reaches approximately 176° (MM2+polarization) in agreement with the LDA reference (174.2°). The Fe atom is computed by MMX to lie in the energy minimum rather close to the porphyrin N‑atoms plane, with a deviation of 0.037Å, always towards the C atom. This is again in agreement with the experiment and reference calculations.
Table 6.
Selected geometric parameters (Å and degrees) from the geometry optimization of Fe(mdi)2(py)(CN) with NLDFT [16].a
| Parameter | MM2 | MM2+NIDb | MM2+IDc | MMXd | MMXd+NIDb | MMXd+IDc | ||
|---|---|---|---|---|---|---|---|---|
| Fe–Nporphe | 1.878 | 1.871 | 1.867 | 1.897 | 1.893 | 1.894 | ||
| Fe–NIm | 1.868 | 1.860 | 1.858 | 1.874 | 1.901 | 1.883 | ||
| Fe–C | 1.944 | 1.942 | 1.941 | 1.981 | 1.963 | 1.977 | ||
| C–N | 1.164 | 1.163 | 1.164 | 1.190 | 1.202 | 1.193 | ||
| Fe–C–N | 179.8 | 179.8 | 179.6 | 172.9 | 176.8 | 179.4 | ||
| Fe-planef | -0.318 | -0.246 | -0.318 | -0.188 | -0.202 | -0.199 | ||
| Parameter | Experiment | NLDFT | ||||||
| Fe–Nporph | 1.980 | 1.917 | ||||||
| Fe–NIm | 2.087 | 2.159 | ||||||
| Fe–C | 1.934 | 1.903 | ||||||
| C–N | 1.150 | 1.173 | ||||||
| Fe–C–N | 179.1 | 180.0 | ||||||
| Fe-plane | -0.010 | – | ||||||
a Experimental data on the [Fe(OEP)(py)(CN)] system are also provided for comparison [60].b NID: polarization by non‑interacting induced dipoles.c ID: polarization by interacting induced dipoles.d Scaling factor D = 0.05.e Average values.f Distance of the Fe atom to the mean plane of the porphyrin N atoms.
Selected bond lengths and angles of Fe(P)(Im)(CN) are indicated in Table 6. The geometric parameters concerning the coordination of CN are well reproduced. The computed values for the Fe–C distance, 1.9–2.0Å, are in agreement with the experimental value of 1.934Å. The calculated values for the C–N distance, 1.16–1.20Å, are close to the experimental report of 1.150Å. The C–N distance increases from 1.147Å in the free molecule (AM1) to 1.202Å in Fe(P)(Im)(CN) (MMX+NID). The interpretation is that electronic charge is transferred from FeP to CN. This is in agreement with the experimental observation that the Fe–CN bond can be formally described as FeIII–(CN)– [9]. The most significant feature of the present structure is the linear Fe–C–N bond. The global energy minimum is linear for Fe–C–N (MM2/MMX+polarization). These calculations are in agreement with the experiment and calculation references. The Fe atom is computed by MMX to lie in the energy minimum rather close to the porphyrin N‑atoms plane, with a deviation of 0.188Å, always towards the C atom. This distance is hardly observed in the experiment.
Table 7.
Molecular mechanics (MMX+ID) results for heme‑IX adducts: distance of the Fe atom to the porphyrin N‑atoms plane.
| Adduct | D = 0.2 | D = 0.1 | D = 0.05 | Experiment | Pure QM | QM/MM |
|---|---|---|---|---|---|---|
| heme | 0.105 | 0.105 | 0.105 | 0.000 | 0.000 | 0.000 |
| heme(–His) | 0.269 | 0.728 | 0.191 | 0.310 | 0.284 | 0.379 |
| heme(–His)–O2 | -0.105 | -0.323 | -0.044 | -0.030 | -0.034 | -0.076 |
| heme(–His)–CO | -0.199 | -0.178 | -0.064 | -0.020 | – | – |
| heme(–His)–CN | -0.174 | -0.334 | -0.199 | -0.010 | – | – |
The MMX+ID displacement of the Fe atom to the porphyrin N‑atoms plane is shown in Table 7 for the heme-IX adducts as a function of different scaling factors D. The average unsigned error for D = 0.05 (0.094Å) is smaller than for D = 0.1 (0.260Å) and D = 0.2 (0.113Å). The results for D = 0.05 lie in or are closer to the range of the three references.
