Epigenetic Regulation of Cancer-Associated Genes in Ovarian Cancer



Abstract
:
1. Introduction
2. Epigenetic Changes in Cancer
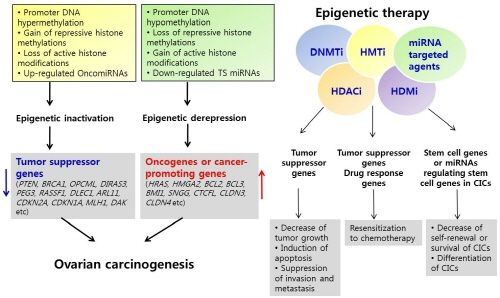
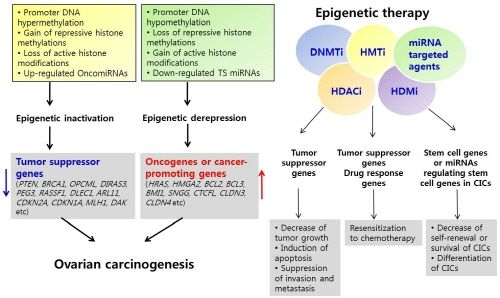
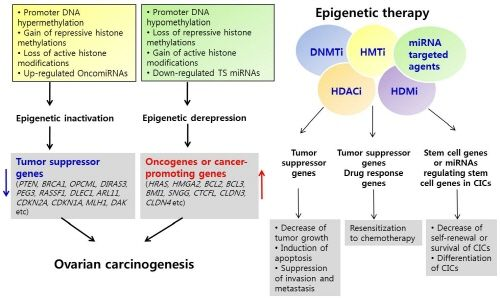
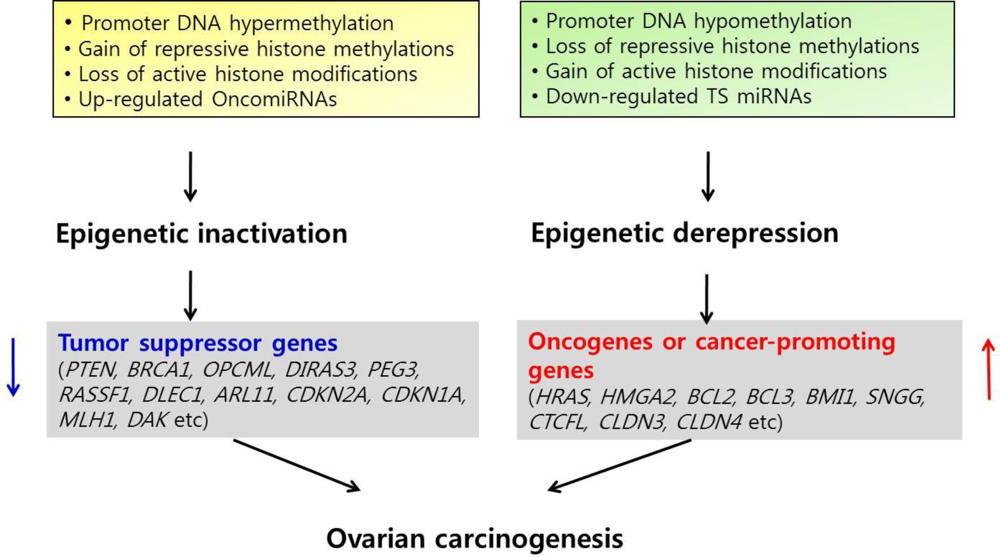
3. Epigenetic Inactivation of Tumor Suppressor Genes or Cancer-Associated Genes in Ovarian Cancer
4. Epigenetic Derepression of Oncogenes or Cancer-Promoting Genes in Ovarian Cancer
4.1. Activation of Oncogenes or Cancer-Promoting Genes by DNA Hypomethylation
4.2. Activation of Oncogenes or Cancer-Promoting Genes by Chromatin Modification
4.3. Regulation of Oncogenes or Cancer-Promoting Genes by miRNAs in Ovarian Cancer
5. Epigenetics of Cancer-Initiating Cells in Ovarian Cancer
6. Epigenetics and Drug Resistance in Ovarian Cancer
7. Epigenetic Therapy
8. Epigenetic Therapy in Ovarian Cancer
9. Conclusions
Acknowledgments
References
- Jemal, A; Siegel, R; Xu, J; Ward, E. Cancer statistics, 2010. CA Cancer J Clin 2010, 60(5), 277–300. [Google Scholar]
- Hennessy, BT; Coleman, RL; Markman, M. Ovarian cancer. Lancet 2009, 374(9698), 1371–1382. [Google Scholar]
- Cho, KR; Shih, IM. Ovarian cancer. Annu. Rev. Pathol 2009, 4, 287–313. [Google Scholar]
- Bast, RC, Jr; Hennessy, B; Mills, GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer 2009, 9(6), 415–428. [Google Scholar]
- Yap, TA; Carden, CP; Kaye, SB. Beyond chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer 2009, 9(3), 167–181. [Google Scholar]
- Feinberg, AP; Ohlsson, R; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat Rev Genet 2006, 7(1), 21–33. [Google Scholar]
- Sharma, S; Kelly, TK; Jones, PA. Epigenetics in cancer. Carcinogenesis 2010, 31(1), 27–36. [Google Scholar]
- Balch, C; Fang, F; Matei, DE; Huang, TH; Nephew, KP. Epigenetic Changes in Ovarian Cancer. Endocrinology 2009, 150, 4003–4011. [Google Scholar]
- Vogelstein, B; Kinzler, KW. Cancer genes and the pathways they control. Nat Med 2004, 10(8), 789–799. [Google Scholar]
- Croce, CM. Oncogenes and cancer. N Engl J Med 2008, 358(5), 502–511. [Google Scholar]
- Iacobuzio-Donahue, CA. Epigenetic changes in cancer. Annu. Rev. Pathol 2009, 4, 229–249. [Google Scholar]
- Esteller, M. Epigenetics in cancer. N Engl J Med 2008, 358(11), 1148–1159. [Google Scholar]
- Garzon, R; Calin, GA; Croce, CM. MicroRNAs in Cancer. Annu. Rev. Med 2009, 60, 167–179. [Google Scholar]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet 2007, 8(4), 286–298. [Google Scholar]
- Lamprecht, B; Walter, K; Kreher, S; Kumar, R; Hummel, M; Lenze, D; Kochert, K; Bouhlel, MA; Richter, J; Soler, E; et al. Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nat Med 2010, 16(5), 571–579. [Google Scholar]
- Turner, BM. Cellular memory and the histone code. Cell 2002, 111(3), 285–291. [Google Scholar]
- Turner, BM. Reading signals on the nucleosome with a new nomenclature for modified histones. Nat Struct Mol Biol 2005, 12(2), 110–112. [Google Scholar]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128(4), 693–705. [Google Scholar]
- Lan, F; Nottke, AC; Shi, Y. Mechanisms involved in the regulation of histone lysine demethylases. Curr Opin Cell Biol 2008, 20(3), 316–325. [Google Scholar]
- Bernstein, BE; Mikkelsen, TS; Xie, X; Kamal, M; Huebert, DJ; Cuff, J; Fry, B; Meissner, A; Wernig, M; Plath, K; Jaenisch, R; Wagschal, A; Feil, R; Schreiber, SL; Lander, ES. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125(2), 315–326. [Google Scholar]
- Golebiewska, A; Atkinson, SP; Lako, M; Armstrong, L. Epigenetic landscaping during hESC differentiation to neural cells. Stem Cells 2009, 27(6), 1298–1308. [Google Scholar]
- Fuks, F. DNA methylation and histone modifications: teaming up to silence genes. Curr Opin Genet Dev 2005, 15(5), 490–495. [Google Scholar]
- Simon, JA; Lange, CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res 2008, 647(1–2), 21–29. [Google Scholar]
- Vaissiere, T; Sawan, C; Herceg, Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res 2008, 659(1–2), 40–48. [Google Scholar]
- Abbosh, PH; Montgomery, JS; Starkey, JA; Novotny, M; Zuhowski, EG; Egorin, MJ; Moseman, AP; Golas, A; Brannon, KM; Balch, C; Huang, TH; Nephew, KP. Dominant-negative histone H3 lysine 27 mutant derepresses silenced tumor suppressor genes and reverses the drug-resistant phenotype in cancer cells. Cancer Res 2006, 66(11), 5582–5591. [Google Scholar]
