Comparison of Free Energy Surfaces Calculations from Ab Initio Molecular Dynamic Simulations at the Example of Two Transition Metal Catalyzed Reactions

Abstract
:1. Introduction
2. Methodological Section
2.1. Computational Details
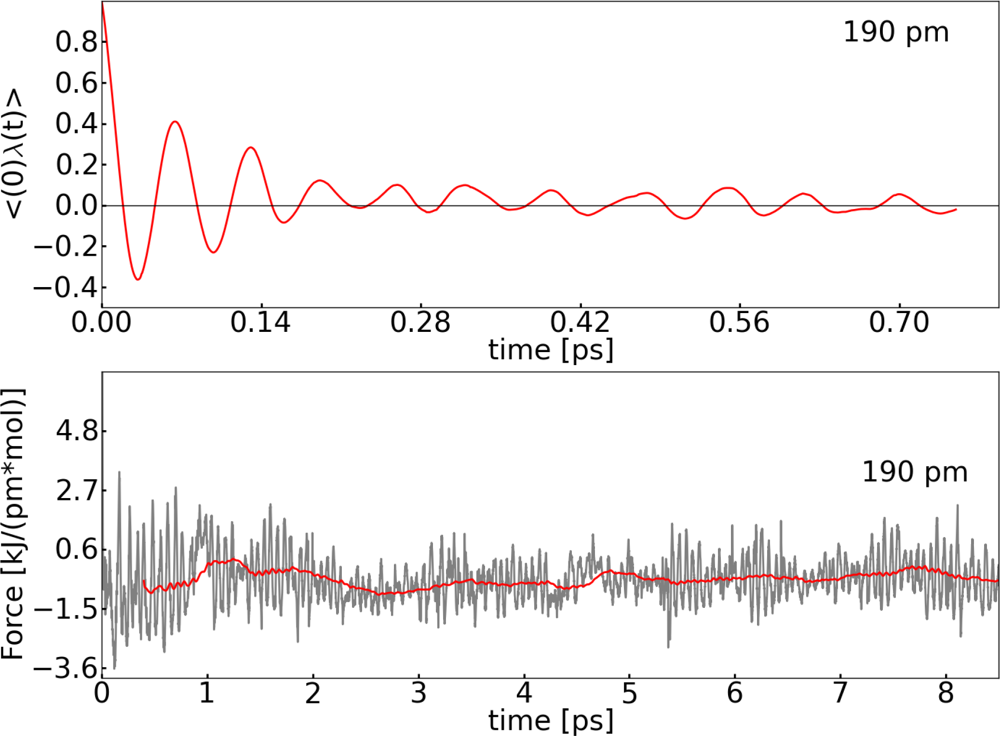
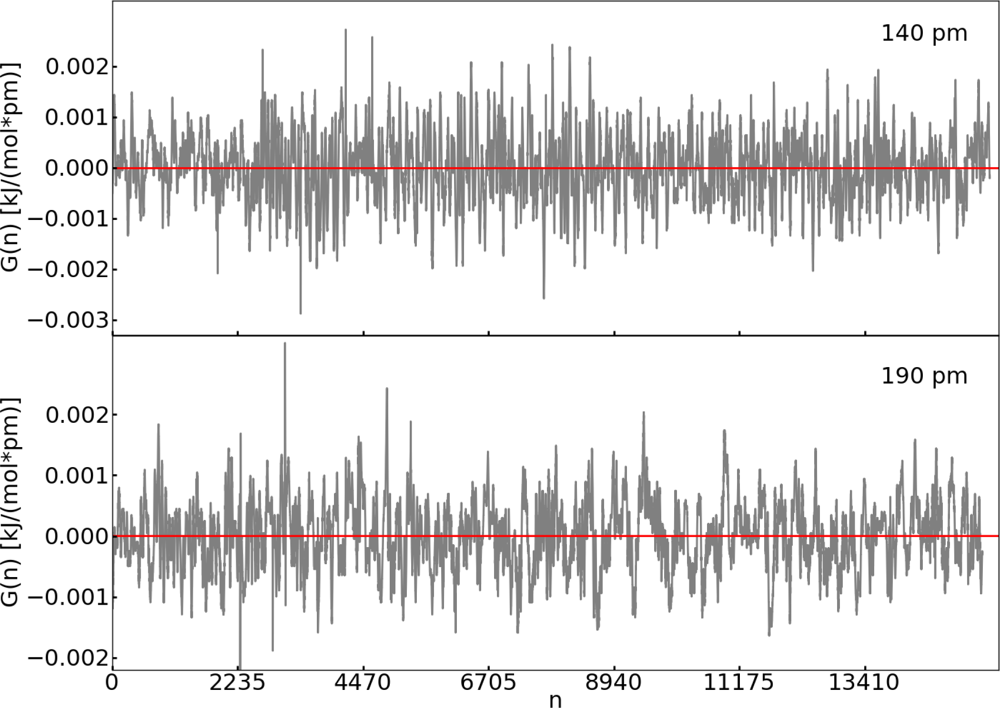
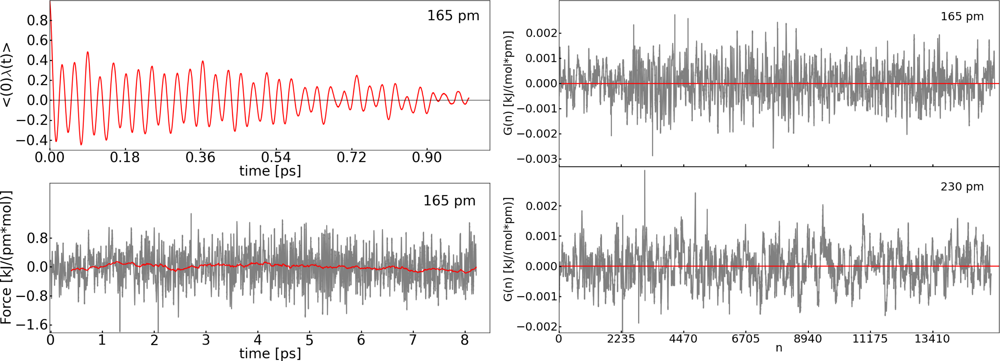
2.2. Thermodynamic Integration
2.3. Metadynamics
3. Results and Discussion
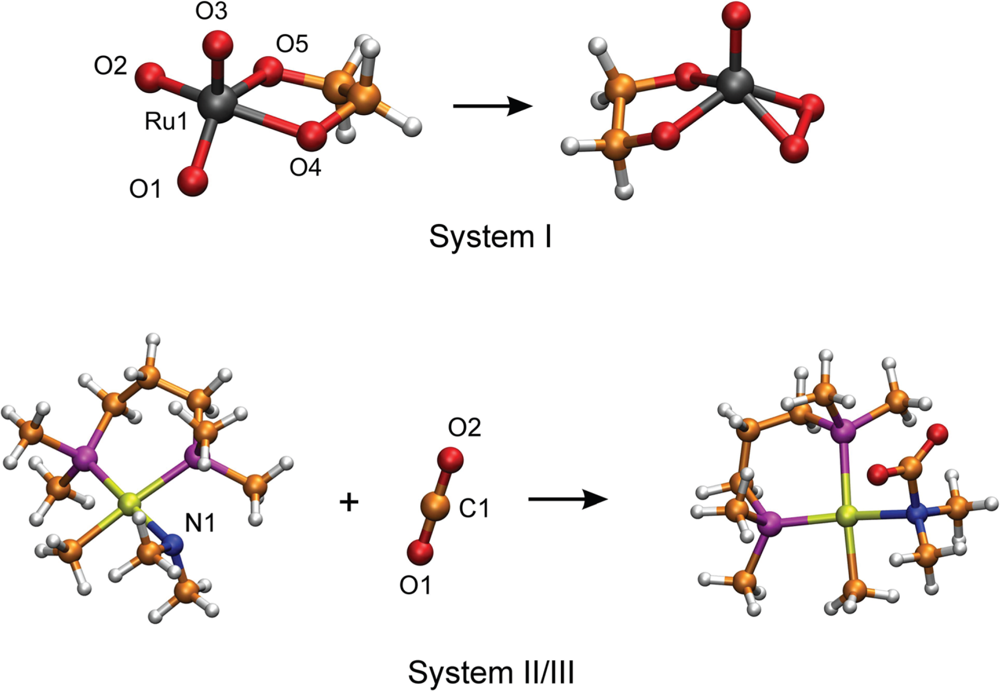
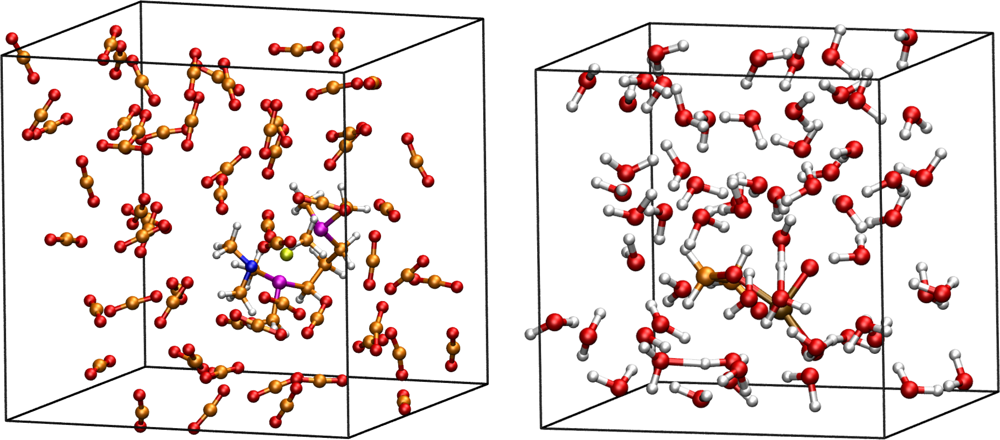
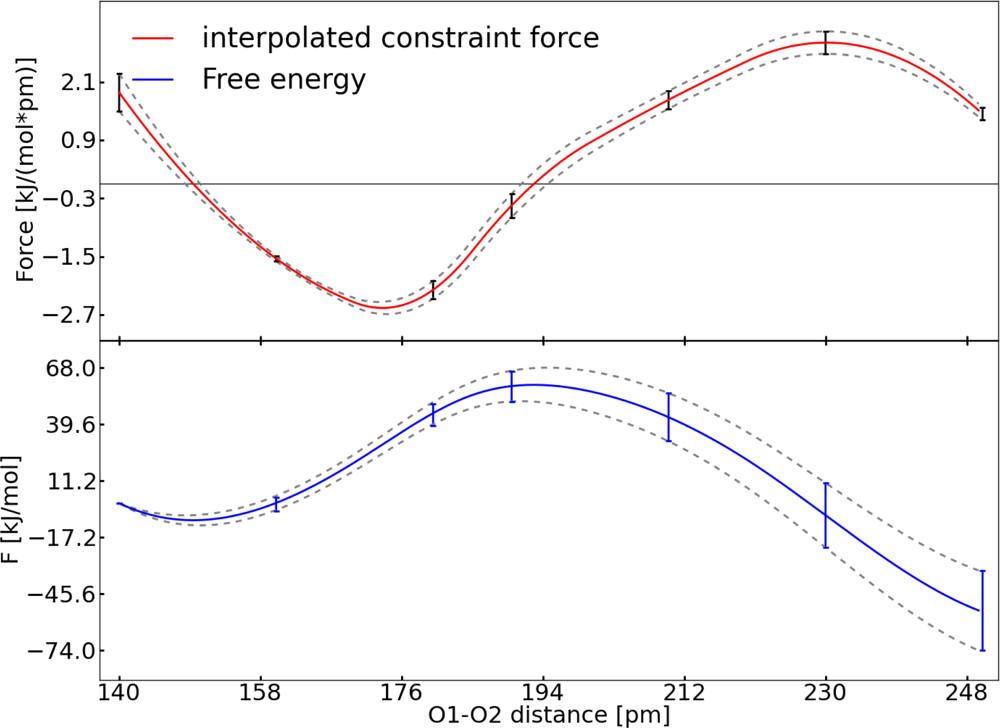
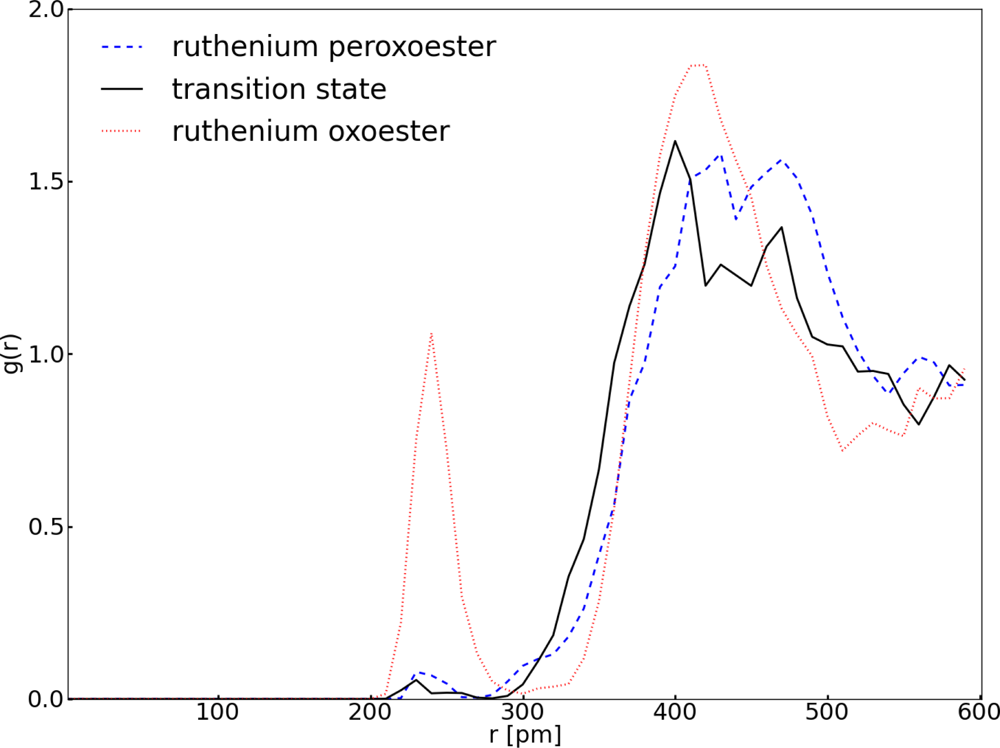
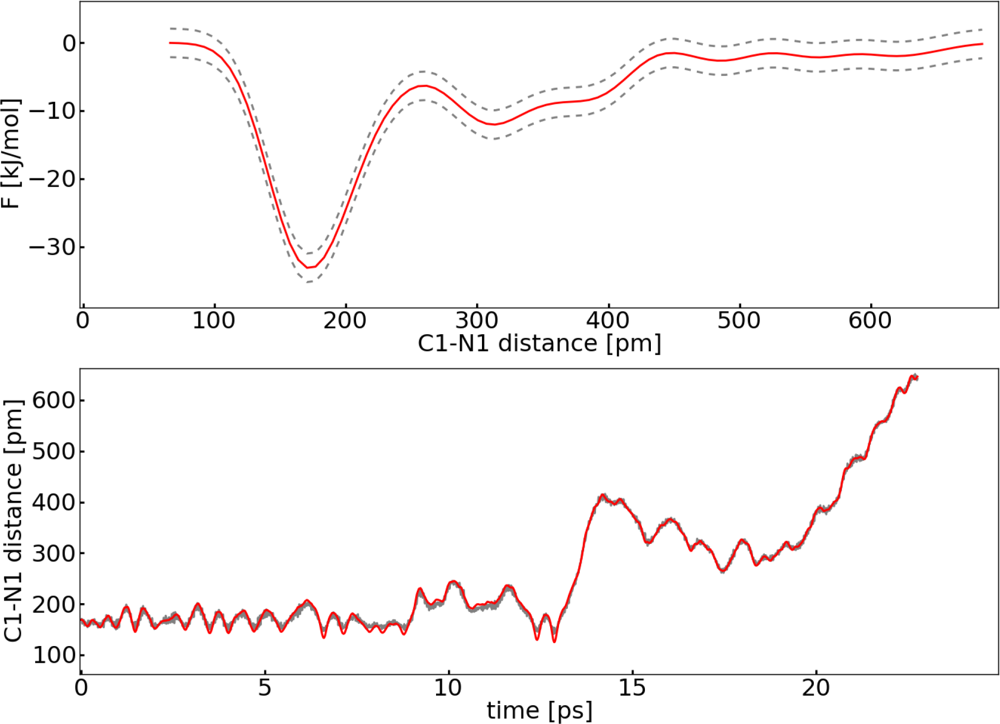
3.1. Thermodynamic Integration of the Ruthenium Peroxo Rearrangement with a Water Box (System I)
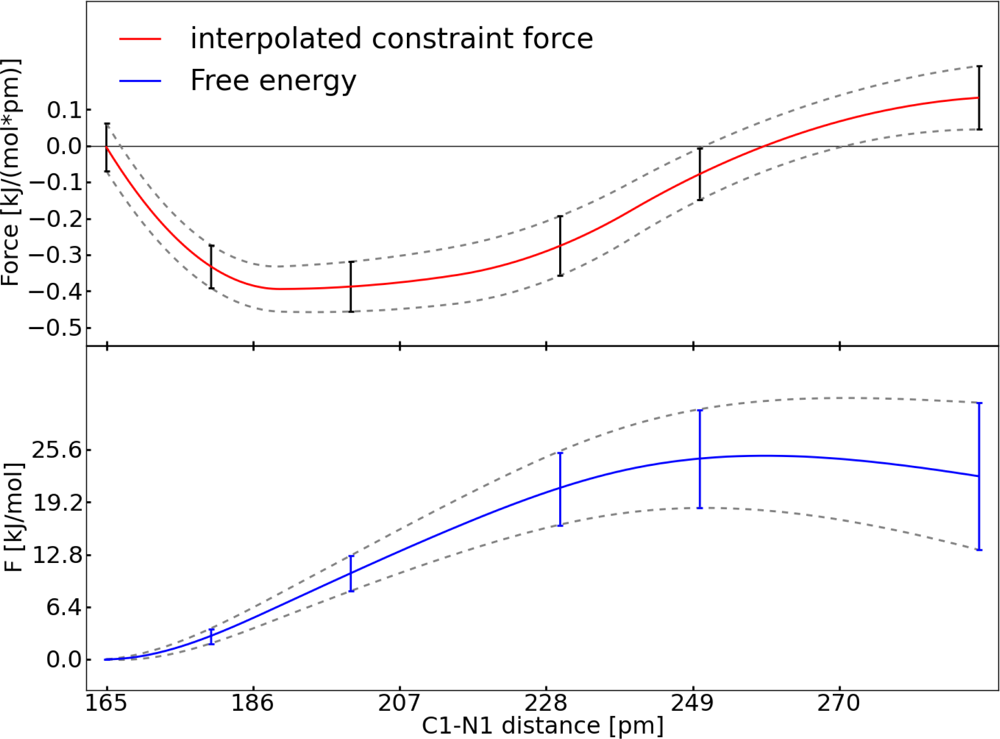
3.2. Thermodynamic Integration of Carbon Dioxide Addition to the Palladium Complex without Solvent (System II)
