Metabolomics of Oxidative Stress in Recent Studies of Endogenous and Exogenously Administered Intermediate Metabolites

Abstract
:1. Introduction
2. Results and Discussion
2.1. What is Metabolomics? What is Metabonomics?
2.2. Oxidative Stress Comes from Excited Electrons that Provide the Chemical Energy for Life
3. Reactive Oxygen Species and Oxidative Stress
4. Investigations of Two Metabolites as Treatments in Oxidative Stress
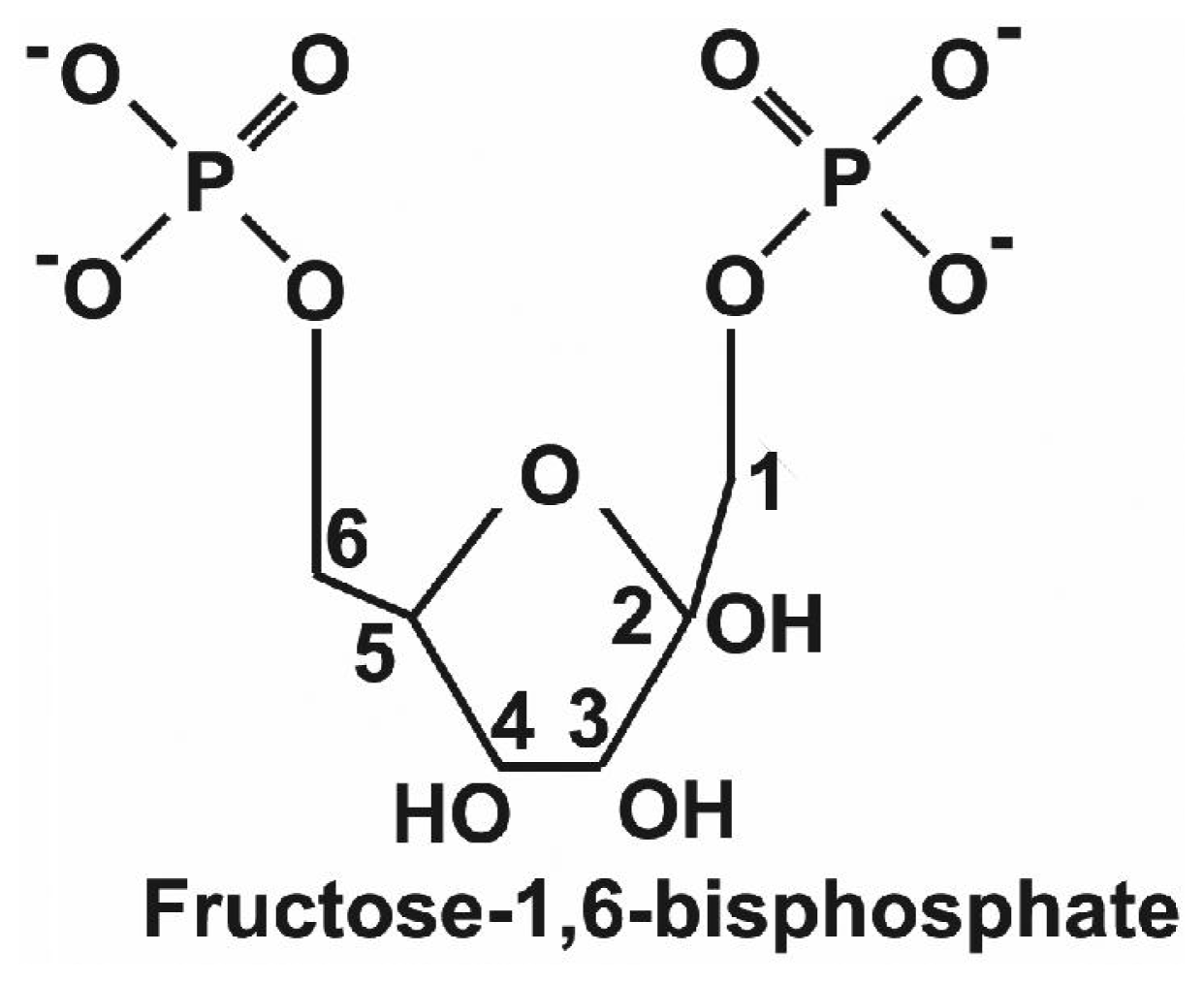
4.1. Fructose-1,6-bisphosphate
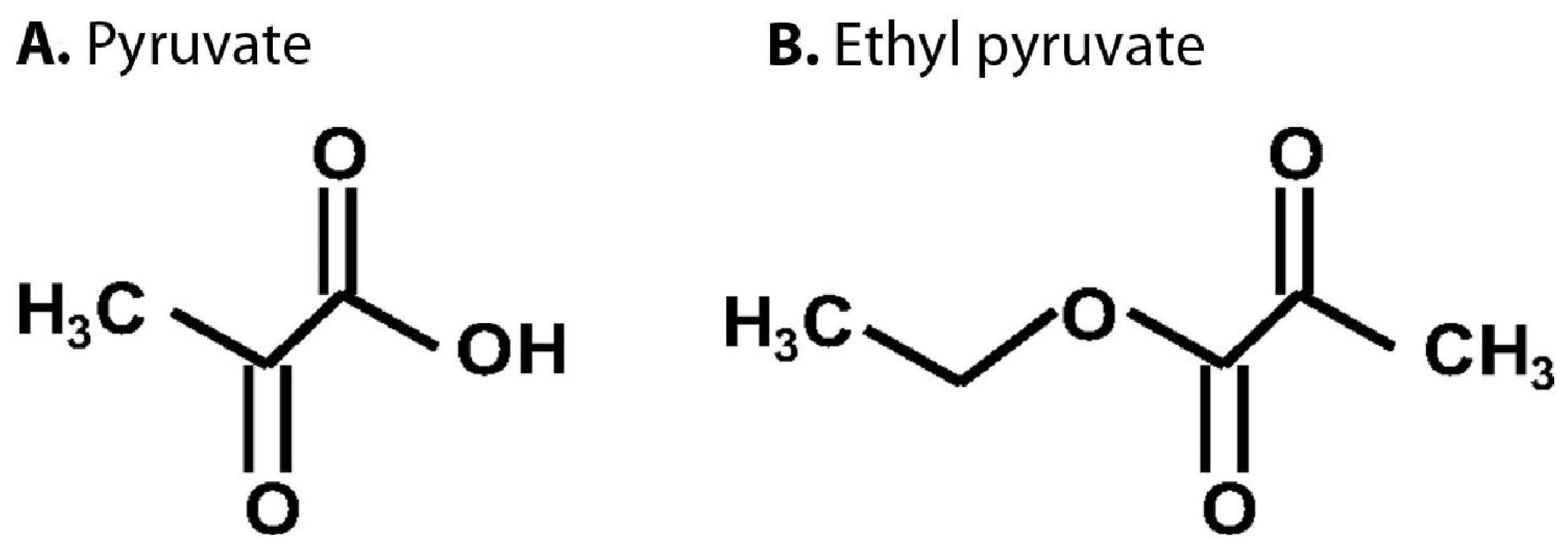
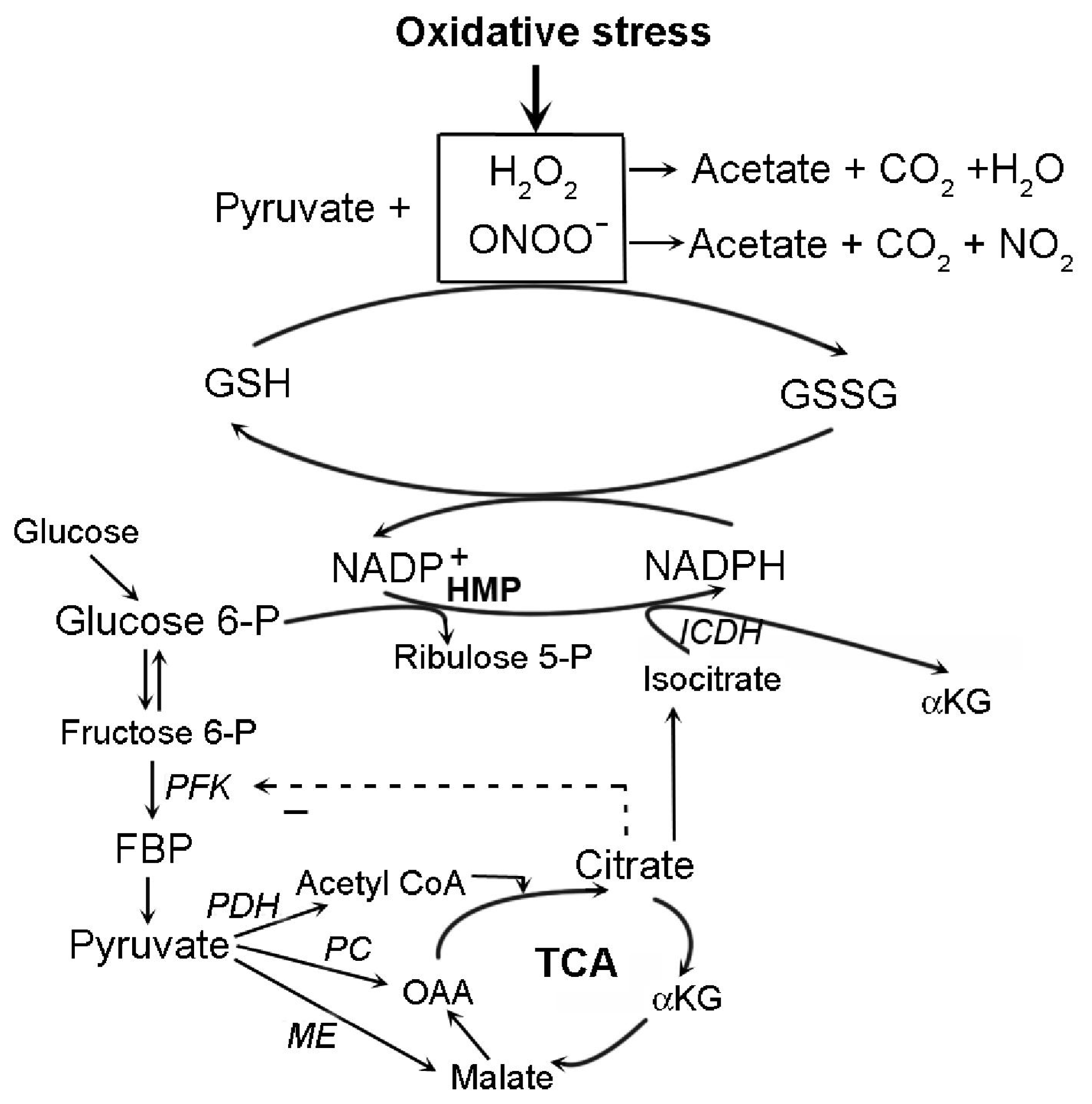
4.2. Pyruvate
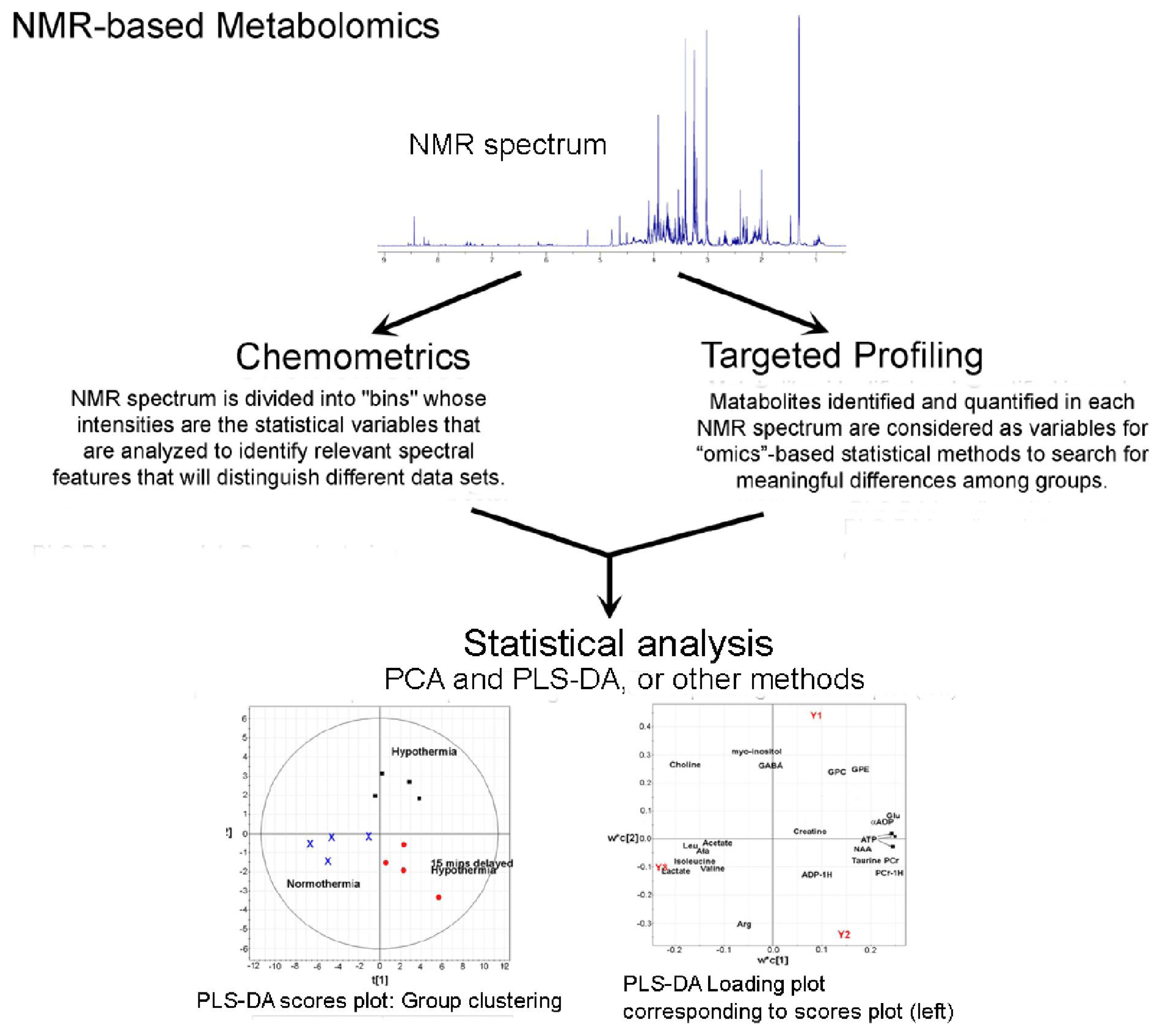
5. The Mechanics of Metabolomics
6. Brain Metabolomics and Oxidative Stress
6.1. Schizophrenia
6.2. Neonatal Asphyxia
6.3. Parkinson’s Disease
6.4. Traumatic Brain Injury
7. Diabetes and Kidney-Metabolomics and Oxidative Stress
8. Liver Metabolomics-Metabolomics and Oxidative Stress
9. Cardiac and Vascular Metabolomics and Oxidative Stress
10. Cancer Metabolomics and Oxidative Stress
11. Conclusions
Acknowledgements
References
- Griffiths, WJ; Koal, T; Wang, Y; Kohl, M; Enot, DP; Deigner, HP. Targeted metabolomics for biomarker discovery. Angew. Chem. Int. Ed. Engl 2010, 49, 5426–5445. [Google Scholar]
- Serkova, NJ; Niemann, CU. Pattern recognition and biomarker validation using quantitative H-1-NMR-based metabolomics. Expert Rev. Mol. Diagn 2006, 6, 717–731. [Google Scholar]
- Serkova, NJ; Niemann, CU. Biochemical mechanisms of nephrotoxicity: Application for metabolomics. Expert Opin. Drug Metab. Toxicol 2007, 3, 527–544. [Google Scholar]
- Serkova, NJ; Reisdorph, NA; van Patot, MCT. Metabolic markers of hypoxia: Systems biology application in biomedicine. Toxicol. Mech. Methods 2008, 18, 81–95. [Google Scholar]
- Serkova, NJ; Spratlin, JL; Eckhardt, SG. NMR-based metabolomics: Translational application and treatment of cancer. Curr. Opin. Mol. Ther 2007, 9, 572–585. [Google Scholar]
- Barderas, MG; Laborde, CM; Posada, M; de la Cuesta, F; Zubiri, I; Vivanco, F; Alvarez-Llamas, G. Metabolomic profiling for identification of novel potential biomarkers in cardiovascular diseases. J Biomed Biotechnol 2011, 2011, 790132:1–790132:9. [Google Scholar]
- Christians, U; Klawitter, J; Brunner, N; Schmitz, V. Biomarkers of immunosuppressant organ toxicity after transplantation: Status, concepts and misconceptions. Expert Opin. Drug Metab. Toxicol 2011, 7, 175–200. [Google Scholar]
- Davis, VW; Bathe, OF; Schiller, DE; Slupsky, CM; Sawyer, MB. Metabolomics and surgical oncology: Potential role for small molecule biomarkers. J. Surg. Oncol 2011, 103, 451–459. [Google Scholar]
- Lane, AN; Fan, TW; Bousamra, M; Higashi, RM; Yan, J; Miller, DM. Stable isotope-resolved metabolomics (SIRM) in cancer research with clinical application to nonsmall cell lung cancer. OMICS 2011, 15, 173–182. [Google Scholar]
- Li, N; Liu, JY; Qiu, H; Harris, TR; Sirish, P; Hammock, BD; Chiamvimonvat, N. Use of metabolomic profiling in the study of arachidonic acid metabolism in cardiovascular disease. Congest Heart Fail 2011, 17, 42–46. [Google Scholar]
- Nagrath, D; Caneba, C; Karedath, T; Bellance, N. Metabolomics for mitochondrial and cancer studies. Biochim. Biophys. Acta 2011, 1807, 650–663. [Google Scholar]
- Rubakhin, SS; Romanova, EV; Nemes, P; Sweedler, JV. Profiling metabolites and peptides in single cells. Nat. Methods 2011, 8, S20–S29. [Google Scholar]
- Sofia, M; Maniscalco, M; de Laurentiis, G; Paris, D; Melck, D; Motta, A. Exploring airway diseases by NMR-based metabonomics: A review of application to exhaled breath condensate. J Biomed Biotechnol 2011, 2011, 403260:1–403260:7. [Google Scholar]
- Nicholson, JK; Lindon, JC; Holmes, E. “Metabonomics”: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data”. Xenobiotica 1999, 29, 1181–1189. [Google Scholar]
- Lindon, JC; Holmes, E; Bollard, ME; Stanley, EG; Nicholson, JK. Metabonomics technologies and their applications in physiological monitoring, drug safety assessment and disease diagnosis. Biomarkers 2004, 9, 1–31. [Google Scholar]
- Robertson, DG. Metabonomics in toxicology: A review. Toxicol. Sci 2005, 85, 809–822. [Google Scholar]
- Fiehn, O. Combining genomics, metabolome analysis, and biochemical modelling to understand metabolic networks. Comp. Funct. Genomics 2001, 2, 155–168. [Google Scholar]
- Bren, L. Metabolomics: Working toward personalized medicine. FDA Consum 2005, 39, 28–33. [Google Scholar]
- Castresana, J; Lubben, M; Saraste, M; Higgins, DG. Evolution of cytochrome oxidase, an enzyme older than atmospheric oxygen. EMBO J 1994, 13, 2516–2525. [Google Scholar]
- Castresana, J; Saraste, M. Evolution of energetic metabolism: the respiration-early hypothesis. Trends Biochem. Sci 1995, 20, 443–448. [Google Scholar]