Conclusions
From the preceding results the following conclusions can be drawn:
- For the heme‑IX adducts, NID or ID polarization energy represents 73% of the total energy MM2+polarization. EPR corresponds to 68% of the total energy MMX+polarization.
- The optimized structure of the 5‑coordinated complexes is a square-base pyramid with the Fe atom 0.3–0.4Å out of the porphyrin plane. This result is in good agreement with the structures of the biomimetic systems that have a sterically unvoluminous axial ligand, but differs from the position of Fe in the α‑subunit of deoxy‑Hb (0.6Å).
- Three different Fe-binding models are proposed for O2, CO and CN: bent superoxide FeIII–O2–, linear FeII–CO and linear FeIII–CN–. The nature of O2 binding in Hb, Mb and simple Fe–porphyrin models is becoming clear. When O2 is bound as a bent, rather than a triangular, ligand, it is best described as bound superoxide. This bent geometry may be critical to biological functioning because it permits discrimination between O2 and CO.
- The distal His protects the heme FeII acting as a proton trap. The distal His has a pKa of ca. 5.5; at neutral pH it is protonated only at Nδ, which faces the solvent. Any proton entering the heme pocket of Hb would be bound by Nε, and simultaneously Nδ would release its proton to the solvent. When the His side chain swings out of the hemo pocket, the protons would interchange, restoring the previous state. No other amino acid side chain could function in this way.
- The fact that in a close analogue of the CO complexes of the hemoproteins the Fe–C–O linkage is linear strongly suggests that another interpretation of the results of the protein studies is in order. The allegedly bent Fe–C–O linkage in these proteins was derived from Fourier maps on which the C and O atoms remained unresolved. These maps were interpreted on the assumption that the Fe–C vector was perpendicular to the porphyrin plane. It is much more reasonable to expect that owing to the fixed nature of the globin pocket, bending will occur at the Fe atom, leading to a linear Fe–C–O bond, which is not perpendicular to the porphyrin plane.
- Unlike Fe–CO, the Fe–O2 complex is highly polar and the bound O2 is selectively stabilized by H‑bonding to distal His in Mb and Hb. The geometry optimizations performed on the experimental structures are in good agreement with X‑ray results, in spite of the fact that only certain residues of the fixing centre have been taken into account. In Hb–O2, the distal His H‑bonds with the O directly linked with Fe. The H‑bond in the fixing centres of Hb differs from that customarily observed in the biomimetic systems and Mb, in which the distal His H‑bonds the ending O. The electronic density of the binding O is lower than that of the ending O, suggesting that the H‑bond between O and the distal His is weaker than in Mb.
Acknowledgements
The author acknowledges financial support from the Spanish MCT (Plan Nacional I+D+I, Project No. BQU2001‑2935‑C02‑01) and Generalitat Valenciana (DGEUI INF01‑051 and INFRA03‑047, and OCYT GRUPOS03‑173).