3.3. Metadynamic Simulations of the Carbon Dioxide Addition to the Palladium Complex without Solvent (system II) and one Collective Variable
3.4. Metadynamics Simulations of the Carbon Dioxide Addition to the Palladium Complex without Solvent (System II) and Two Collective Variables
3.5. Metadynamic Simulations of the Carbon Dioxide Addition to the Palladium Complex with a Carbon Dioxide Box (System III) and One Collective Variable
4. Summary and Conclusions
Acknowledgments
References
- Chipot, C; Pohorille, A. Free Energy Calculations Theorey and Applications in Chemistry and Biology; Springer: Berlin, Heidelberg, Germany, 2007. [Google Scholar]
- Frenkel, D; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications; Academic Press: Orlando, FL, USA, 2002. [Google Scholar]
- Tuckerman, ME. Statistical Mechanics: Theory and Molecular Simulations; Oxford University Press: New York, NY, USA, 2010. [Google Scholar]
- Ciccotti, G; Kapral, R; Vanden-Eijnden, E. Blue moon sampling, vectorial reaction coordinates, and unbiased constrained dynamics. Chem. Phys. Chem 2005, 6, 1809–1814. [Google Scholar]
- Coluzza, I; Sprik, M; Ciccotti, G. Constrained reaction coordinate dynamics for systems with constraints. Phys. Lett 2003, 101, 2885–2894. [Google Scholar]
- Carter, E; Ciccotti, G; Hynes, J; Kapral, R. Constrained reaction coordinate dynamics for the simulation of rare events. Chem. Phys. Lett 1989, 156, 472–477. [Google Scholar]
- Laio, A; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar]
- Michel, C; Laio, A; Mohamed, F; Krack, M; Parrinello, M; Milet, A. Free energy ab initio metadynamics: A new tool for the theoretical study of organometallic reactivity? Example of the C–C and C–H reductive eliminations from platinum(IV) complexes. Organometeallics 2007, 26, 1241–1249. [Google Scholar]