- Maxwell, SR; Thomason, H; Sandler, D; Leguen, C; Baxter, MA; Thorpe, GH; Jones, AF; Barnett, AH. Antioxidant status in patients with uncomplicated insulin-dependent and non-insulin-dependent diabetes mellitus. Eur. J. Clin. Invest 1997, 27, 484–490. [Google Scholar]
- Moncada, S. Nitric oxide: Discovery and impact on clinical medicine. J. R. Soc. Med 1999, 92, 164–169. [Google Scholar]
- Koppenol, WH; Moreno, JJ; Pryor, WA; Ischiropoulos, H; Beckman, JS. Peroxynitrite, a cloaked oxidant formed by nitric oxide and superoxide. Chem. Res. Toxicol 1992, 5, 834–842. [Google Scholar]
- Beckman, JS; Koppenol, WH. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol 1996, 271, C1424–C1437. [Google Scholar]
- Pacher, P; Beckman, JS; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev 2007, 87, 315–424. [Google Scholar]
- Wink, DA; Mitchell, JB. Chemical biology of nitric oxide: Insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic. Biol. Med 1998, 25, 434–456. [Google Scholar]
- Janssen-Heininger, YM; Persinger, RL; Korn, SH; Pantano, C; McElhinney, B; Reynaert, NL; Langen, RC; Ckless, K; Shrivastava, P; Poynter, ME. Reactive nitrogen species and cell signaling: implications for death or survival of lung epithelium. Am. J. Respir. Crit. Care Med 2002, 166, S9–S16. [Google Scholar]
- Xu, QB; Mayr, M; Metzler, B; Chung, YL; McGregor, E; Mayr, U; Troy, H; Hu, YH; Leitges, M; Pachinger, O; Griffiths, JR; Dunn, MJ. Ischemic preconditioning exaggerates cardiac damage in PKC-delta null mice. Am. J. Physiol.-Heart Circ. Physiol 2004, 287, H946–H956. [Google Scholar]
- Markov, AK; Oglethorpe, NC; Blake, TM; Lehan, PH; Hellems, HK. Hemodynamic, electrocardiographic, and metabolic effects of fructose diphosphate on acute myocardial ischemia. Am. Heart J 1980, 100, 639–646. [Google Scholar]
- Markov, AK. Hemodynamics and metabolic effects of fructose 1–6 diphosphate in ischemia and shock—Experimental and clinical observations. Ann. Emerg. Med 1986, 15, 1470–1477. [Google Scholar]
- Farias, LA; Willis, M; Gregory, GA. Effects of fructose-1,6-diphosphate, glucose, and saline on cardiac resuscitation. Anesthesiology 1986, 65, 595–601. [Google Scholar]
- Kuluz, JW; Gregory, GA; Han, Y; Dietrich, WD; Schleien, CL. Fructose-1,6-bisphosphate reduces infarct volume after reversible middle cerebral artery occlusion in rats. Stroke 1993, 24, 1576–1583. [Google Scholar]
- Kelleher, JA; Gregory, GA; Chan, PH. Effect of fructose-1,6-bisphosphate on glutamate uptake and glutamine synthetase activity in hypoxic astrocyte cultures. Neurochem. Res 1994, 19, 209–215. [Google Scholar]
- Kelleher, JA; Chan, TY; Chan, PH; Gregory, GA. Protection of astrocytes by fructose 1,6-bisphosphate and citrate ameliorates neuronal injury under hypoxic conditions. Brain Res 1996, 726, 167–173. [Google Scholar]
- Gobbel, GT; Chan, TY; Gregory, GA; Chan, PH. Response of cerebral endothelial cells to hypoxia: Modification by fructose-1,6-bisphosphate but not glutamate receptor antagonists. Brain Res 1994, 653, 23–30. [Google Scholar]
- Markov, AK; Warren, ET; Cohly, HH; Sauls, DJ; Skelton, TN. Influence of fructose-1,6-diphosphate on endotoxin-induced lung injuries in sheep. J. Surg. Res 2007, 138, 45–50. [Google Scholar]
- Kaakinen, T; Heikkinen, J; Dahlbacka, S; Alaoja, H; Laurila, P; Kiviluoma, K; Salomaki, T; Romsi, P; Tuominen, H; Biancari, F; et al. Fructose-1,6-bisphosphate supports cerebral energy metabolism in pigs after ischemic brain injury caused by experimental particle embolization. Heart Surg. Forum 2006, 9, E828–E835. [Google Scholar]
- Trimarchi, GR; Arcadi, FA; de Luca, R; Imperatore, C; Santoro, G; Trimarchi, F; Costa, G. Neuroprotective activity of fructose-1,6-bisphosphate following transient forebrain ischemia in the Mongolian gerbil. Jpn. J. Pharmacol 1993, 62, 215–222. [Google Scholar]
- Bickler, PE; Buck, LT. Effects of fructose-1,6-bisphosphate on glutamate release and ATP loss from rat brain slices during hypoxia. J. Neurochem 1996, 67, 1463–1468. [Google Scholar]
- Bickler, PE; Kelleher, JA. Fructose-1,6-bisphosphate stabilizes brain intracellular calcium during hypoxia in rats. Stroke 1992, 23, 1617–1622. [Google Scholar]
- LeBlanc, MH; Farias, LA; Markov, AK; Evans, OB; Smith, B; Smith, EE; Brown, EG. Fructose-1,6-diphosphate, when given five minutes after injury, does not ameliorate hypoxic ischemic injury to the central nervous system in the newborn pig. Biol. Neonate 1991, 59, 98–108. [Google Scholar]
- LeBlanc, MH; Farias, LA; Evans, OB; Vig, V; Smith, EE; Markov, AK. Fructose-1,6-bisphosphate, when given immediately before reoxygenation, or before injury, does not ameliorate hypoxic ischemic injury to the central nervous system in the newborn pig. Crit. Care Med 1991, 19, 75–83. [Google Scholar]
