Molecular Diagnosis of Analbuminemia: A New Case Caused by a Nonsense Mutation in the Albumin Gene

Abstract
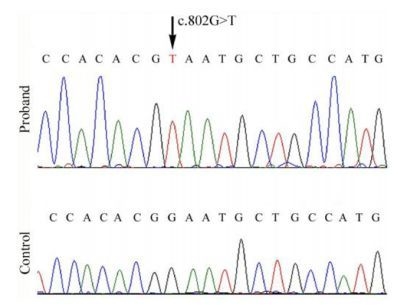
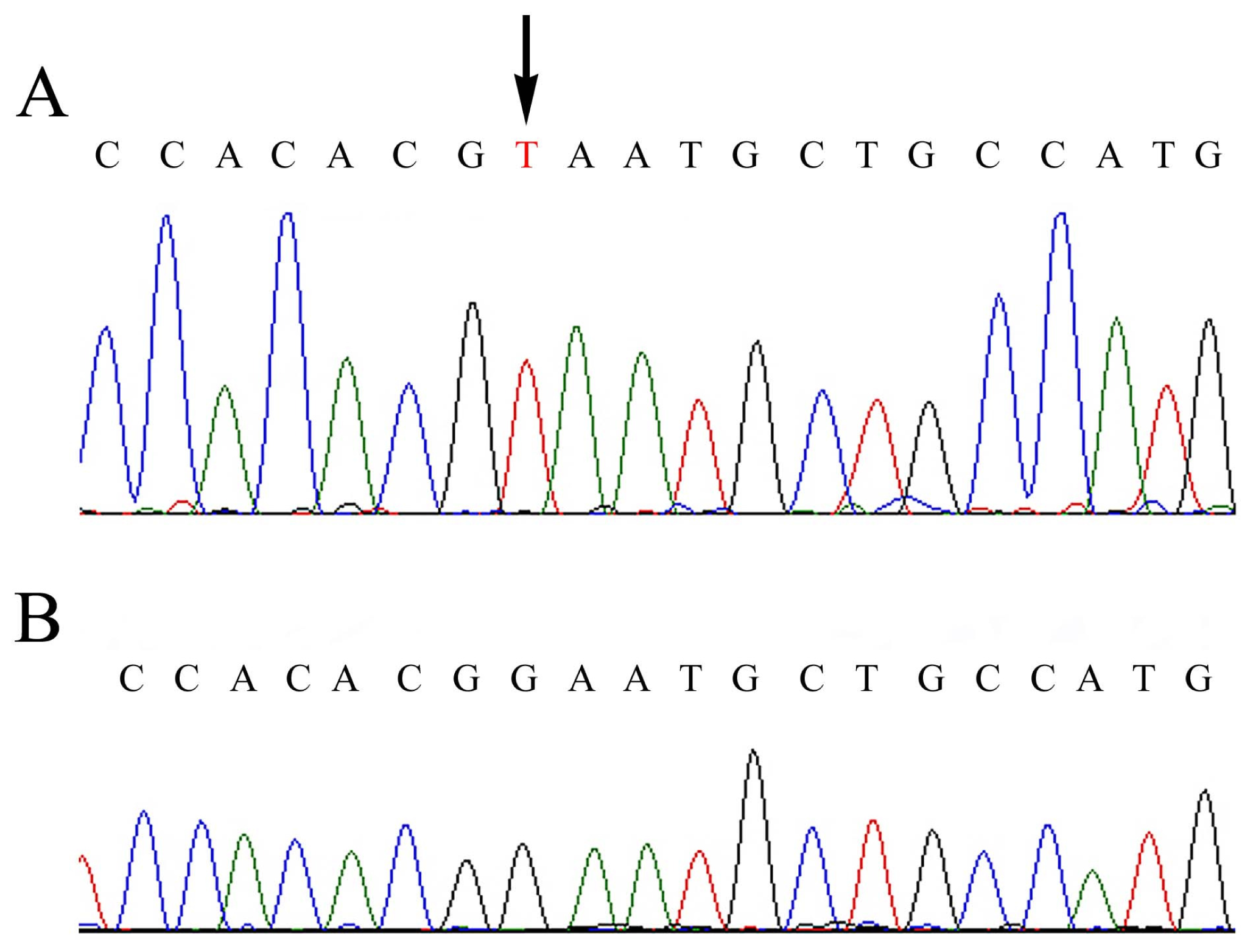
:
1. Introduction
2. Results and Discussion
2.1. Patient