- Urakawa, A; Iannuzzi, M; Hutter, J; Baiker, A. Towards a rational design of ruthenium CO2 hydrogenation catalysts by ab initio metadynamics. Chem. Eur. J 2007, 13, 6828–6840. [Google Scholar]
- Buehl, M; Schreckenbach, G. Oxygen Exchange in Uranyl Hydroxide via Two “Nonclassical” Ions. Inorg. Chem 2010, 49, 3821–3827. [Google Scholar]
- Straatsma, T; Berendsen, H. Free energy of ionic hydration: Analysis of a thermodynamic integration technique to evaluate free energy differences by molecular dynamics simulations. J. Chem. Phys 1988, 89, 5876–5886. [Google Scholar]
- Carlsen, P; Katsuki, T; Martin, V; Sharpless, K. A greatly improved procedure for ruthenium tetraoxide catalyzed oxisations of organic-compounds. J. Org. Chem 1981, 46, 3936–3938. [Google Scholar]
- Djerassi, C; Engle, R. Oxidations with ruthenium tetraoxide. J. Am. Chem. Soc 1953, 75, 3838–3840. [Google Scholar]
- Shing, T; Tai, V; Tam, E. Practical and rapid vicinal hydroxylation of alkenes by catalytic ruthenium tetraoxide. Angew. Chem. Int. Ed 1994, 33, 2312–2313. [Google Scholar]
- di Dio, PJ; Zahn, S; Stark, CBW; Kirchner, B. Understanding Selectivities in Ligand-free Oxidative Cyclizations of 1,5-and 1,6-Dienes with RuO4 from Density Functional Theory. Z. Naturforsch 2010, 65b, 367–375. [Google Scholar]
- Sakamoto, M; Shimizu, I; Yamamoto, A. Synthesis of the 1st carbon-dioxide coordinated palladium(0) complex, Pd(ETA-2-CO2)(PMEPH2)2. Organometallics 1994, 13, 407. [Google Scholar]
- Tolman, WB. Activation of Small Molecules; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Aresta, M. Carbon Dioxide as Chemical Feedstock; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Brüssel, M; Zahn, S; Hey-Hawkins, E; Kirchner, B. Theoretical Investigation of Solvent Effects and Complex Systems: Toward the calculations of bioinorganic systems from ab initio molecular dynamics simulations and static quantum chemistry. Adv. Inorg. Chem 2010, 62, 111–142. [Google Scholar]
- Huber, H; Dyson, AJ; Kirchner, B. Calculation of bulk properties of liquids and supercritical fluids from pure theory. Chem. Soc. Rev 1999, 28, 121–133. [Google Scholar]
- Kirchner, B. Theory of complicated liquids. Phys Rep 2007, (1–3), 1–111. [Google Scholar]
- Kirchner, B. Cooperative versus dispersion effects: What is more important in an associated liquid such as water? J. Chem. Phys 2005, 123, 204116. [Google Scholar]
- Lippert, G; Hutter, J; Parrinello, M. The Gaussian and augmented-plane-wave density functional method for ab initio molecular dynamics simulations. Theor. Chem. Acc 1999, 103, 124–140. [Google Scholar]
- CP2k Developers Home Page. CP2k developers group under the terms of the GNU General Public License. Avaiable online: http://cp2k.berlios.de (accessed on May 24 2010).