- Fujii, E; Kodama, Y; Takahashi, N; Roman, C; Ferriero, D; Gregory, G; Parer, JT. Fructose-1,6-bisphosphate did not affect hippocampal neuronal damage caused by 10 min of complete umbilical cord occlusion in fetal sheep. Neurosci. Lett 2001, 309, 49–52. [Google Scholar]
- Vexler, ZS; Wong, A; Francisco, C; Manabat, C; Christen, S; Tauber, M; Ferriero, DM; Gregory, G. Fructose-1,6-bisphosphate preserves intracellular glutathione and protects cortical neurons against oxidative stress. Brain Res 2003, 960, 90–98. [Google Scholar]
- Mazzio, EA; Soliman, KF. Cytoprotection of pyruvic acid and reduced beta-nicotinamide adenine dinucleotide against hydrogen peroxide toxicity in neuroblastoma cells. Neurochem. Res 2003, 28, 733–741. [Google Scholar]
- Park, JY; Kim, EJ; Kwon, KJ; Jung, YS; Moon, CH; Lee, SH; Baik, EJ. Neuroprotection by fructose-1,6-bisphosphate involves ROS alterations via p38 MAPK/ERK. Brain Res 2004, 1026, 295–301. [Google Scholar]
- Fahn, S; Cohen, G. The oxidant stress hypothesis in Parkinson’s disease: Evidence supporting it. Ann. Neurol 1992, 32, 804–812. [Google Scholar]
- Ying, W; Chen, Y; Alano, CC; Swanson, RA. Tricarboxylic acid cycle substrates prevent PARP-mediated death of neurons and astrocytes. J. Cereb. Blood Flow Metab 2002, 22, 774–779. [Google Scholar]
- Zeng, J; Hirai, K; Yang, GY; Ying, W; Swanson, RA; Kelly, M; Mayer, M; James, TL; Litt, L. Using 31P NMR spectroscopy at 14.1 Tesla to investigate PARP-1 associated energy failure and metabolic rescue in cerebrocortical slices. J. Bioenerg. Biomembr 2004, 36, 415–419. [Google Scholar]
- Fink, MP. Ringer’s ethyl pyruvate solution: A novel resuscitation fluid. Minerva Anestesiol 2001, 67, 190–192. [Google Scholar]
- Fink, MP. Reactive oxygen species as mediators of organ dysfunction caused by sepsis, acute respiratory distress syndrome, or hemorrhagic shock: Potential benefits of resuscitation with Ringer’s ethyl pyruvate solution. Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 167–174. [Google Scholar]
- Varma, SD; Devamanoharan, PS; Ali, AH. Formation of advanced glycation end (AGE) products in diabetes: Prevention by pyruvate and alpha-keto glutarate. Mol. Cell. Biochem 1997, 171, 23–28. [Google Scholar]
- Varma, SD; Devamanoharan, PS; Ali, AH. Prevention of intracellular oxidative stress to lens by pyruvate and its ester. Free Radic. Res 1998, 28, 131–135. [Google Scholar]
- Varma, SD; Hegde, KR; Kovtun, S. Oxidative damage to lens in culture: Reversibility by pyruvate and ethyl pyruvate. Ophthalmologica 2006, 220, 52–57. [Google Scholar]
- Liu, J; Segal, M; Yoo, S; Yang, GY; Kelly, M; James, TL; Litt, L. Antioxidant effect of ethyl pyruvate in respiring neonatal cerebrocortical slices after H(2)O(2) stress. Neurochem. Int 2009, 54, 106–110. [Google Scholar]
- Huh, SH; Chung, YC; Piao, Y; Jin, MY; Son, HJ; Yoon, NS; Hong, JY; Pak, YK; Kim, YS; Hong, JK; et al. Ethyl pyruvate rescues nigrostriatal dopaminergic neurons by regulating glial activation in a mouse model of Parkinson’s disease. J. Immunol 2011, 187, 960–969. [Google Scholar]
- Varma, SD; Hegde, KR. Kynurenine-induced photo oxidative damage to lens in vitro: Protective effect of caffeine. Mol. Cell. Biochem 2010, 340, 49–54. [Google Scholar]
- Varma, SD; Kovtun, S; Hegde, KR. Role of ultraviolet irradiation and oxidative stress in cataract formation-medical prevention by nutritional antioxidants and metabolic agonists. Eye Contact Lens 2011, 37, 233–245. [Google Scholar]
- Mouchiroud, L; Molin, L; Kasturi, P; Triba, MN; Dumas, ME; Wilson, MC; Halestrap, AP; Roussel, D; Masse, I; Dalliere, N; et al. Pyruvate imbalance mediates metabolic reprogramming and mimics lifespan extension by dietary restriction in Caenorhabditis elegans. Aging Cell 2011, 10, 39–54. [Google Scholar]
- Mallet, RT; Sun, J. Antioxidant properties of myocardial fuels. Mol. Cell. Biochem 2003, 253, 103–111. [Google Scholar]
- Ryou, MG; Flaherty, DC; Hoxha, B; Gurji, H; Sun, J; Hodge, LM; Olivencia-Yurvati, AH; Mallet, RT. Pyruvate-enriched cardioplegia suppresses cardiopulmonary bypass-induced myocardial inflammation. Ann. Thorac. Surg 2010, 90, 1529–1535. [Google Scholar]
- Mazzio, EA; Reams, RR; Soliman, KF. The role of oxidative stress, impaired glycolysis and mitochondrial respiratory redox failure in the cytotoxic effects of 6-hydroxydopamine in vitro. Brain Res 2004, 1004, 29–44. [Google Scholar]
- Kang, YH; Chung, SJ; Kang, IJ; Park, JH; Bunger, R. Intramitochondrial pyruvate attenuates hydrogen peroxide-induced apoptosis in bovine pulmonary artery endothelium. Mol. Cell. Biochem 2001, 216, 37–46. [Google Scholar]