- Schlesinger, Y; Straussman, R; Keshet, I; Farkash, S; Hecht, M; Zimmerman, J; Eden, E; Yakhini, Z; Ben-Shushan, E; Reubinoff, BE; Bergman, Y; Simon, I; Cedar, H. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet 2007, 39(2), 232–236. [Google Scholar]
- Vire, E; Brenner, C; Deplus, R; Blanchon, L; Fraga, M; Didelot, C; Morey, L; Van Eynde, A; Bernard, D; Vanderwinden, JM; Bollen, M; Esteller, M; Di Croce, L; de Launoit, Y; Fuks, F. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439(7078), 871–874. [Google Scholar]
- Fuks, F; Burgers, WA; Brehm, A; Hughes-Davies, L; Kouzarides, T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet 2000, 24(1), 88–91. [Google Scholar]
- Rountree, MR; Bachman, KE; Baylin, SB. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat Genet 2000, 25(3), 269–277. [Google Scholar]
- Smallwood, A; Esteve, PO; Pradhan, S; Carey, M. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev 2007, 21(10), 1169–1178. [Google Scholar]
- van der Vlag, J; Otte, AP. Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nat Genet 1999, 23(4), 474–478. [Google Scholar]
- Fraga, MF; Ballestar, E; Villar-Garea, A; Boix-Chornet, M; Espada, J; Schotta, G; Bonaldi, T; Haydon, C; Ropero, S; Petrie, K; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 2005, 37(4), 391–400. [Google Scholar]
- Ventura, A; Jacks, T. MicroRNAs and cancer: short RNAs go a long way. Cell 2009, 136(4), 586–591. [Google Scholar]
- Johnson, SM; Grosshans, H; Shingara, J; Byrom, M; Jarvis, R; Cheng, A; Labourier, E; Reinert, KL; Brown, D; Slack, FJ. RAS is regulated by the let-7 microRNA family. Cell 2005, 120(5), 635–647. [Google Scholar]
- Lee, YS; Dutta, A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev 2007, 21(9), 1025–1030. [Google Scholar]
- Sampson, VB; Rong, NH; Han, J; Yang, Q; Aris, V; Soteropoulos, P; Petrelli, NJ; Dunn, SP; Krueger, LJ. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res 2007, 67(20), 9762–9770. [Google Scholar]
- Cimmino, A; Calin, GA; Fabbri, M; Iorio, MV; Ferracin, M; Shimizu, M; Wojcik, SE; Aqeilan, RI; Zupo, S; Dono, M; Rassenti, L; Alder, H; Volinia, S; Liu, CG; Kipps, TJ; Negrini, M; Croce, CM. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA 2005, 102(39), 13944–13949. [Google Scholar]
- Meng, F; Henson, R; Wehbe-Janek, H; Ghoshal, K; Jacob, ST; Patel, T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007, 133(2), 647–658. [Google Scholar]
- He, L; He, X; Lim, LP; de Stanchina, E; Xuan, Z; Liang, Y; Xue, W; Zender, L; Magnus, J; Ridzon, D; Jackson, AL; Linsley, PS; Chen, C; Lowe, SW; Cleary, MA; Hannon, GJ. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar]
- Raver-Shapira, N; Marciano, E; Meiri, E; Spector, Y; Rosenfeld, N; Moskovits, N; Bentwich, Z; Oren, M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell 2007, 26(5), 731–743. [Google Scholar]
- Chang, TC; Wentzel, EA; Kent, OA; Ramachandran, K; Mullendore, M; Lee, KH; Feldmann, G; Yamakuchi, M; Ferlito, M; Lowenstein, CJ; Arking, DE; Beer, MA; Maitra, A; Mendell, JT. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 2007, 26(5), 745–752. [Google Scholar]
- O’Donnell, KA; Wentzel, EA; Zeller, KI; Dang, CV; Mendell, JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005, 435(7043), 839–843. [Google Scholar]
- Chang, TC; Yu, D; Lee, YS; Wentzel, EA; Arking, DE; West, KM; Dang, CV; Thomas-Tikhonenko, A; Mendell, JT. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet 2008, 40(1), 43–50. [Google Scholar]
- Fabbri, M; Garzon, R; Cimmino, A; Liu, Z; Zanesi, N; Callegari, E; Liu, S; Alder, H; Costinean, S; Fernandez-Cymering, C; Volinia, S; Guler, G; Morrison, CD; Chan, KK; Marcucci, G; Calin, GA; Huebner, K; Croce, CM. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci USA 2007, 104(40), 15805–15810. [Google Scholar]
- Friedman, JM; Liang, G; Liu, CC; Wolff, EM; Tsai, YC; Ye, W; Zhou, X; Jones, PA. The putative tumor suppressor microRNA-101 modulates the cancer epigenome by repressing the polycomb group protein EZH2. Cancer Res 2009, 69(6), 2623–2629. [Google Scholar]
- Ma, L; Teruya-Feldstein, J; Weinberg, RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007, 449(7163), 682–688. [Google Scholar]
- Huang, Q; Gumireddy, K; Schrier, M; le Sage, C; Nagel, R; Nair, S; Egan, DA; Li, A; Huang, G; Klein-Szanto, AJ; Gimotty, PA; Katsaros, D; Coukos, G; Zhang, L; Pure, E; Agami, R. The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat Cell Biol 2008, 10(2), 202–210. [Google Scholar]