- Nose, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys 1984, 81, 511–519. [Google Scholar]
- Martyna, G; Klein, M; Tuckerman, M. Nose–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys 1992, 97, 2635–2643. [Google Scholar]
- Hoover, W. Canonical dynamics: Equilibrium phase space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar]
- Goedecker, S; Teter, M; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar]
- Hartwigsen, C; Goedecker, S; Hutter, J. Relativistic separable dual-space Gaussian pseudopotentials from H to Rn. Phys. Rev. B 1998, 58, 3641–3662. [Google Scholar]
- van de Vondele, J; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys 2007, 127, 114105. [Google Scholar] [Green Version]
- Lee, C; Yang, W; Parr, RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Becke, AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comp. Chem 2006, 27, 1787–1799. [Google Scholar]
- Lindahl, E; Hess, B; van der Spoel, D. A package for molecular simulation and trajectory analysis. J. Mol. Mod 2001, 7, 306–317. [Google Scholar]
- Perdew, J. Density-Functional approximation for the correlation energy of the inhomogenous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys 2006, 8, 1057–1065. [Google Scholar]
- Baerends, EJ; Ellis, DE; Ros, P. Self-consistent molecular Hartree-Fock-Slater calculations—I. The computational procedure. Chem. Phys 1973, 2, 41–51. [Google Scholar]
- Dunlap, B; Connolly, J; Sabin, J. Some approximations in applications of X-alpha theory. J. Chem. Phys 1979, 71, 3396–3402. [Google Scholar]
- Andrae, D; Haussermann, U; Dolg, M; Stoll, H; Preuss, H. Energy-adjusted ab initio pseudopotentials for the 2nd-row and 3rd-row transition-elements Molecular test for Ag2, Au2 and RuH, OsH. Theor. Chim. Acta 1991, 78, 247–266. [Google Scholar]
- Weigend, F; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys 2005, 7, 3297–3305. [Google Scholar]
- Neugebauer, J; Reiher, M; Kind, C; Hess, B. Quantum chemical calculation of vibrational spectra of large molecules—Raman and IR spectra for buckminsterfullerene. J. Comp. Chem 2002, 23, 895–910. [Google Scholar]
- Hunter, JD. Matplotlib: A 2D graphics environment. Comput. Sci. Eng 2007, 9, 90–95. [Google Scholar]
- Sprik, M; Ciccotti, G. Free energy from constrained molecular dynamics. J. Chem. Phys 1998, 109, 7737–7744. [Google Scholar]
- Iannuzzi, M; Laio, A; Parrinello, M. Efficient exploration of reactive potential energy surfaces using Car-Parrinello molecular dynamics. Phys. Rev. Lett 2003, 90, 238302. [Google Scholar]
- Ensing, B; Laio, A; Parrinello, M; Klein, M. A recipe for the computation of the free energy barrier and the lowest free energy path of concerted reactions. J. Phys. Chem. B 2005, 109, 6676–6687. [Google Scholar]
- Laio, A; Rodriguez-Fortea, A; Gervasio, F; Ceccarelli, M; Parrinello, M. Assessing the accuracy of metadynamics. J. Phys. Chem. B 2005, 109, 6714–6721. [Google Scholar]