- Pellerin, L; Magistretti, PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar]
- Berthet, C; Lei, H; Thevenet, J; Gruetter, R; Magistretti, PJ; Hirt, L. Neuroprotective role of lactate after cerebral ischemia. J. Cereb. Blood Flow Metab 2009, 29, 1780–1789. [Google Scholar]
- Castro, MA; Beltran, FA; Brauchi, S; Concha, II. A metabolic switch in brain: Glucose and lactate metabolism modulation by ascorbic acid. J. Neurochem 2009, 110, 423–440. [Google Scholar]
- Prieto, R; Tavazzi, B; Taya, K; Barrios, L; Amorini, AM; di Pietro, V; Pascual, JM; Marmarou, A; Marmarou, CR. Brain energy depletion in a rodent model of diffuse traumatic brain injury is not prevented with administration of sodium lactate. Brain Res 2011, 1404, 39–49. [Google Scholar]
- Kimelberg, HK. The role of hypotheses in current research, illustrated by hypotheses on the possible role of astrocytes in energy metabolism and cerebral blood flow: From Newton to now. J. Cereb. Blood Flow Metab 2004, 24, 1235–1239. [Google Scholar]
- Van den Berg, RA; van der Werf, MJ. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genomics 2006, 7, 142. [Google Scholar]
- Eriksson, L; Antti, H; Gottfries, J; Holmes, E; Johansson, E; Lindgren, F; Long, I; Lundstedt, T; Trygg, J; Wold, S. Using chemometrics for navigating in the large data sets of genomics, proteomics, and metabonomics (gpm). Anal. Bioanal. Chem 2004, 380, 419–429. [Google Scholar]
- Alam, TM; Alam, MK. Chemometric analysis of NMR spectroscopy data: A review. Ann. Rep. NMR Spectrosc 2004, 54, 41–80. [Google Scholar]
- Weljie, AM; Newton, J; Mercier, P; Carlson, E; Slupsky, CM. Targeted profiling: Quantitative analysis of 1H NMR metabolomics data. Anal. Chem 2006, 78, 4430–4442. [Google Scholar]
- Wishart, DS. Quantitative metabolomics using NMR. Trends Anal. Chem 2008, 27, 228–237. [Google Scholar]
- Provencher, SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn. Reson. Med 1993, 30, 672–679. [Google Scholar]
- Eriksson, L; Johansson, E; Kettaneh-Wold, N; Trygg, J; Wikström, C; Wold, S. Multi- and Megavariate Data Analysis Part I: Basic Principles and Applications, 2nd revised and enlarged edition ed; Umetrics Academy: Umeå, Sweden, 2006; Volume 1. [Google Scholar]
- Kemp, ML; Wille, L; Lewis, CL; Nicholson, LB; Lauffenburger, DA. Quantitative network signal combinations downstream of TCR activation can predict IL-2 production response. J. Immunol 2007, 178, 4984–4992. [Google Scholar]
- Parsons, HM; Ludwig, C; Viant, MR. Line-shape analysis of J-resolved NMR spectra: application to metabolomics and quantification of intensity errors from signal processing and high signal congestion. Magn. Reson. Chem 2009, 47, S86–S95. [Google Scholar]
- Ludwig, C; Viant, MR. Two-dimensional J-resolved NMR spectroscopy: Review of a key methodology in the metabolomics toolbox. Phytochem. Anal 2010, 21, 22–32. [Google Scholar]
- Viant, MR; Bundy, JG; Pincetich, CA; de Ropp, JS; Tjeerdema, RS. NMR-derived developmental metabolic trajectories: An approach for visualizing the toxic actions of trichloroethylene during embryogenesis. Metabolomics 2005, 1, 149–158. [Google Scholar]
- Lane, AN; Fan, TW-M. Quantification and identification of isotopomer distributions of metabolites in crude cell extracts using 1H TOCSY. Metabolomics 2007, 3, 79–86. [Google Scholar]
- Lewis, IA; Karsten, RH; Norton, ME; Tonelli, M; Westler, WM; Markley, JL. NMR method for measuring carbon-13 isotopic enrichment of metabolites in complex solutions. Anal. Chem 2010, 82, 4558–4563. [Google Scholar]
- Fan, TW; Lane, AN. NMR-based stable isotope resolved metabolomics in systems biochemistry. J. Biomol. NMR 2011, 49, 267–280. [Google Scholar]
- Pontoizeau, C; Herrmann, T; Toulhoat, P; Elena-Herrmann, B; Emsley, L. Targeted projection NMR spectroscopy for unambiguous metabolic profiling of complex mixtures. Magn. Reson. Chem 2010, 48, 727–733. [Google Scholar]
- Gowda, GA; Zhang, S; Gu, H; Asiago, V; Shanaiah, N; Raftery, D. Metabolomics-based methods for early disease diagnostics. Expert Rev. Mol. Diagn 2008, 8, 617–633. [Google Scholar]
- Ezzati, M; Hoorn, SV; Lopez, AD; Danaei, G; Rodgers, A; Mathers, CD; Murray, CJL. Comparative Quantification of Health Risks: Global and Regional Burden of Disease Attributable to Selected Major Risk Factors; Ezzati, M, Lopez, AD, Rodgers, A, Murray, CJL, Eds.; WHO: Geneva, Switzerland, 2006. [Google Scholar]
- Stephan, KE; Baldeweg, T; Friston, KJ. Synaptic plasticity and dysconnection in schizophrenia. Biol. Psychiat 2006, 59, 929–939. [Google Scholar]
- Munoz Maniega, S; Lymer, GK; Bastin, ME; Marjoram, D; Job, DE; Moorhead, TW; Owens, DG; Johnstone, EC; McIntosh, AM; Lawrie, SM. A diffusion tensor MRI study of white matter integrity in subjects at high genetic risk of schizophrenia. Schizophr. Res 2008, 106, 132–139. [Google Scholar]