- Schondorf, T; Ebert, MP; Hoffmann, J; Becker, M; Moser, N; Pur, S; Gohring, UJ; Weisshaar, MP. Hypermethylation of the PTEN gene in ovarian cancer cell lines. Cancer Lett 2004, 207(2), 215–220. [Google Scholar]
- Yang, H; Kong, W; He, L; Zhao, JJ; O’Donnell, JD; Wang, J; Wenham, RM; Coppola, D; Kruk, PA; Nicosia, SV; Cheng, JQ. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res 2008, 68(2), 425–433. [Google Scholar]
- Baldwin, RL; Nemeth, E; Tran, H; Shvartsman, H; Cass, I; Narod, S; Karlan, BY. BRCA1 promoter region hypermethylation in ovarian carcinoma: a population-based study. Cancer Res 2000, 60(19), 5329–5333. [Google Scholar]
- Feng, W; Marquez, RT; Lu, Z; Liu, J; Lu, KH; Issa, JP; Fishman, DM; Yu, Y; Bast, RC. Imprinted tumor suppressor genes ARHI and PEG3 are the most frequently down-regulated in human ovarian cancers by loss of heterozygosity and promoter methylation. Cancer 2008, 112(7), 1489–1502. [Google Scholar]
- Chan, MW; Huang, YW; Hartman-Frey, C; Kuo, CT; Deatherage, D; Qin, H; Cheng, AS; Yan, PS; Davuluri, RV; Huang, TH; Nephew, KP; Lin, HJ. Aberrant transforming growth factor beta1 signaling and SMAD4 nuclear translocation confer epigenetic repression of ADAM19 in ovarian cancer. Neoplasia 2008, 10(9), 908–919. [Google Scholar]
- Caslini, C; Capo-chichi, CD; Roland, IH; Nicolas, E; Yeung, AT; Xu, XX. Histone modifications silence the GATA transcription factor genes in ovarian cancer. Oncogene 2006, 25(39), 5446–5461. [Google Scholar]
- Iorio, MV; Visone, R; Di Leva, G; Donati, V; Petrocca, F; Casalini, P; Taccioli, C; Volinia, S; Liu, CG; Alder, H; Calin, GA; Menard, S; Croce, CM. MicroRNA signatures in human ovarian cancer. Cancer Res 2007, 67(18), 8699–6707. [Google Scholar]
- Nam, EJ; Yoon, H; Kim, SW; Kim, H; Kim, YT; Kim, JH; Kim, JW; Kim, S. MicroRNA expression profiles in serous ovarian carcinoma. Clin Cancer Res 2008, 14(9), 2690–2695. [Google Scholar]
- Bendoraite, A; Knouf, EC; Garg, KS; Parkin, RK; Kroh, EM; O’Briant, KC; Ventura, AP; Godwin, AK; Karlan, BY; Drescher, CW; Urban, N; Knudsen, BS; Tewari, M. Regulation of miR-200 family microRNAs and ZEB transcription factors in ovarian cancer: evidence supporting a mesothelial-to-epithelial transition. Gynecol Oncol 2010, 116(1), 117–125. [Google Scholar]
- Sellar, GC; Watt, KP; Rabiasz, GJ; Stronach, EA; Li, L; Miller, EP; Massie, CE; Miller, J; Contreras-Moreira, B; Scott, D; Brown, I; Williams, AR; Bates, PA; Smyth, JF; Gabra, H. OPCML at 11q25 is epigenetically inactivated and has tumor-suppressor function in epithelial ovarian cancer. Nat Genet 2003, 34(3), 337–343. [Google Scholar]
- Lu, Z; Luo, RZ; Peng, H; Rosen, DG; Atkinson, EN; Warneke, C; Huang, M; Nishmoto, A; Liu, J; Liao, WS; Yu, Y; Bast, RC. Transcriptional and posttranscriptional down-regulation of the imprinted tumor suppressor gene ARHI (DRAS3) in ovarian cancer. Clin Cancer Res 2006, 12(8), 2404–2413. [Google Scholar]
- Tobias, ES; Hurlstone, AF; MacKenzie, E; McFarlane, R; Black, DM. The TES gene at 7q31.1 is methylated in tumours and encodes a novel growth-suppressing LIM domain protein. Oncogene 2001, 20(22), 2844–2853. [Google Scholar]
- Qiu, H; Zhu, J; Yuan, C; Yan, S; Yang, Q; Kong, B. Frequent hypermethylation and loss of heterozygosity of the testis derived transcript gene in ovarian cancer. Cancer Sci 2010, 101(5), 1255–1260. [Google Scholar]
- Yanaihara, N; Nishioka, M; Kohno, T; Otsuka, A; Okamoto, A; Ochiai, K; Tanaka, T; Yokota, J. Reduced expression of MYO18B, a candidate tumor-suppressor gene on chromosome arm 22q, in ovarian cancer. Int J Cancer 2004, 112(1), 150–154. [Google Scholar]
- Ibanez de Caceres, I; Battagli, C; Esteller, M; Herman, JG; Dulaimi, E; Edelson, MI; Bergman, C; Ehya, H; Eisenberg, BL; Cairns, P. Tumor cell-specific BRCA1 and RASSF1A hypermethylation in serum, plasma, and peritoneal fluid from ovarian cancer patients. Cancer Res 2004, 64(18), 6476–6481. [Google Scholar]
- Kwong, J; Lee, JY; Wong, KK; Zhou, X; Wong, DT; Lo, KW; Welch, WR; Berkowitz, RS; Mok, SC. Candidate tumor-suppressor gene DLEC1 is frequently downregulated by promoter hypermethylation and histone hypoacetylation in human epithelial ovarian cancer. Neoplasia 2006, 8(4), 268–278. [Google Scholar]
- Petrocca, F; Iliopoulos, D; Qin, HR; Nicoloso, MS; Yendamuri, S; Wojcik, SE; Shimizu, M; Di Leva, G; Vecchione, A; Trapasso, F; Godwin, AK; Negrini, M; Calin, GA; Croce, CM. Alterations of the tumor suppressor gene ARLTS1 in ovarian cancer. Cancer Res 2006, 66(21), 10287–10291. [Google Scholar]