- Isayev, O; Gorb, L; Leszczynski, J. Theoretical calculations: Can Gibbs free energy for intermolecular complexes be predicted efficiently and accurately? J. Comp. Chem 2007, 28, 1598–1609. [Google Scholar]
- Barducci, A; Bussi, G; Parrinello, M. Well-tempered metadynamics: A smoothly converging and tunable free-energy method. Phys. Rev. Lett 2008, 100, 020603. [Google Scholar]
- Bonomi, M; Barducci, A; Parrinello, M. Reconstructing the Equilibrium Boltzmann Distribution from Well-Tempered Metadynamics. J. Comp. Chem 2009, 30, 1615–1621. [Google Scholar]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distance (pm) | 〈M(t)〉 (kJ pm−1 mol−1) | 〈G(n)〉 (kJ pm−1 mol−1) |
|---|---|---|
| 140 | 1.9 | 5.6 × 10−5 |
| 160 | −1.5 | −1.9 × 10−6 |
| 180 | −2.2 | −3.6 × 10−5 |
| 190 | −0.5 | 3.2 × 10−6 |
| 210 | 1.7 | 8.6 × 10−5 |
| 230 | 2.9 | 9.5 × 10−6 |
| 250 | 1.4 | 5.5 × 10−6 |
| Distance (pm) | Ru1–O1 | Ru1–O2 | Ru1–O3 | Ru1–O4 | Ru1–O5 |
|---|---|---|---|---|---|
| Ruthenium peroxoester | 194 | 195 | 168 | 192 | 192 |
| Transition state | 190 | 193 | 169 | 191 | 193 |
| Ruthenium oxoester | 172 | 172 | 174 | 202 | 201 |
| Angle (deg) | O4–Ru1–O5 | O1–Ru1–O3 | O2–Ru1–O3 | ||
| Ruthenium peroxoester | 84 | 119 | 118 | ||
| Transition state | 83 | 114 | 113 | ||
| Ruthenium oxoester | 77 | 105 | 105 | ||
| 〈(sm − Sm(Ri))2〉 (pm2) | 〈(Sm(Ri) − 〈Sm(Ri)〉)〉 (pm2) | (pm2) | (kJ mol−1 pm−1) | FSm (kJ mol−1 pm−1) | |
|---|---|---|---|---|---|
| Set I | 25.4 | 44.5 | 33.3 | 7.5 × 10−3 | 9.5 × 10−1 |
| Set II | 57.6 | 51.9 | 2659.1 | 7.5 × 10−3 | 7.5 × 10−1 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Brüssel, M.; Di Dio, P.J.; Muñiz, K.; Kirchner, B. Comparison of Free Energy Surfaces Calculations from Ab Initio Molecular Dynamic Simulations at the Example of Two Transition Metal Catalyzed Reactions. Int. J. Mol. Sci. 2011, 12, 1389-1409. https://doi.org/10.3390/ijms12021389
Brüssel M, Di Dio PJ, Muñiz K, Kirchner B. Comparison of Free Energy Surfaces Calculations from Ab Initio Molecular Dynamic Simulations at the Example of Two Transition Metal Catalyzed Reactions. International Journal of Molecular Sciences. 2011; 12(2):1389-1409. https://doi.org/10.3390/ijms12021389
Chicago/Turabian StyleBrüssel, Marc, Philipp J. Di Dio, Kilian Muñiz, and Barbara Kirchner. 2011. "Comparison of Free Energy Surfaces Calculations from Ab Initio Molecular Dynamic Simulations at the Example of Two Transition Metal Catalyzed Reactions" International Journal of Molecular Sciences 12, no. 2: 1389-1409. https://doi.org/10.3390/ijms12021389
APA StyleBrüssel, M., Di Dio, P. J., Muñiz, K., & Kirchner, B. (2011). Comparison of Free Energy Surfaces Calculations from Ab Initio Molecular Dynamic Simulations at the Example of Two Transition Metal Catalyzed Reactions. International Journal of Molecular Sciences, 12(2), 1389-1409. https://doi.org/10.3390/ijms12021389