References
- Perutz, M. F. Stereochemical Mechanism of Oxygen Transport by Haemoglobin. Proc. R. Soc. London, B 1980, 208, 135–162. [Google Scholar]
- Perutz, M. F.; Fermi, G.; Luisi, B.; Shaanan, B.; Liddington, R. C. Stereochemistry of Cooperative Mechanisms in Hemoglobin. Acc. Chem. Res. 1987, 20, 309–321. [Google Scholar]
- Welinder, K. G. Superfamily of Plant, Fungal and Bacterial Peroxidases. Curr. Opin. Struct. Biol. 1992, 2, 388–393. [Google Scholar]
- Malmström, B. G. Cytochrome C Oxidase as a Redox-Linked Proton Pump. Chem. Rev. 1990, 90, 1247–1260. [Google Scholar]
- Sono, M.; Roach, M. P.; Coulter, E. D.; Dawson, J. H. Heme-Containing Oxygenases. Chem. Rev. 1996, 96, 2841–2888. [Google Scholar]
- Barber, J.; Andersson, B. Revealing the Blueprint of Photosynthesis. Nature (London) 1994, 370, 31–34. [Google Scholar]
- Drennan, C. L.; Huang, S.; Drummond, J. T.; Matthews, R. G.; Ludwig, M. L. How a Protein Binds B12: A 3.0 Å X–Ray Structure of B12–Binding Domains of Methionine Synthase. Science 1994, 266, 1669–1674. [Google Scholar]
- Halcrow, M. A.; Christou, G. Biomimetic Chemistry of Nickel. Chem. Rev. 1994, 94, 2421–2481. [Google Scholar]
- Momenteau, M.; Reed, C. A. Synthetic Heme-Dioxygen Complexes. Chem. Rev. 1994, 94, 659–698. [Google Scholar]
- Dedieu, A.; Rohmer, M.-M.; Veillard, A. Ab-Initio Calculations of Metalloporphyrins. Adv. Quantum Chem. 1982, 16, 43–95. [Google Scholar]
- Rohmer, M.-M.; Dedieu, A.; Veillard, A. Structure and Properties of a Model of Deoxyheme, an ab Initio SCF Calculation. Chem. Phys. 1983, 77, 449–462. [Google Scholar]
- Rohmer, M.‑M. Electronic Ground State of Iron(II)porphyrin. Ab Initio SCF and CI Calculations and Computed Electron Deformation Densities. Chem. Phys. Lett. 1985, 116, 44–49. [Google Scholar]
- Li, N.; Su, Z.; Coppens, P.; Landrum, J. X-Ray Diffraction Study of the Electronic Ground State of (meso-Tetraphenylporphinato)iron(II). J. Am. Chem. Soc. 1990, 112, 7294–7298. [Google Scholar]
- Spiro, T. G.; Kozlowski, P. M. Will the Real FeCO Please Stand up? J. Biol. Inorg. Chem. 1997, 2, 516–520. [Google Scholar]
- Spiro, T. G.; Kozlowski, P. M. Discordant Results on FeCO Deformability in Heme Proteins Reconciled by Density Functional Theory. J. Am. Chem. Soc. 1998, 120, 4524–4525. [Google Scholar]
- Vangberg, T.; Bocian, D. F.; Ghosh, A. Deformability of Fe(II)CO and Fe(III)CN Groups in Heme Protein Models: Nonlocal Density Functional Theory Calculations. J. Biol. Inorg. Chem. 1997, 2, 526–530. [Google Scholar]
- Jewsbury, P.; Yamamoto, S.; Minato, T.; Saito, M.; Kitagawa, T. The Proximal Residue Largely Determines the CO Distortion in Carbonmonoxy Globin Proteins. An ab Initio Study of a Heme Prosthetic Unit. J. Am. Chem. Soc. 1994, 116, 11586–11587. [Google Scholar]
- Jewsbury, P.; Yamamoto, S.; Minato, T.; Saito, M.; Kitagawa, T. The Proximal Residue Largely Determines the CO Distortion in Carbon Monoxy Globin Proteins. An ab Initio Study of a Heme Prosthetic Unit. J. Phys. Chem. 1995, 99, 12677–12685. [Google Scholar]