- Sussmann, JE; Lymer, GK; McKirdy, J; Moorhead, TW; Munoz Maniega, S; Job, D; Hall, J; Bastin, ME; Johnstone, EC; Lawrie, SM; McIntosh, AM. White matter abnormalities in bipolar disorder and schizophrenia detected using diffusion tensor magnetic resonance imaging. Bipolar Disord 2009, 11, 11–18. [Google Scholar]
- Yao, JK; Reddy, R. Oxidative stress in schizophrenia: Pathogenetic and therapeutic implications. Antioxid. Redox Signal 2011, 15, 1999–2002. [Google Scholar]
- Martins-de-Souza, D; Harris, LW; Guest, PC; Bahn, S. The role of energy metabolism dysfunction and oxidative stress in schizophrenia revealed by proteomics. Antioxid. Redox Signal 2011, 15, 2067–2079. [Google Scholar]
- Kodavali, CV; Mikhil, BN; Vishwajit, NL. Genetic association studies of antioxidant pathway genes and schizophrenia. Antioxid. Redox Signal 2011, 15, 2037–2045. [Google Scholar]
- Prabakaran, S; Swatton, JE; Ryan, MM; Huffaker, SJ; Huang, JT; Griffin, JL; Wayland, M; Freeman, T; Dudbridge, F; Lilley, KS; et al. Mitochondrial dysfunction in schizophrenia: Evidence for compromised brain metabolism and oxidative stress. Mol. Psychiat 2004, 9, 684–697. [Google Scholar]
- Quinones, MP; Kaddurah-Daouk, R. Metabolomics tools for identifying biomarkers for neuropsychiatric diseases. Neurobiol. Dis 2009, 35, 165–176. [Google Scholar]
- Reddy, R. Antioxidant therapeutics for schizophrenia. Antioxid. Redox Signal 2011, 15, 2047–2055. [Google Scholar]
- Hoehn, T; Hansmann, G; Buhrer, C; Simbruner, G; Gunn, AJ; Yager, J; Levene, M; Hamrick, SE; Shankaran, S; Thoresen, M. Therapeutic hypothermia in neonates. Review of current clinical data, ILCOR recommendations and suggestions for implementation in neonatal intensive care units. Resuscitation 2008, 78, 7–12. [Google Scholar]
- Gancia, P; Pomero, G. Brain cooling therapy. Minerva Pediatr 2010, 62, 173–175. [Google Scholar]
- Gluckman, PD; Wyatt, JS; Azzopardi, D; Ballard, R; Edwards, AD; Ferriero, DM; Polin, RA; Robertson, CM; Thoresen, M; Whitelaw, A; et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: Multicentre randomised trial. Lancet 2005, 365, 663–670. [Google Scholar]
- Shankaran, S; Laptook, AR; Ehrenkranz, RA; Tyson, JE; McDonald, SA; Donovan, EF; Fanaroff, AA; Poole, WK; Wright, LL; Higgins, RD; et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N. Engl. J. Med 2005, 353, 1574–1584. [Google Scholar]
- Seppelt, I. Hypothermia does not improve outcome from traumatic brain injury. Crit. Care Resusc 2005, 7, 233–237. [Google Scholar]
- Hindman, BJ; Todd, MM; Gelb, AW; Loftus, CM; Craen, RA; Schubert, A; Mahla, ME; Torner, JC. Mild hypothermia as a protective therapy during intracranial aneurysm surgery: A randomized prospective pilot trial. Neurosurgery 1999, 44, 23–32. [Google Scholar]
- Ramani, R. Hypothermia for brain protection and resuscitation. Curr. Opin. Anaesthesiol 2006, 19, 487–491. [Google Scholar]
- Bernard, SA; Gray, TW; Buist, MD; Jones, BM; Silvester, W; Gutteridge, G; Smith, K. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N. Engl. J. Med 2002, 346, 557–563. [Google Scholar]
- Nolan, JP; Morley, PT; Hoek, TL; Hickey, RW. Therapeutic hypothermia after cardiac arrest. An advisory statement by the Advancement Life support Task Force of the International Liaison committee on Resuscitation. Resuscitation 2003, 57, 231–235. [Google Scholar]
- Sugerman, NT; Abella, BS. Hospital-based use of therapeutic hypothermia after cardiac arrest in adults. J. Neurotrauma 2009, 26, 371–376. [Google Scholar]
- Sanders, AB. Therapeutic hypothermia after cardiac arrest. Curr. Opin. Crit. Care 2006, 12, 213–217. [Google Scholar]
- Chu, CY; Xiao, X; Zhou, XG; Lau, TK; Rogers, MS; Fok, TF; Law, LK; Pang, CP; Wang, CC. Metabolomic and bioinformatic analyses in asphyxiated neonates. Clin. Biochem 2006, 39, 203–209. [Google Scholar]
- Atzori, L; Xanthos, T; Barberini, L; Antonucci, R; Murgia, F; Lussu, M; Aroni, F; Varsami, M; Papalois, A; Lai, A; et al. A metabolomic approach in an experimental model of hypoxia-reoxygenation in newborn piglets: Urine predicts outcome. J. Matern.-Fetal Neonatal Med 2010, 23, 134–137. [Google Scholar]
- Liu, J; Litt, L; Segal, MR; Kelly, MJ; Yoshihara, HA; James, TL. Outcome-related metabolomic patterns from 1H/31P NMR after mild hypothermia treatments of oxygen-glucose deprivation in a neonatal brain slice model of asphyxia. J. Cereb. Blood Flow Metab 2011, 31, 547–559. [Google Scholar]
- Lang, AE; Lozano, AM. Parkinson’s disease. Second of two parts. N. Engl. J. Med 1998, 339, 1130–1143. [Google Scholar]
- Lang, AE; Lozano, AM. Parkinson’s disease. First of two parts. N. Engl. J. Med 1998, 339, 1044–1053. [Google Scholar]