- Milde-Langosch, K; Ocon, E; Becker, G; Loning, T. p16/MTS1 inactivation in ovarian carcinomas: high frequency of reduced protein expression associated with hyper-methylation or mutation in endometrioid and mucinous tumors. Int J Cancer 1998, 79(1), 61–65. [Google Scholar]
- Katsaros, D; Cho, W; Singal, R; Fracchioli, S; Rigault De La Longrais, IA; Arisio, R; Massobrio, M; Smith, M; Zheng, W; Glass, J; Yu, H. Methylation of tumor suppressor gene p16 and prognosis of epithelial ovarian cancer. Gynecol Oncol 2004, 94(3), 685–692. [Google Scholar]
- Richon, VM; Sandhoff, TW; Rifkind, RA; Marks, PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci USA 2000, 97(18), 10014–10019. [Google Scholar]
- Zhang, H; Zhang, S; Cui, J; Zhang, A; Shen, L; Yu, H. Expression and promoter methylation status of mismatch repair gene hMLH1 and hMSH2 in epithelial ovarian cancer. Aust N Z J Obstet Gynaecol 2008, 48(5), 505–509. [Google Scholar]
- Meng, CF; Dai, DQ; Guo, KJ. Effects of 5-Aza-2′-deoxycytidine and trichostatin A on DNA methylation and expression of hMLH1 in ovarian cancer cell line COC1/DDP. Ai Zheng 2008, 27(12), 1251–1255. (in Chinese). [Google Scholar]
- Bai, T; Tanaka, T; Yukawa, K; Maeda, M; Umesaki, N. Reduced expression of death-associated protein kinase in human uterine and ovarian carcinoma cells. Oncol Rep 2004, 11(3), 661–665. [Google Scholar]
- Yuecheng, Y; Hongmei, L; Xiaoyan, X. Clinical evaluation of E-cadherin expression and its regulation mechanism in epithelial ovarian cancer. Clin Exp Metastasis 2006, 23(1), 65–74. [Google Scholar]
- Chou, JL; Su, HY; Chen, LY; Liao, YP; Hartman-Frey, C; Lai, YH; Yang, HW; Deatherage, DE; Kuo, CT; Huang, YW; et al. Promoter hypermethylation of FBXO32, a novel TGF-beta/SMAD4 target gene and tumor suppressor, is associated with poor prognosis in human ovarian cancer. Lab Invest 2010, 90(3), 414–425. [Google Scholar]
- Kikuchi, R; Tsuda, H; Kanai, Y; Kasamatsu, T; Sengoku, K; Hirohashi, S; Inazawa, J; Imoto, I. Promoter hypermethylation contributes to frequent inactivation of a putative conditional tumor suppressor gene connective tissue growth factor in ovarian cancer. Cancer Res 2007, 67(15), 7095–7105. [Google Scholar]
- Kikuchi, R; Tsuda, H; Kozaki, K; Kanai, Y; Kasamatsu, T; Sengoku, K; Hirohashi, S; Inazawa, J; Imoto, I. Frequent inactivation of a putative tumor suppressor, angiopoietin-like protein 2, in ovarian cancer. Cancer Res 2008, 68(13), 5067–5075. [Google Scholar]
- Arnold, JM; Cummings, M; Purdie, D; Chenevix-Trench, G. Reduced expression of intercellular adhesion molecule-1 in ovarian adenocarcinomas. Br J Cancer 2001, 85(9), 1351–1358. [Google Scholar]
- Fu, Y; Campbell, EJ; Shepherd, TG; Nachtigal, MW. Epigenetic regulation of proprotein convertase PACE4 gene expression in human ovarian cancer cells. Mol Cancer Res 2003, 1(8), 569–576. [Google Scholar]
- Czekierdowski, A; Czekierdowska, S; Wielgos, M; Smolen, A; Kaminski, P; Kotarski, J. The role of CpG islands hypomethylation and abnormal expression of neuronal protein synuclein-gamma (SNCG) in ovarian cancer. Neuro Endocrinol Lett 2006, 27(3), 381–386. [Google Scholar]
- Gupta, A; Godwin, AK; Vanderveer, L; Lu, A; Liu, J. Hypomethylation of the synuclein gamma gene CpG island promotes its aberrant expression in breast carcinoma and ovarian carcinoma. Cancer Res 2003, 63(3), 664–673. [Google Scholar]
- Woloszynska-Read, A; James, SR; Link, PA; Yu, J; Odunsi, K; Karpf, AR. DNA methylation-dependent regulation of BORIS/CTCFL expression in ovarian cancer. Cancer Immun 2007, 7, 21. [Google Scholar]
- Woloszynska-Read, A; James, SR; Song, C; Jin, B; Odunsi, K; Karpf, AR. BORIS/CTCFL expression is insufficient for cancer-germline antigen gene expression and DNA hypomethylation in ovarian cell lines. Cancer Immun 2010, 10, 6. [Google Scholar]
- Strathdee, G; Davies, BR; Vass, JK; Siddiqui, N; Brown, R. Cell type-specific methylation of an intronic CpG island controls expression of the MCJ gene. Carcinogenesis 2004, 25(5), 693–701. [Google Scholar]
- Strathdee, G; Vass, JK; Oien, KA; Siddiqui, N; Curto-Garcia, J; Brown, R. Demethylation of the MCJ gene in stage III/IV epithelial ovarian cancer and response to chemotherapy. Gynecol Oncol 2005, 97(3), 898–903. [Google Scholar]
- Lee, PS; Teaberry, VS; Bland, AE; Huang, Z; Whitaker, RS; Baba, T; Fujii, S; Secord, AA; Berchuck, A; Murphy, SK. Elevated MAL expression is accompanied by promoter hypomethylation and platinum resistance in epithelial ovarian cancer. Int J Cancer 2010, 126(6), 1378–1389. [Google Scholar]