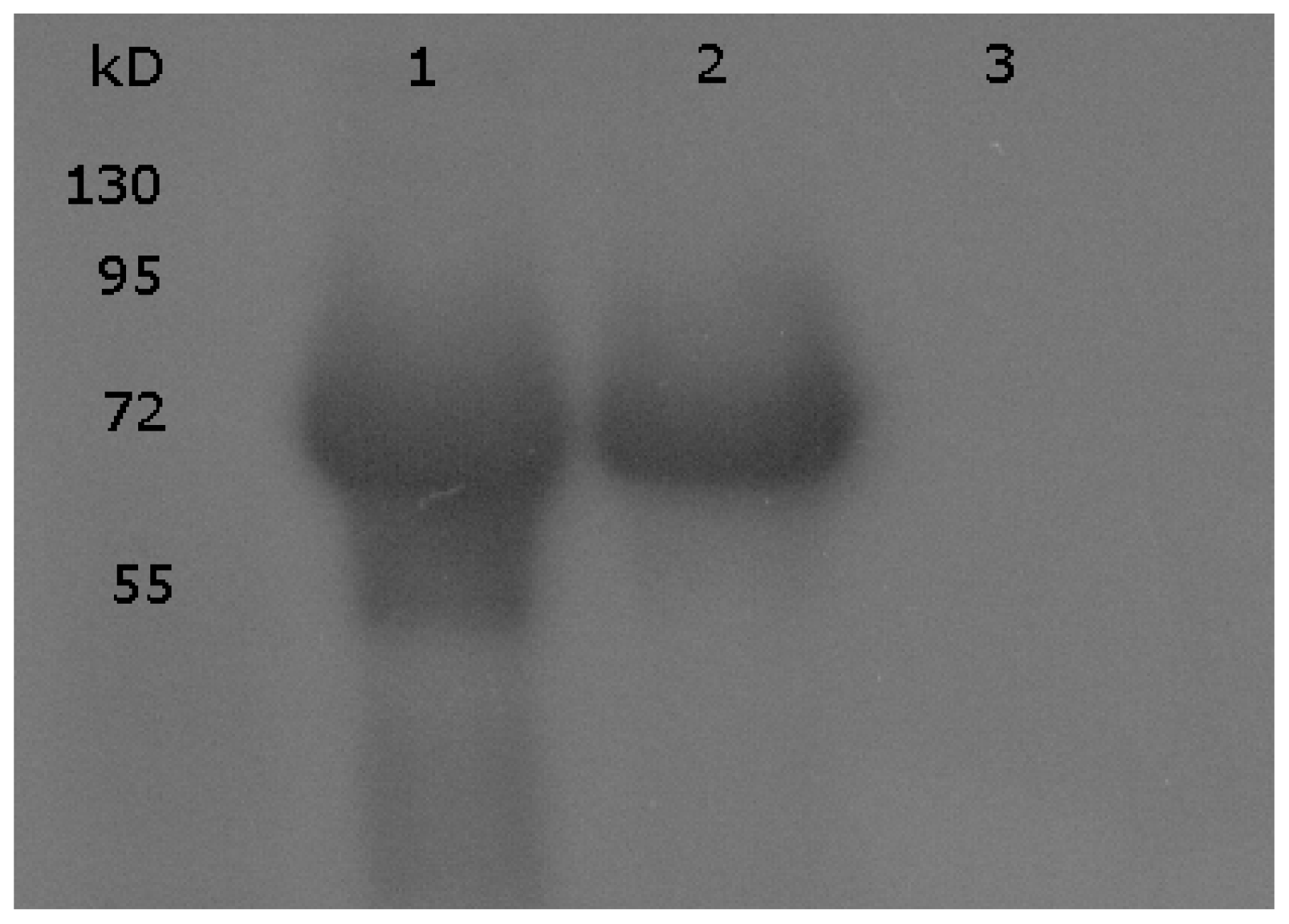
2.2. Mutational Analysis
3. Experimental Section
3.1. Immunoblotting
3.2. Mutational Analysis
3.3. Two-Dimensional Electrophoresis
4. Conclusions
Acknowledgements
- Conflict of InterestThe authors declare no conflict of interest.