- Beal, MF; Bogdanov, M; Matson, WR; Wang, L; Matson, T; Saunders-Pullman, R; Bressman, SS. Metabolomic profiling to develop blood biomarkers for Parkinson’s disease. Brain 2008, 131, 389–396. [Google Scholar]
- Fasano, M; Alberio, T; Lopiano, L. Peripheral biomarkers of Parkinson’s disease as early reporters of central neurodegeneration. Biomarkers Med 2008, 2, 465–478. [Google Scholar]
- Caudle, WM; Bammler, TK; Lin, Y; Pan, S; Zhang, J. Using “omics” to define pathogenesis and biomarkers of Parkinson’s disease. Expert Rev. Neurother 2010, 10, 925–942. [Google Scholar]
- Viant, MR; Lyeth, BG; Miller, MG; Berman, RF. An NMR metabolomic investigation of early metabolic disturbances following traumatic brain injury in a mammalian model. NMR Biomed 2005, 18, 507–516. [Google Scholar]
- Kagan, VE; Tyurin, VA; Tyurina, YY; Borisenko, GG; Sokolova, TV; Ritov, VB; Quinn, PJ; Rose, M; Kochanek, P; Graham, SH. Oxidative stress following traumatic brain injury in rats: Quantitation of biomarkers and detection of free radical intermediates. J. Neurochem 2000, 75, 2178–2189. [Google Scholar]
- Palmer, AM; Marion, DW; Botscheller, ML; Swedlow, PE; Styren, SD; Dekosky, ST. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J. Neurochem 1993, 61, 2015–2024. [Google Scholar]
- Oberley, LW. Free radicals and diabetes. Free Radic. Biol. Med 1988, 5, 113–124. [Google Scholar]
- Wolff, SP; Jiang, ZY; Hunt, JV. Protein glycation and oxidative stress in diabetes mellitus and ageing. Free Radic. Biol. Med 1991, 10, 339–352. [Google Scholar]
- Baynes, JW. Role of oxidative stress in development of complications in diabetes. Diabetes 1991, 40, 405–412. [Google Scholar]
- Barba, I; Garcia-Ramirez, M; Hernandez, C; Alonso, MA; Masmiquel, L; Garcia-Dorado, D; Simo, R. Metabolic fingerprints of proliferative diabetic retinopathy: An 1H-NMR-based metabonomic approach using vitreous humor. Invest. Ophthalmol. Vis. Sci 2010, 51, 4416–4421. [Google Scholar]
- Zhang, S; Zheng, C; Lanza, IR; Nair, KS; Raftery, D; Vitek, O. Interdependence of signal processing and analysis of urine 1H NMR spectra for metabolic profiling. Anal. Chem 2009, 81, 6080–6088. [Google Scholar]
- Klawitter, J; Haschke, M; Kahle, C; Dingmann, C; Leibfritz, D; Christians, U. Toxicodynamic effects of ciclosporin are reflected by metabolite profiles in the urine of healthy individuals after a single dose. Br. J. Clin. Pharmacol 2010, 70, 241–251. [Google Scholar]
- Serkova, N; Fuller, TF; Klawitter, J; Freise, CE; Niemann, CU. H-NMR-based metabolic signatures of mild and severe ischemia/reperfusion injury in rat kidney transplants. Kidney Int 2005, 67, 1142–1151. [Google Scholar]
- Lanz, C; Patterson, AD; Slavik, J; Krausz, KW; Ledermann, M; Gonzalez, FJ; Idle, JR. Radiation metabolomics. 3. Biomarker discovery in the urine of gamma-irradiated rats using a simplified metabolomics protocol of gas chromatography-mass spectrometry combined with random forests machine learning algorithm. Radiat. Res 2009, 172, 198–212. [Google Scholar]
- Chen, C; Krausz, KW; Idle, JR; Gonzalez, FJ. Identification of novel toxicity-associated metabolites by metabolomics and mass isotopomer analysis of acetaminophen metabolism in wild-type and Cyp2e1-null mice. J. Biol. Chem 2008, 283, 4543–4559. [Google Scholar]
- Wei, L; Liao, P; Wu, H; Li, X; Pei, F; Li, W; Wu, Y. Toxicological effects of cinnabar in rats by NMR-based metabolic profiling of urine and serum. Toxicol. Appl. Pharmacol 2008, 227, 417–429. [Google Scholar]
- Ohta, T; Masutomi, N; Tsutsui, N; Sakairi, T; Mitchell, M; Milburn, MV; Ryals, JA; Beebe, KD; Guo, L. Untargeted metabolomic profiling as an evaluative tool of fenofibrate-induced toxicology in Fischer 344 male rats. Toxicol. Pathol 2009, 37, 521–535. [Google Scholar]
- Sieber, M; Wagner, S; Rached, E; Amberg, A; Mally, A; Dekant, W. Metabonomic study of ochratoxin a toxicity in rats after repeated administration: Phenotypic anchoring enhances the ability for biomarker discovery. Chem. Res. Toxicol 2009, 22, 1221–1231. [Google Scholar]
- Kalhan, SC; Guo, L; Edmison, J; Dasarathy, S; McCullough, AJ; Hanson, RW; Milburn, M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism 2011, 60, 404–413. [Google Scholar]
- Viant, MR; Hrydziuszko, O; Silva, MA; Perera, MTPR; Richards, DA; Murphy, N; Mirza, D. Application of metabolomics to investigate the process of human orthotopic liver transplantation: A proof-of-principle study. OMICS: J. Integr. Biol 2010, 14, 143–150. [Google Scholar]
- Moazzami, AA; Andersson, R; Kamal-Eldin, A. Changes in the metabolic profile of rat liver after alpha-tocopherol deficiency as revealed by metabolomics analysis. NMR Biomed 2011, 24, 499–505. [Google Scholar]