- Ghosh, A.; Bocian, D. F. Carbonyl Tilting and Bending Potential Energy Surface of Carbon Monoxyhemes. J. Phys. Chem. 1996, 100, 6363–6367. [Google Scholar]
- Sigfridsson, E.; Ryde, U. On the Significance of Hydrogen Bonds for the Discrimination between CO and O2 by Myoglobin. J. Biol. Inorg. Chem. 1999, 4, 99–110. [Google Scholar]
- Rovira, C.; Ballone, P.; Parrinello, M. A Density Functional Study of Iron-Porphyrin Complexes. Chem. Phys. Lett. 1997, 271, 247–250. [Google Scholar]
- Rovira, C.; Kunc, K.; Hutter, J.; Ballone, P.; Parrinello, M. Equilibrium Geometries and Electronic Structure of Iron-Porphyrin Complexes: A Density Functional Study. J. Phys. Chem. A 1997, 101, 8914–8925. [Google Scholar]
- Rovira, C.; Kunc, K.; Hutter, J.; Ballone, P.; Parrinello, M. A Comparative Study of O2, CO and NO Binding to Iron–Porphyrin. Int. J. Quantum Chem. 1998, 69, 31–35. [Google Scholar]
- Rovira, C.; Parrinello, M. Factors Influencing Ligand-Binding Properties of Heme Models: A First Principles Study of Picket-Fence and Protoheme Complexes. Chem. Eur. J. 1999, 5, 250–262. [Google Scholar]
- Rovira, C.; Carloni, P.; Parrinello, M. The Iron-Sulfur Bond in Cytochrome C. J. Phys. Chem. B 1999, 103, 7031–7035. [Google Scholar]
- Salzmann, R.; McMahon, M. T.; Godbout, N.; Sanders, L. K.; Wojdelski, M.; Oldfield, E. Solid-State NMR, Crystallographyc and Density Functional Theory Investigation of Fe–CO and Fe–CO Analogue Metalloporphyrins and Metalloproteins. J. Am. Chem. Soc. 1999, 121, 3818–3828. [Google Scholar]
- Godbout, N.; Sanders, L. K.; Salzmann, R.; Havlin, R. H.; Wojdelski, M.; Oldfield, E. Solid-State NMR, Mössbauer, Crystallographyc, and Density Functional Theory Investigation of Fe–O2 and Fe–O2 Analogue Metalloporphyrins and Metalloproteins. J. Am. Chem. Soc. 1999, 121, 3829–3844. [Google Scholar]
- Loew, G.; Dupuis, M. Structure of a Model Transient Peroxide Intermediate of Peroxidases by ab Initio Methods. J. Am. Chem. Soc. 1996, 118, 10584–10587. [Google Scholar]
- Harris, D. L.; Loew, G. H. Identification of Putative Peroxide Intermediates of Peroxidases by Electronic Structure and Spectra Calculations. J. Am. Chem. Soc. 1996, 118, 10588–10594. [Google Scholar]
- Harris, D.; Loew, G.; Waskell, L. Structure and Spectra of Ferrous Dioxygen and Reduced Ferrous Dioxygen Model Cytochrome P450. J. Am. Chem. Soc. 1998, 120, 4308–4318. [Google Scholar]
- Woon, D. E.; Loew, G. H. An ab Initio Model System Investigation of the Proposed Mechanism for Activation of Peroxidases: Cooperative Catalytic Contributions from the Ion and Microsolvent Water. J. Phys. Chem. A 1998, 102, 10380–10384. [Google Scholar]
- Zakharieva, O.; Grodzicki, M.; Trautwein, A. X.; Veeger, C.; Rietgens, I. M. C. M. Molecular Orbital Study of the Hydroxylation of Benzene and Monofluorobenzene Catalysed by Iron-Oxo Porphyrin π Cation Radical Complexes. J. Biol. Inorg. Chem. 1996, 1, 192–204. [Google Scholar]