Abbreviations
| ALB | serum albumin (protein) |
| ALB | albumin gene |
| FcRn | neonatal Fc receptor |
References
- Peters, T., Jr. All about Albumin: Biochemistry, Genetics and Medical Applications; Academic Press: San Diego, CA USA, 1996; pp. 1–432. [Google Scholar]
- The Albumin Home Page. Available online: http://albumin.org accessed on 20 July 2011.
- Minchiotti, L.; Galliano, M.; Kragh-Hansen, U.; Peters, T., Jr. Mutations and polymorphisms of the gene of the major human blood protein, serum albumin. Hum. Mutat 2008, 29, 1007–1016. [Google Scholar]
- Watkins, S.; Madison, J.; Galliano, M.; Minchiotti, L.; Putnam, F.W. A nucleotide insertion and frameshift cause analbuminemia in an Italian family. Proc. Natl. Acad. Sci. USA 1994, 91, 2275–2279. [Google Scholar]
- Minghetti, P.P.; Ruffner, D.E.; Kuang, W.J.; Dennison, O.E.; Hawkins, J.W.; Beattie, W.G.; Dugaiczyk, A. Molecular structure of the human albumin gene is revealed by nucleotide sequence within q11–22 of chromosome 4. J. Biol. Chem 1986, 261, 6747–6757. [Google Scholar]
- Campagnoli, M.; Sala, A.; Romano, A.; Rossi, A.; Nauta, J.; Koot, B.G.; Minchiotti, L.; Galliano, M. Novel nonsense mutation causes analbuminemia in a Moroccan family. Clin. Chem 2005, 51, 227–229. [Google Scholar]
- del Ben, M.; Burattin, M.; Arca, M.; Ceci, F.; Violi, F.; Angelico, F. Treatment of severe hypercholesterolemia with atorvastatin in congenital analbuminemia. Am. J. Med 2004, 117, 803–804. [Google Scholar]
- Koot, B.G.; Houwen, R.; Pot, D.J.; Nauta, J. Congenital analbuminemia: biochemical and clinical implications. A case report and literature review. Eur. J. Pediatr 2004, 163, 664–670. [Google Scholar]
- Newstead, J.; Card, S.; Lyon, A. Low serum albumin and abnormal body shape in a young Canadian First Nations woman. Lab. Med 2004, 35, 350–356. [Google Scholar]
- Ruhoff, M.S.; Greene, M.W.; Peters, T. Location of the mutation site in the first two reported cases of analbuminemia. Clin. Biochem 2010, 43, 525–527. [Google Scholar]
- Dagnino, M.; Caridi, G.; Marsciani, M.; Bettocchi, I.; Tassinari, D; Bernardi, F; Chiodo, F; Campagnoli, M; Galliano, M; Minchiotti, L. A novel frame-shift deletion causing analbuminemia in an Italian paediatric patient. Eur. J. Clin. Invest 2010, 40, 281–284. [Google Scholar]
- Dagnino, M.; Caridi, G.; Aydin, Z.; Ozturk, S.; Karaali, Z.; Kazancioglu, R.; Cefle, K.; Gursu, M.; Campagnoli, M.; Galliano, M.; Minchiotti, L. A novel frameshift deletion in the albumin gene causes analbuminemia in a young Turkish woman. Clin. Chim. Acta 2010, 411, 1711–1715. [Google Scholar]
- Andersen, J.T.; Sandlie, I. A receptor-mediated mechanism to support clinical observation of altered albumin variants. Clin. Chem 2007, 53, 1549–1552. [Google Scholar]
- Andersen, J.T.; Daba, M.B.; Sandlie, I. FcRn binding properties of an abnormal truncated analbuminemic albumin variant. Clin. Biochem 2010, 43, 367–372. [Google Scholar]
- Lyon, A.W.; Meinert, P.; Bruce, G.A.; Laxdal, V.A.; Salkie, M.L. Influence of methodology on the detection and diagnosis of congenital analbuminemia. Clin. Chem 1998, 44, 2365–2367. [Google Scholar]