- Cheng, W; Jiang, Y; Liu, C; Shen, O; Tang, W; Wang, X. Identification of aberrant promoter hypomethylation of HOXA10 in ovarian cancer. J Cancer Res Clin Oncol 2010, 136(8), 1221–1227. [Google Scholar]
- Izutsu, N; Maesawa, C; Shibazaki, M; Oikawa, H; Shoji, T; Sugiyama, T; Masuda, T. Epigenetic modification is involved in aberrant expression of class III beta-tubulin, TUBB3, in ovarian cancer cells. Int J Oncol 2008, 32(6), 1227–1235. [Google Scholar]
- Kwon, MJ; Kim, SS; Choi, YL; Jung, HS; Balch, C; Kim, SH; Song, YS; Marquez, VE; Nephew, KP; Shin, YK. Derepression of CLDN3 and CLDN4 during ovarian tumorigenesis is associated with loss of repressive histone modifications. Carcinogenesis 2010, 31(6), 974–983. [Google Scholar]
- Zhang, L; Volinia, S; Bonome, T; Calin, GA; Greshock, J; Yang, N; Liu, CG; Giannakakis, A; Alexiou, P; Hasegawa, K; et al. Genomic and epigenetic alterations deregulate microRNA expression in human epithelial ovarian cancer. Proc Natl Acad Sci USA 2008, 105(19), 7004–7009. [Google Scholar]
- Dahiya, N; Sherman-Baust, CA; Wang, TL; Davidson, B; Shih Ie, M; Zhang, Y; Wood, W, III; Becker, KG; Morin, PJ. MicroRNA expression and identification of putative miRNA targets in ovarian cancer. PLoS One 2008, 3(6), e2436. [Google Scholar]
- Bhattacharya, R; Nicoloso, M; Arvizo, R; Wang, E; Cortez, A; Rossi, S; Calin, GA; Mukherjee, P. MiR-15a and MiR-16 control Bmi-1 expression in ovarian cancer. Cancer Res 2009, 69(23), 9090–9095. [Google Scholar]
- Guo, LM; Pu, Y; Han, Z; Liu, T; Li, YX; Liu, M; Li, X; Tang, H. MicroRNA-9 inhibits ovarian cancer cell growth through regulation of NF-kappaB1. FEBS J 2009, 276(19), 5537–5546. [Google Scholar]
- Guan, Y; Yao, H; Zheng, Z; Qiu, G; Sun, K. MiR-125b targets BCL3 and suppresses ovarian cancer proliferation. Int J Cancer 2010. [Google Scholar] [CrossRef]
- Cowden Dahl, KD; Dahl, R; Kruichak, JN; Hudson, LG. The epidermal growth factor receptor responsive miR-125a represses mesenchymal morphology in ovarian cancer cells. Neoplasia 2009, 11(11), 1208–1215. [Google Scholar]
- Yang, N; Kaur, S; Volinia, S; Greshock, J; Lassus, H; Hasegawa, K; Liang, S; Leminen, A; Deng, S; Smith, L; Johnstone, CN; Chen, XM; Liu, CG; Huang, Q; Katsaros, D; Calin, GA; Weber, BL; Butzow, R; Croce, CM; Coukos, G; Zhang, L. MicroRNA microarray identifies Let-7i as a novel biomarker and therapeutic target in human epithelial ovarian cancer. Cancer Res 2008, 68(24), 10307–10314. [Google Scholar]
- Honda, H; Pazin, MJ; D’Souza, T; Ji, H; Morin, PJ. Regulation of the CLDN3 Gene in Ovarian Cancer Cells. Cancer Biol Ther 2007, 6(11), 1733–1742. [Google Scholar]
- Litkouhi, B; Kwong, J; Lo, CM; Smedley, JG, III; McClane, BA; Aponte, M; Gao, Z; Sarno, JL; Hinners, J; Welch, WR; Berkowitz, RS; Mok, SC; Garner, EI. Claudin-4 overexpression in epithelial ovarian cancer is associated with hypomethylation and is a potential target for modulation of tight junction barrier function using a C-terminal fragment of Clostridium perfringens enterotoxin. Neoplasia 2007, 9(4), 304–314. [Google Scholar]
- Honda, H; Pazin, MJ; Ji, H; Wernyj, RP; Morin, PJ. Crucial roles of Sp1 and epigenetic modifications in the regulation of the CLDN4 promoter in ovarian cancer cells. J Biol Chem 2006, 281(30), 21433–21444. [Google Scholar]
- Clarke, MF; Dick, JE; Dirks, PB; Eaves, CJ; Jamieson, CH; Jones, DL; Visvader, J; Weissman, IL; Wahl, GM. Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 2006, 66(19), 9339–9344. [Google Scholar]
- Bonnet, D; Dick, JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997, 3(7), 730–737. [Google Scholar]
- O’Brien, CA; Kreso, A; Jamieson, CH. Cancer stem cells and self-renewal. Clin Cancer Res 2010, 16(12), 3113–3120. [Google Scholar]
- Bapat, SA; Mali, AM; Koppikar, CB; Kurrey, NK. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res 2005, 65(8), 3025–3029. [Google Scholar]
- Zhang, S; Balch, C; Chan, MW; Lai, HC; Matei, D; Schilder, JM; Yan, PS; Huang, TH; Nephew, KP. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res 2008, 68(11), 4311–4320. [Google Scholar]
- Gao, MQ; Choi, YP; Kang, S; Youn, JH; Cho, NH. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene 2010, 29(18), 2672–2680. [Google Scholar]
- Baba, T; Convery, PA; Matsumura, N; Whitaker, RS; Kondoh, E; Perry, T; Huang, Z; Bentley, RC; Mori, S; Fujii, S; Marks, JR; Berchuck, A; Murphy, SK. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene 2009, 28(2), 209–218. [Google Scholar]