- Green, M. T. Role of the Axial Ligand in Determining the Spin State of Resting Cytochrome P450. J. Am. Chem. Soc. 1998, 120, 10772–10773. [Google Scholar]
- Jameson, G. B.; Rodley, G. A.; Robinson, W. T.; Gagne, R. R.; Reed, C. A.; Collman, J. P. Structure of a Dioxygen Adduct of (1-Methylimidazole)-meso-tetrakis(α,α,α,α-o-pivalamido-phenyl)porphinatoiron(II). An Iron Dioxygen Model for the heme Component of Oxymyoglobin. Inorg. Chem. 1978, 17, 850–857. [Google Scholar]
- Jameson, G. B.; Molinaro, F. S.; Ibers, J. A.; Collman, J. P.; Brauman, J. I.; Rose, E.; Suslick, K. S. Models for the Active Site of Oxygen-Binding Hemoproteins. Dioxygen Binding Properties and the Structures of (2-Methylimidazole)-meso-tetra(α,α,α,α–o-pivalamidophenyl)porphyrinato-iron(II)–Ethanol and Its Dioxygen Adduct. J. Am. Chem. Soc. 1980, 102, 3224–3237. [Google Scholar]
- Maseras, F. Binding of Dioxygen in a Picket-Fence Porphyrin Complex of Iron. A Theoretical QM/MM Study. New J. Chem. 1998, 22, 327–332. [Google Scholar]
- Maréchal, J.‑D.; Barea, G.; Maseras, F.; Lledós, A.; Mouawad, L.; Pérahia, D. Theoretical Modeling of the Heme Group with a Hybrid QM/MM Method. J. Comput. Chem. 2000, 21, 282–294. [Google Scholar]
- Salzmann, R.; Ziegler, C. J.; Godbout, N.; McMahon, M. T.; Suslick, K. S.; Oldfield, E. Carbonyl Complexes of Iron(II), Ruthenium(II), and Osmium(II) 5,10,15,20-Tetraphenylporphyrinates: A Comparative Investigation by X-Ray Crystallography, Solid-State NMR Spectroscopy, and Density Functional Theory. J. Am. Chem. Soc. 1998, 120, 11323–11334. [Google Scholar]
- Han, S.; Cho, K.; Ihm, J. Ab Initio Study on the Molecular Recognition by Metalloporphyrins: CO Interaction with Iron Porphyrin. Phys. Rev. E 1999, 59, 2218–2221. [Google Scholar]
- Kim, K.; Fettinger, J.; Sessler, J. L.; Cyr, M.; Hugdahl, J.; Collman, J. P.; Ibers, J. A. Structural Characterization of a Sterically Encumbered Iron(II) Porphyrin Carbonyl Complex. J. Am. Chem. Soc. 1989, 111, 403–405. [Google Scholar]
- Johansson, M. P.; Blomberg, M. R. A.; Sundholm, D.; Wikström, M. Change in Electron and Spin Density upon Electron Transfer to Haem. Biochim. Biophys. Acta 2002, 1553, 183–187. [Google Scholar]
- Johansson, M. P.; Sundholm, D.; Gerfen, G.; Wikström, M. The Spin Distribution of Low-Spin Iron Porphyrins. J. Am. Chem. Soc. 2002, 124, 11771–11780. [Google Scholar]
- Allinger, N. L. Conformational Analysis. 130. MM2. A Hydrocarbon Force Field Utilizing V1 and V2 Torsional Terms. J. Am. Chem. Soc. 1977, 99, 8127–8134. [Google Scholar]
- Torrens, F.; Ruiz-López, M.; Cativiela, C.; García, J. I.; Mayoral, J. A. Conformational Aspects of Some Asymmetric Diels-Alder Reactions. A Molecular Mechanics + Polarization Study. Tetrahedron 1992, 48, 5209–5218. [Google Scholar]
- Torrens, F. Polarization Force Fields for Peptides Implemented in ECEPP2 and MM2. Mol. Simul. 2000, 24, 391–410. [Google Scholar]