- Becker-Cohen, R.; Belostotsky, R.; Ben-Shalom, E.; Feinstein, S.; Rinat, C.; Frishberg, Y. Congenital analbuminemia with acute glomerulonephritis: A diagnostic challenge. Pediatr. Nephrol 2009, 24, 403–406. [Google Scholar]
- Galliano, M.; Campagnoli, M.; Rossi, A.; Wirsing von König, C.H.; Lyon, AW; Cefle, K; Yildiz, A; Palanduz, S; Ozturk, S; Minchiotti, L. Molecular diagnosis of analbuminemia: A novel mutation identified in two Amerindian and two Turkish families. Clin. Chem 2002, 48, 844–849. [Google Scholar]
- Gianazza, E.; Astrua-Testori, S.; Giacon, P.; Righetti, P.G. An improved protocol for 2D maps of serum proteins with immobilized pH gradients in the first dimension. Electrophoresis 1985, 6, 332–339. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Analyte | Proband | Units | Normal Reference Range |
|---|---|---|---|
| albumin (relative) | 1.7 | % | 56.8–66.2 |
| albumin (absolute) | 0.9 | g/L | 35.0–52.0 |
| α1 globulins (relative) | 12.3 | % | 3.0–5.3 |
| α1 globulins (absolute) | 6.4 | g/L | 2.3–3.7 |
| α2 globulins (relative) | 29.0 | % | 7.2–11.4 |
| α2 globulins (absolute) | 15.0 | g/L | 5.2–8.6 |
| β globulins (relative) | 25.5 | % | 8.7–12.7 |
| β globulins (absolute) | 13.2 | g/L | 6.4–10.0 |
| γ globulins (relative) | 31.5 | % | 11.0–18.7 |
| γ globulins (absolute) | 16.3 | g/L | 3.8–7.5 |
| Total protein (absolute) | 51,8 | g/L | 60.0–81.0 |
| Total cholesterol | 8.43 | mmol/L | <5.0 |
| LDL cholesterol | 5.91 | mmol/L | <3.0 |
| HDL cholesterol | 1.87 | mmol/L | >1.0 |
| Triglycerides | 0.84 | mmol/L | <2.0 |
| Calcium | 1.94 | mmol/L | 2.20–2.65 |
| Phosphate | 1.72 | mmol/L | 0.81–1.61 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dagnino, M.; Caridi, G.; Haenni, U.; Duss, A.; Aregger, F.; Campagnoli, M.; Galliano, M.; Minchiotti, L. Molecular Diagnosis of Analbuminemia: A New Case Caused by a Nonsense Mutation in the Albumin Gene. Int. J. Mol. Sci. 2011, 12, 7314-7322. https://doi.org/10.3390/ijms12117314
Dagnino M, Caridi G, Haenni U, Duss A, Aregger F, Campagnoli M, Galliano M, Minchiotti L. Molecular Diagnosis of Analbuminemia: A New Case Caused by a Nonsense Mutation in the Albumin Gene. International Journal of Molecular Sciences. 2011; 12(11):7314-7322. https://doi.org/10.3390/ijms12117314
Chicago/Turabian StyleDagnino, Monica, Gianluca Caridi, Ueli Haenni, Adrian Duss, Fabienne Aregger, Monica Campagnoli, Monica Galliano, and Lorenzo Minchiotti. 2011. "Molecular Diagnosis of Analbuminemia: A New Case Caused by a Nonsense Mutation in the Albumin Gene" International Journal of Molecular Sciences 12, no. 11: 7314-7322. https://doi.org/10.3390/ijms12117314
APA StyleDagnino, M., Caridi, G., Haenni, U., Duss, A., Aregger, F., Campagnoli, M., Galliano, M., & Minchiotti, L. (2011). Molecular Diagnosis of Analbuminemia: A New Case Caused by a Nonsense Mutation in the Albumin Gene. International Journal of Molecular Sciences, 12(11), 7314-7322. https://doi.org/10.3390/ijms12117314