- Curley, MD; Therrien, VA; Cummings, CL; Sergent, PA; Koulouris, CR; Friel, AM; Roberts, DJ; Seiden, MV; Scadden, DT; Rueda, BR; Foster, R. CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells 2009, 27(12), 2875–2883. [Google Scholar]
- Alvero, AB; Chen, R; Fu, HH; Montagna, M; Schwartz, PE; Rutherford, T; Silasi, DA; Steffensen, KD; Waldstrom, M; Visintin, I; Mor, G. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle 2009, 8(1), 158–166. [Google Scholar]
- Wei, X; Dombkowski, D; Meirelles, K; Pieretti-Vanmarcke, R; Szotek, PP; Chang, HL; Preffer, FI; Mueller, PR; Teixeira, J; MacLaughlin, DT; Donahoe, PK. Mullerian inhibiting substance preferentially inhibits stem/progenitors in human ovarian cancer cell lines compared with chemotherapeutics. Proc Natl Acad Sci USA 2010, 107(44), 18874–18879. [Google Scholar]
- Landen, CN, Jr; Goodman, B; Katre, AA; Steg, AD; Nick, AM; Stone, RL; Miller, LD; Mejia, PV; Jennings, NB; Gershenson, DM; Bast, RC, Jr; Coleman, RL; Lopez-Berestein, G; Sood, AK. Targeting aldehyde dehydrogenase cancer stem cells in ovarian cancer. Mol Cancer Ther 2010, 9(12), 3186–3199. [Google Scholar]
- Surani, MA; Hayashi, K; Hajkova, P. Genetic and epigenetic regulators of pluripotency. Cell 2007, 128(4), 747–762. [Google Scholar]
- Ohm, JE; McGarvey, KM; Yu, X; Cheng, L; Schuebel, KE; Cope, L; Mohammad, HP; Chen, W; Daniel, VC; Yu, W; Berman, DM; Jenuwein, T; Pruitt, K; Sharkis, SJ; Watkins, DN; Herman, JG; Baylin, SB. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 2007, 39(2), 237–242. [Google Scholar]
- DeSano, JT; Xu, L. MicroRNA regulation of cancer stem cells and therapeutic implications. AAPS J 2009, 11(4), 682–692. [Google Scholar]
- Gifford, G; Paul, J; Vasey, PA; Kaye, SB; Brown, R. The acquisition of hMLH1 methylation in plasma DNA after chemotherapy predicts poor survival for ovarian cancer patients. Clin Cancer Res 2004, 10(13), 4420–4426. [Google Scholar]
- Su, HY; Lai, HC; Lin, YW; Liu, CY; Chen, CK; Chou, YC; Lin, SP; Lin, WC; Lee, HY; Yu, MH. Epigenetic silencing of SFRP5 is related to malignant phenotype and chemoresistance of ovarian cancer through Wnt signaling pathway. Int J Cancer 2010, 127(3), 555–567. [Google Scholar]
- Nicholson, LJ; Smith, PR; Hiller, L; Szlosarek, PW; Kimberley, C; Sehouli, J; Koensgen, D; Mustea, A; Schmid, P; Crook, T. Epigenetic silencing of argininosuccinate synthetase confers resistance to platinum-induced cell death but collateral sensitivity to arginine auxotrophy in ovarian cancer. Int J Cancer 2009, 125(6), 1454–1463. [Google Scholar]
- Li, M; Balch, C; Montgomery, JS; Jeong, M; Chung, JH; Yan, P; Huang, TH; Kim, S; Nephew, KP. Integrated analysis of DNA methylation and gene expression reveals specific signaling pathways associated with platinum resistance in ovarian cancer. BMC Med. Genomics 2009, 2, 34. [Google Scholar]
- Boren, T; Xiong, Y; Hakam, A; Wenham, R; Apte, S; Chan, G; Kamath, SG; Chen, DT; Dressman, H; Lancaster, JM. MicroRNAs and their target messenger RNAs associated with ovarian cancer response to chemotherapy. Gynecol Oncol 2009, 113(2), 249–255. [Google Scholar]
- Ellis, L; Atadja, PW; Johnstone, RW. Epigenetics in cancer: targeting chromatin modifications. Mol Cancer Ther 2009, 8(6), 1409–1420. [Google Scholar]
- Gal-Yam, EN; Saito, Y; Egger, G; Jones, PA. Cancer epigenetics: modifications, screening, and therapy. Annu. Rev. Med 2008, 59, 267–280. [Google Scholar]
- Szyf, M. Epigenetics, DNA methylation, and chromatin modifying drugs. Annu. Rev. Pharmacol. Toxicol 2009, 49, 243–263. [Google Scholar]
- Xu, WS; Parmigiani, RB; Marks, PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007, 26(37), 5541–5552. [Google Scholar]
- Tan, J; Yang, X; Zhuang, L; Jiang, X; Chen, W; Lee, PL; Karuturi, RK; Tan, PB; Liu, ET; Yu, Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev 2007, 21(9), 1050–1063. [Google Scholar]
- Greiner, D; Bonaldi, T; Eskeland, R; Roemer, E; Imhof, A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9. Nat Chem Biol 2005, 1(3), 143–145. [Google Scholar]
- Kubicek, S; O’Sullivan, RJ; August, EM; Hickey, ER; Zhang, Q; Teodoro, ML; Rea, S; Mechtler, K; Kowalski, JA; Homon, CA; Kelly, TA; Jenuwein, T. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell 2007, 25(3), 473–481. [Google Scholar]
- Huang, Y; Greene, E; Murray Stewart, T; Goodwin, AC; Baylin, SB; Woster, PM; Casero, RA, Jr. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc Natl Acad Sci USA 2007, 104(19), 8023–8028. [Google Scholar]