- Thole, B. T. Molecular Polarizabilities Calculated with a Modified Dipole Interaction. Chem. Phys. 1981, 59, 341–350. [Google Scholar]
- Hill, T. L. Steric Effects. I. Van der Waals Potential Energy Curves. J. Chem. Phys. 1948, 16, 399–404. [Google Scholar]
- Deiters, J. A.; Gallucci, J. C.; Clark, T. E.; Holmes, R. R. Computer Simulation of Phosphoane Structures. J. Am. Chem. Soc. 1977, 99, 5461–5471. [Google Scholar]
- Yow, H.; Bartell, L. S. Molecular Structure of (CH3)3PF2. An Electron Diffraction Study of an Analogue of ArF2. J. Mol. Struct. 1973, 15, 209–215. [Google Scholar]
- Rappé, A. K.; Casewit, C. J.; Colwell, K. S.; Goddard III, W. A.; Skiff, W. M. UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar]
- Torrens, F. Molecular Polarizability of Fullerenes and Endohedral Metallofullerenes. J. Phys. Org. Chem. 2002, 15, 742–749. [Google Scholar]
- Weiss, J. J. Nature of the Iron-Oxygen Bond in Oxyhemoglobin. Nature (London) 1964, 202, 83–84. [Google Scholar]
- Misra, H. P.; Fridovich, I. The Generation of Superoxide Radical During the Autoxidation of Hemoglobin. J. Biol. Chem. 1972, 247, 6960–6962. [Google Scholar]
- Pauling, L. Magnetic Properties and Structure of Oxyhemoglobin. Proc. Natl. Acad. Sci. USA 1977, 74, 2612–2613. [Google Scholar]
- Dewar, M. J. S.; Zoebisch, E. G.; Healy, E. F.; Stewart, J. J. P. AM1: A New General Purpose Quantum Mechanical Molecular Model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar]
- Huber, R.; Epp, O.; Formanek, H. The Structures of Deoxy-and Carbonmonoxy-Erythrocruorin. J. Mol. Biol. 1970, 52, 349–354. [Google Scholar]
- Padlan, E. A.; Love, W. E. Three-Dimensional Structure of Hemoglobin from the Polychaete Annelid, Glycera dibranchiata, at 2.5 A Resolution. J. Biol. Chem. 1974, 249, 4067–4078. [Google Scholar]
- Norvell, J. C.; Nunes, A. C.; Schoenborn, B. P. Neutron Diffraction Analysis of Myoglobin: Structure of the Carbon Monoxide Derivative. Science 1975, 190, 568–570. [Google Scholar]
- Momenteau, M.; Scheidt, W. R.; Eigenbrot, C. W.; Reed, C. A. A Deoxymyoglobin Model with a Sterically Unhindered Axial Imidazole. J. Am. Chem. Soc. 1988, 110, 1207–1215. [Google Scholar]
- Scheidt, W. R.; Hatano, K. Structure of (Cyano)(2,3,7,8,12,13,17,18–octaethylporphinato) (pyridine)iron(III) Chloroform Solvate. Acta Crystallogr., Sect. C 1991, 47, 2201–2203. [Google Scholar]
- Sample availability: Not applicable.
© 2004 by MDPI (http:www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Torrens, F. A Comparative Study of O2, CO and CN Binding to Heme IX Protein Models. Molecules 2004, 9, 632-649. https://doi.org/10.3390/90800632
AMA Style
Torrens F. A Comparative Study of O2, CO and CN Binding to Heme IX Protein Models. Molecules. 2004; 9(8):632-649. https://doi.org/10.3390/90800632
Chicago/Turabian StyleTorrens, Francisco. 2004. "A Comparative Study of O2, CO and CN Binding to Heme IX Protein Models" Molecules 9, no. 8: 632-649. https://doi.org/10.3390/90800632