- Ellinger-Ziegelbauer, H; Adler, M; Amberg, A; Brandenburg, A; Callanan, JJ; Connor, S; Fountoulakis, M; Gmuender, H; Gruhler, A; Hewitt, P; et al. The enhanced value of combining conventional and “omics” analyses in early assessment of drug-induced hepatobiliary injury. Toxicol. Appl. Pharmacol 2011, 252, 97–111. [Google Scholar]
- Niemann, CU; Serkova, NJ; Jackman, M; Brown, JL; Liu, T; Hirose, R; Roberts, JP; Maher, JJ. Metabolic profiling of livers and blood from obese Zucker rats. J. Hepatol 2006, 44, 956–962. [Google Scholar]
- Soga, T; Baran, R; Suematsu, M; Ueno, Y; Ikeda, S; Sakurakawa, T; Kakazu, Y; Ishikawa, T; Robert, M; Nishioka, T; Tomita, M. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem 2006, 281, 16768–16776. [Google Scholar]
- Lindon, JC; Keun, HC; Ebbels, TM; Pearce, JM; Holmes, E; Nicholson, JK. The Consortium for Metabonomic Toxicology (COMET): Aims, activities and achievements. Pharmacogenomics 2005, 6, 691–699. [Google Scholar]
- Gao, P; Lin, HL; Zhang, J. Silent myocardial ischemia is associated with altered plasma phospholipids. J. Clin. Lab. Anal 2009, 23, 45–50. [Google Scholar]
- Tissot van Patot, MC; Serkova, NJ; Haschke, M; Kominsky, DJ; Roach, RC; Christians, U; Henthorn, TK; Honigman, B. Enhanced leukocyte HIF-1alpha and HIF-1 DNA binding in humans after rapid ascent to 4300 m. Free Radic. Biol. Med 2009, 46, 1551–1557. [Google Scholar]
- Ciborowski, M; Martin-Ventura, JL; Meilhac, O; Michel, JB; Ruperez, FJ; Tunon, J; Egido, J; Barbas, C. Metabolites secreted by human atherothrombotic aneurysms revealed through a metabolomic approach. J. Proteome Res 2011, 10, 1374–1382. [Google Scholar]
- Mayr, M; Chung, YL; Mayr, U; Yin, XK; Ly, L; Troy, H; Fredericks, S; Hu, YH; Griffiths, JR; Xu, QB. Proteomic and metabolomic analyses of atherosclerotic vessels from apolipoprotein E-deficient mice reveal alterations in inflammation, oxidative stress, and energy metabolism. Arterioscler. Thromb. Vasc. Biol 2005, 25, 2135–2142. [Google Scholar]
- De Souza, AI; Cardin, S; Wait, R; Chung, YL; Vijayakumar, M; Maguy, A; Camm, AJ; Nattel, S. Proteomic and metabolomic analysis of atrial profibrillatory remodelling in congestive heart failure. J. Mol. Cell. Cardiol 2010, 49, 851–863. [Google Scholar]
- Xu, QB; Mayr, M; Siow, R; Chung, YL; Mayr, U; Griffiths, JR. Proteomic and metabolomic analysis of vascular smooth muscle cells - Role of PKC delta. Circ. Res 2004, 94, E87–E96. [Google Scholar]
- Cascante, M; Benito, A; Zanuy, M; Vizan, P; Marin, S; de Atauri, P. Metabolic network adaptations in cancer as targets for novel therapies. Biochem. Soc. Trans 2010, 38, 1302–1306. [Google Scholar]
- Britz-McKibbin, P; Lee, R. Metabolomic studies of radiation-induced apoptosis of human leukocytes by capillary electrophoresis-mass spectrometry and flow cytometry: Adaptive cellular responses to ionizing radiation. Electrophoresis 2010, 31, 2328–2337. [Google Scholar]
- Pacak, K. Phaeochromocytoma: A catecholamine and oxidative stress disorder. Endocr. Regul 2011, 45, 65–90. [Google Scholar]
- Bahn, S; Holmes, E; Tsang, TM; Huang, JTJ; Leweke, FM; Koethe, D; Gerth, CW; Nolden, BM; Gross, S; Schreiber, D; Nicholson, JK. Metabolic profiling of CSF: Evidence that early intervention may impact on disease progression and outcome in schizophrenia. PLoS Med 2006, 3, 1420–1428. [Google Scholar]
- Kaddurah-Daouk, R; McEvoy, J; Baillie, RA; Lee, D; Yao, JK; Doraiswamy, PM; Krishnan, KRR. Metabolomic mapping of atypical antipsychotic effects in schizophrenia. Mol. Psychiat 2007, 12, 934–945. [Google Scholar]
- Johansen, KK; Wang, L; Aasly, JO; White, LR; Matson, WR; Henchcliffe, C; Beal, MF; Bogdanov, M. Metabolomic profiling in LRRK2-related Parkinson’s disease. PLoS one 2009, 4, 1–9. [Google Scholar]
- Zhang, S; Nagana Gowda, GA; Asiago, V; Shanaiah, N; Barbas, C; Raftery, D. Correlative and quantitative 1H NMR-based metabolomics reveals specific metabolic pathway disturbances in diabetic rats. Anal. Biochem 2008, 383, 76–84. [Google Scholar]
- Portilla, D; Schnackenberg, L; Beger, RD. Metabolomics as an extension of proteomic analysis: Study of acute kidney injury. Semin. Nephrol 2007, 27, 609–620. [Google Scholar]
- Andreadou, I; Papaefthimiou, M; Zira, A; Constantinou, M; Sigala, F; Skaltsounis, AL; Tsantili-Kakoulidou, A; Iliodromitis, EK; Kremastinos, DT; Mikros, E. Metabonomic identification of novel biomarkers in doxorubicin cardiotoxicity and protective effect of the natural antioxidant oleuropein. NMR Biomed 2009, 22, 585–592. [Google Scholar]
Appendix

{kind=link}
{kind=link}
{kind=link}
{kind=link}
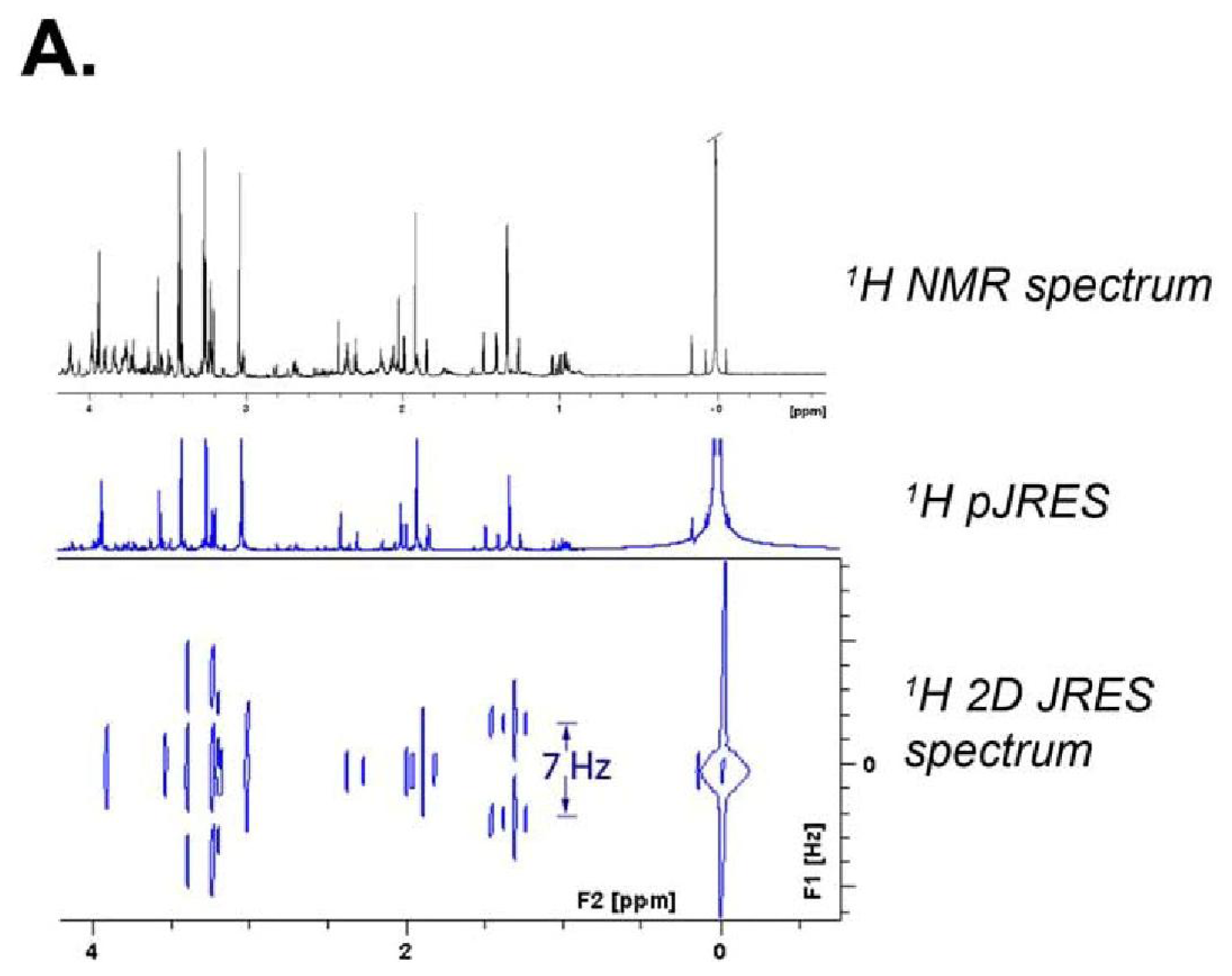{kind=link}
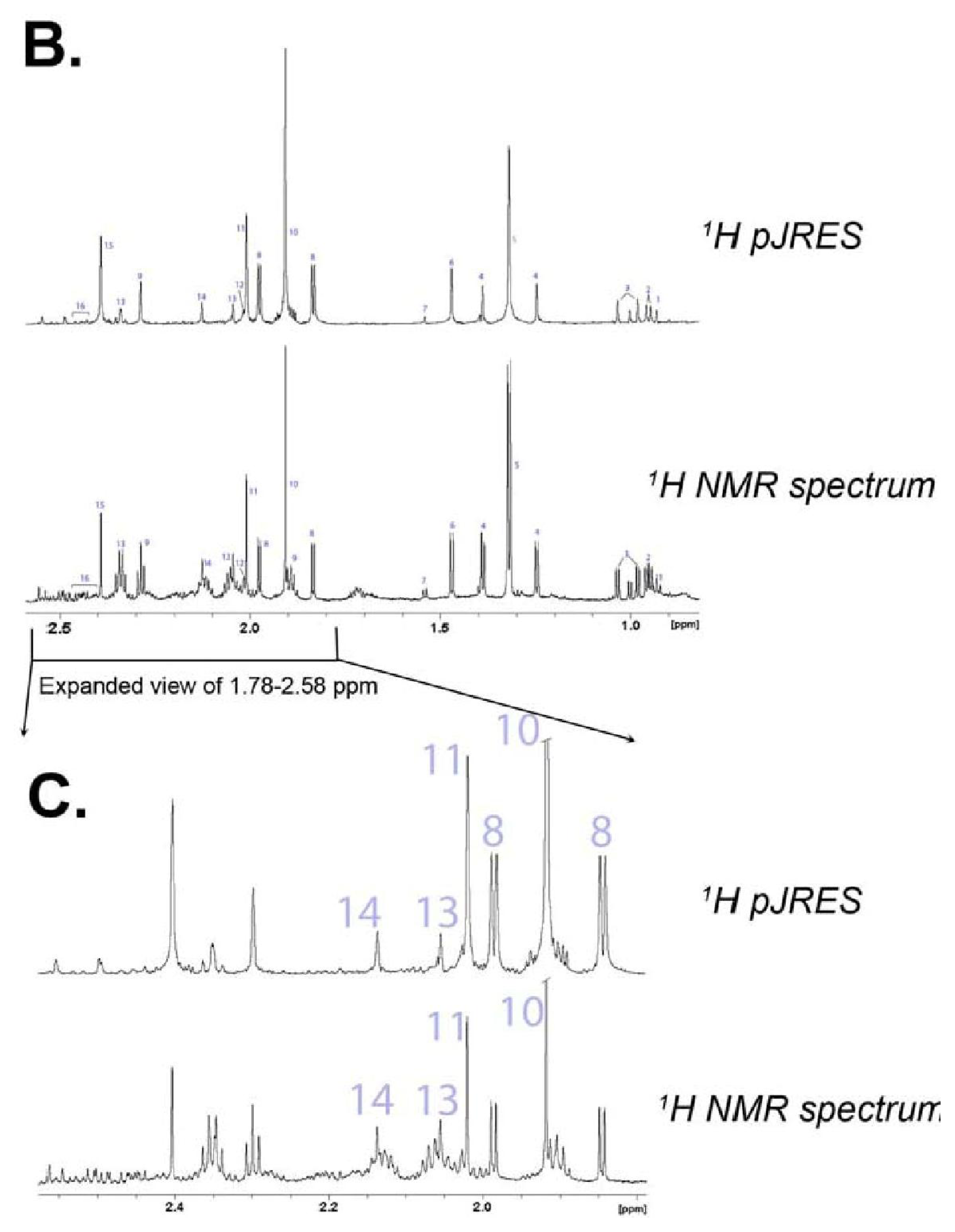{kind=link}