- Huang, Y; Stewart, TM; Wu, Y; Baylin, SB; Marton, LJ; Perkins, B; Jones, RJ; Woster, PM; Casero, RA, Jr. Novel Oligoamine Analogues Inhibit Lysine-Specific Demethylase 1 and Induce Reexpression of Epigenetically Silenced Genes. Clin. Cancer Res 2009, 15, 7217. [Google Scholar]
- Cameron, EE; Bachman, KE; Myohanen, S; Herman, JG; Baylin, SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet 1999, 21(1), 103–107. [Google Scholar]
- Issa, JP. DNA methylation as a therapeutic target in cancer. Clin Cancer Res 2007, 13(6), 1634–1637. [Google Scholar]
- Fiskus, W; Buckley, K; Rao, R; Mandawat, A; Yang, Y; Joshi, R; Wang, Y; Balusu, R; Chen, J; Koul, S; Joshi, A; Upadhyay, S; Atadja, P; Bhalla, KN. Panobinostat treatment depletes EZH2 and DNMT1 levels and enhances decitabine mediated de-repression of JunB and loss of survival of human acute leukemia cells. Cancer Biol Ther 2009, 8(10), 939–950. [Google Scholar]
- Yoo, CB; Jones, PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov 2006, 5(1), 37–50. [Google Scholar]
- Jones, PA; Baylin, SB. The epigenomics of cancer. Cell 2007, 128(4), 683–692. [Google Scholar]
- Gaudet, F; Hodgson, JG; Eden, A; Jackson-Grusby, L; Dausman, J; Gray, JW; Leonhardt, H; Jaenisch, R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300(5618), 489–492. [Google Scholar]
- Eden, A; Gaudet, F; Waghmare, A; Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003, 300(5618), 455. [Google Scholar]
- Plumb, JA; Strathdee, G; Sludden, J; Kaye, SB; Brown, R. Reversal of drug resistance in human tumor xenografts by 2′-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res 2000, 60(21), 6039–6044. [Google Scholar]
- Qian, X; LaRochelle, WJ; Ara, G; Wu, F; Petersen, KD; Thougaard, A; Sehested, M; Lichenstein, HS; Jeffers, M. Activity of PXD101, a histone deacetylase inhibitor, in preclinical ovarian cancer studies. Mol Cancer Ther 2006, 5(8), 2086–2095. [Google Scholar]
- Lin, CT; Lai, HC; Lee, HY; Lin, WH; Chang, CC; Chu, TY; Lin, YW; Lee, KD; Yu, MH. Valproic acid resensitizes cisplatin-resistant ovarian cancer cells. Cancer Sci 2008, 99(6), 1218–1226. [Google Scholar]
- Steele, N; Finn, P; Brown, R; Plumb, JA. Combined inhibition of DNA methylation and histone acetylation enhances gene re-expression and drug sensitivity in vivo. Br J Cancer 2009, 100(5), 758–763. [Google Scholar]
- Asadollahi, R; Hyde, CA; Zhong, XY. Epigenetics of ovarian cancer: from the lab to the clinic. Gynecol Oncol 2010, 118(1), 81–87. [Google Scholar]
- Fu, S; Hu, W; Iyer, R; Kavanagh, JJ; Coleman, RL; Levenback, CF; Sood, AK; Wolf, JK; Gershenson, DM; Markman, M; Hennessy, BT; Kurzrock, R; Bast, RC, Jr. Phase 1b-2a study to reverse platinum resistance through use of a hypomethylating agent, azacitidine, in patients with platinum-resistant or platinum-refractory epithelial ovarian cancer. Cancer 2010. [Google Scholar] [CrossRef]
- Fang, F; Balch, C; Schilder, J; Breen, T; Zhang, S; Shen, C; Li, L; Kulesavage, C; Snyder, AJ; Nephew, KP; Matei, DE. A phase 1 and pharmacodynamic study of decitabine in combination with carboplatin in patients with recurrent, platinum-resistant, epithelial ovarian cancer. Cancer 2010, 116(17), 4043–4053. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene name | Chromosome | Mechanism of Down-Regulation | Reference |
|---|---|---|---|---|
| Tumor Suppressor Genes Down-Regulated by Both Genetic and Epigenetic Changes | ||||
| PTEN | Phosphatase and tensin homolog | 10q23 | LOH, mutation | [4] |
| Promoter DNA methylation miRNA (miR-214) | [48] | |||
| [49] | ||||
| BRCA1 | Breast cancer 1, early onset | 17q21 | Mutation, LOH | [4] |
| Promoter DNA methylation | [50] | |||
| OPCML | Opioid binding protein/cell adhesion molecule-like | 11q25 | LOH, mutation | [4] |
| Promoter DNA methylation | [57] | |||
| DIRAS3 (ARHI) | DIRAS family, GTP-binding RAS-like 3 | 1p31 | Imprinting, LOH, promoter DNA methylation | [51,58] |
| Transcription down-regulated by E2F1 and E2F4 | [4] | |||
| PEG3 | Paternally expressed 3 | 19q13 | Imprinting, LOH, promoter DNA methylation | [51] |
| TES * | Testis-derived transcript (3 LIM domains) | 7q31.2 | LOH, promoter DNA methylation | [59,60] |
| MYO18B * | Myosin XVIIIB | 22q12.1 | Mutation, promoter DNA methylation | [61] |
| Tumor Suppressor Genes Down-Regulated By Epigenetic Changes | ||||
| RASSF1 | Ras association | 3p21 | Promoter DNA methylation | [62] |
| (RASSF1A) | (RalGDS/AF-6) domain family member 1 | Histone methylation (H3K27me3) | [25] | |