| Pathology | Change of metabolites | Samples | Analytical technique | Ref. | Year |
|---|---|---|---|---|---|
| CNS | |||||
| Schizophrenia | Taurine ↑; Glutathione | Prefrontal cortex tissue from patients | 1H NMR | [92] | 2004 |
| Schizophrenia | Glucose ↑; Acetate ↓; Alanine; Glutamine | CSF from human | 1H NMR | [145] | 2006 |
| Schizophrenia | Phosphotidylethanolamine ↓ Phosphotidylcholine ↓ | Lipid extracts of patient’s plasma | HPLC | [146] | 2007 |
| Parkinson’s disease | Uric acid ↓; GSSG ↑ | Plasma from patients | LCECA | [111] | 2008 |
| Parkinson’s disease | Urate ↓ | Plasma from patients | LCECA | [147] | 2009 |
| Trauma Brain Injury | Ascorbate ↓; Glutamate ↓; NAA ↓; Total of PC/GPC ↓ | Brain tissue extracts and plasma from rats | 1H NMR | [114] | 2005 |
| Asphyxia | Glutarate ↑; Methylmalonate ↑; 3-hyroxy-butyrate ↑; Orotate ↑ | Urine from patients | MS | [106] | 2006 |
| Hypoxia-reoxygenati on | Urea; Creatinine; Malonate; Methylguanidine; Hydrooxyisobutyric acid | Urine from piglets 1–4 days old | 1H NMR | [107] | 2010 |
| Oxygen-Glucose deprivation | PCr; ATP ; NAA; Taurine | Rat brain slice extracts | 1H/31P NMR | [108] | 2011 |
| Diabetes and Renal diseases | |||||
| Proliferative diabetic retinopathy | Ascorbate ↓; Galactitol ↓ | Vitreous samples from patients | 1H NMR | [120] | 2010 |
| Type 1 diabetes | Glucose ↑; Alanine ↑; Lactate ↑; Ethanol ↑; Acetate ↑; Fumarate ↑ | Urine and plasma from rats | 1H NMR | [148] | 2008 |
| Cyclosporine nephrotoxicity | 15-F(2t)-isoprostane ↑; Creatinine ↑; Citrate ↓; Hippurate ↑; Phenylalanine | Urine and plasma from human | 1H NMR HPLC-MS | [122] | 2010 |
| Kidney transplantation | Allantoin ↑; Polyunsaturated fatty acids ↓ | Kidney tissue and plasma from rats | 1H NMR | [123] | 2005 |
| Radiation injury | Glyoxylate ↑; Threonate ↑; Thymine ↑; Uracil ↑; Citrate ↓; Adipate ↓; Pimelate ↓; Suberate ↓; 2-oxoglutarate ↓ | Urine from rats | GC-MS | [124] | 2009 |
| Ochratoxin A toxicity | 2-oxoglutarate ↓; Citrate ↓; Glucose ↑; Creatinine ↑; Pseudouridine ↑; 5-oxoproline ↑; Myo-inositol ↑ | Urine from rats | GC-MS 1H NMR LC-MS | [128] | 2009 |
| Cisplatin-induced nephrotoxicity | Glucosuria ↑; Nonesterified fatty acids ↑; Triglycerides ↑ | Kidney tissue, urine, plasma from rats | 1H NMR | [149] | 2007 |
| Cardiovascular diseases | |||||
| Hypoxia | Glutathione ↓; Lactate ↑; Succinate↑ | Plasma and urine from human | 1H NMR | [137] | 2009 |
| Abdominal aortic aneurysms | Fatty acid amides | Aortas tissue from human | LC-MS | [138] | 2011 |
| Atherosclerosis | Alanine ↓; 1-Cys peroxiredoxin (identified in proteomics) | Mouse aortas tissue | 1H NMR | [139] | 2005 |
| H2O2-induced stress | Alanine ↓; Lactate ↑; Carnitine ↑; Glutathione↓ | Mouse C2C12 myotubes | MS | [141] | 2004 |
| Doxorubicin cardiotoxicity | Acetate ↑; Succinate ↑ | Dog heart tissue extracts | 1H NMR | [150] | 2009 |
| Hepatic diseases | |||||
| Fatty liver disease Steatohepatitis | Glycocholate↑; Taurocholate↑ Carnitine↑; Butyrylcarnitine↑; Cysteine/Glutathione↓ | Plasma from patients | GC-MS | [129] | 2011 |
| Acetaminophen-induced hepatotoxicity | Ophthalmate ↓ | Mouse liver extracts and serum | CE-TOFMS | [134] | 2006 |
| Obesity | Hepatic ratios of PUFA/MUFA ↓ Glutathione ↓ | Liver lipid extracts and serum from rat | 1H NMR | [133] | 2006 |
| Cancer | |||||
| Ionizing radiation | Arginine ↑; Glutamine ↑; Creatine ↑; Proline ↑; GSH↑ | Human leukocytes | CE-ESI-MS | [143] | 2010 |






© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, J.; Litt, L.; Segal, M.R.; Kelly, M.J.S.; Pelton, J.G.; Kim, M. Metabolomics of Oxidative Stress in Recent Studies of Endogenous and Exogenously Administered Intermediate Metabolites. Int. J. Mol. Sci. 2011, 12, 6469-6501. https://doi.org/10.3390/ijms12106469
Liu J, Litt L, Segal MR, Kelly MJS, Pelton JG, Kim M. Metabolomics of Oxidative Stress in Recent Studies of Endogenous and Exogenously Administered Intermediate Metabolites. International Journal of Molecular Sciences. 2011; 12(10):6469-6501. https://doi.org/10.3390/ijms12106469
Chicago/Turabian StyleLiu, Jia, Lawrence Litt, Mark R. Segal, Mark J. S. Kelly, Jeffrey G. Pelton, and Myungwon Kim. 2011. "Metabolomics of Oxidative Stress in Recent Studies of Endogenous and Exogenously Administered Intermediate Metabolites" International Journal of Molecular Sciences 12, no. 10: 6469-6501. https://doi.org/10.3390/ijms12106469
APA StyleLiu, J., Litt, L., Segal, M. R., Kelly, M. J. S., Pelton, J. G., & Kim, M. (2011). Metabolomics of Oxidative Stress in Recent Studies of Endogenous and Exogenously Administered Intermediate Metabolites. International Journal of Molecular Sciences, 12(10), 6469-6501. https://doi.org/10.3390/ijms12106469