| DLEC1 | Deleted in lung and esophageal cancer 1 | 3p22.3 | Promoter DNA methylation, histone hypoacetylation | [63] |
| ARL11 (ARLTS1) | ADP-ribosylation factor-like 11 | 13q.14 | Promoter DNA methylation | [64] |
| CDKN2A(p16) | Cyclin-dependent kinase inhibitor 2A (melanoma, p16, inhibits CDK4) | 9p21 | Promoter DNA methylation | [65,66] |
| CDKN1A(p21) | Cyclin-dependent kinase inhibitor 1A (p21, Cip1) | 6p21.2 | Hypoacetylation of H3Ac and H4Ac | [67] |
| MLH1(hMLH1) | MutL homolog 1, colon cancer, nonpolyposis type 2 (E. coli) | 3p21.3 | Promoter DNA methylation | [68,69] |
| DAK(DAK1) | Death-associated protein kinase 1 | 11q12.2 | Promoter DNA methylation | [70] |
| CDH1(E-cadherin) | Cadherin 1, type 1, E-cadherin (epithelial) | 16q22.1 | Promoter DNA methylation | [71] |
| FBXO32 | F-box protein 32 | 8q24.13 | Promoter DNA methylation | [72] |
| CTGF * | Connective tissue growth factor | 6q23.1 | Promoter DNA methylation | [73] |
| ANGPTL2 * | Angiopoietin-like protein 2 | 9q33.3 | Promoter DNA methylation | [74] |
| Cancer-Associated Genes Down-Regulated By Epigenetic Changes | ||||
| ICAM1 | Intercellular adhesion molecule 1 | 19p13.3–p13.2 | Promoter DNA methylation | [75] |
| PCSK6 (PACE4) | Proprotein convertase subtilisin/kexin type 6 | 15q26.3 | Promoter DNA methylation and histone deacetylation | [76] |
| GATA4, GATA6 | GATA binding protein 4 GATA binding protein 6 | 8p23.1 p22 18q11.1 q11.2 | Hypoacetylation of H3Ac and H4Ac, loss of H3K4me3 | [53] |
| ADAM19 | ADAM metallopeptidase domain 19 | 5q33.3 | Repressive histone modifications (H3K27me3 and H3K9me2) | [52] |
| ZEB1 | Zinc finger E-box binding homeobox 1 | 10p11.2 | miRNA (miR-200 family) | [56] |
| ZEB2 | Zinc finger E-box binding homeobox 2 | 2q22.3 | miRNA (miR-200 family) | [56] |
| Gene Symbol | Name | Chromosome | Mechanism of Up-Regulation | Reference |
|---|---|---|---|---|
| Oncogene or Proto-Oncogenes | ||||
| HRAS | v-Ha-ras Harvey rat sarcoma viral oncogene homolog | 11p15.5 | miRNA (Let-7i) | [93] |
| HMGA2 | High mobility group AT-hook 2 | 12q15 | miRNA (Let-7i) | [93] |
| BCL2 | B-cell CLL/lymphoma 2 | 18q21.3 | miRNA (miR-15a and miR-16) | [89] |
| BCL3 | B-cell CLL/lymphoma 3 | 19q13.1–q13.2 | miRNA (miR-125b) | [91] |
| BMI1 | BMI1 polycomb ring finger oncogene | 10p11.23 | miRNA (miR-15a and miR-16) | [89] |
| NFKB1(NF-kappa B1) | Nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 | 4q24 | miRNA (miR-9) | [90] |
| Cancer-Promoting Genes | ||||
| SNCG | Synuclein, gamma (breast cancer-specific protein 1) | 10q23.2–q23.3 | Promoter DNA hypomethylation | [77,78] |
| CTCFL(BORIS) | CCCTC-binding factor (zinc finger protein)-like | 20q13.31 | Promoter DNA hypomethylation | [79] |
| CLDN3 | Claudin-3 | 7q11.23 | Promoter DNA hypomethylation and histone acetylation | [94] |
| Loss of repressive histone methylations | [86] | |||
| CLDN4 | Claudin-4 | 7q11.23 | Promoter DNA hypomethylation, histone acetylation | [95,96] |
| Loss of repressive histone methylations | [86] | |||
| Cancer-Associated Genes | ||||
| DNAJC15(MCJ) | DnaJ (Hsp40) homolog, subfamily C, member 15 | 13q14.1 | Promoter DNA hypomethylation | [81,82] |
| MAL | Mal, T-cell differentiation protein | 2cen-q13 | Promoter DNA hypomethylation | [83] |
| HOXA10 | Homeobox A10 | 7p15.2 | Promoter DNA hypomethylation | [84] |
| TUBB3 | Class III ß-tubulin | 16q24.3 | Promoter DNA hypomethylation and histone acetylation | [85] |
| ARID3B | AT-rich interactive domain 3B | 15q24 | miRNA (miR-125a) | [92] |
| Epigenetic Drugs | Compound (Commercial name) | Target | Status | Indication | Reference | |
|---|---|---|---|---|---|---|
| DNA methylation Inhibitor (DNMTi) | Nucleoside analog | 5-aza-cytidine (Vidaza) | DNMT | FDA approved | MDS | [116–118] |
| 5-aza-2′-deoxy cytidine (decitabine) | DNMT | FDA approved | MDS | |||
| Zebularine | DNMT | |||||
| Non-nucleoside | Hydralazine | DNMT | Phase I | |||
| HDAC inhibitor (HDACi) | Hydroxamate | SAHA (Vorinostat) | Class I, II HDACs | FDA approved | T cell cutaenous lymphoma | [116,118,119] |
| TSA (Tricostatin A) | Class I, II HDACs | Preclinical | ||||
| LBH589 (Panobinostat) | Class I, II HDACs | Phase I/II | ||||
| PXD101 (Belinostat) | Class I, II HDACs | Phase I/II | ||||
| PCI-24781 | Class I, II HDACs | Phase I | ||||
| Aliphatic acid | Sodium phenyl butyrate | Class I, IIa HDACs | Phase I/II | |||
| Valproic acid | Class I, IIa HDACs | Phase I/II | ||||
| Cyclic peptide | FK228 (Romidepsin) | HDAC1, 2 | Phase I/II | |||
| Benzamide | MGCD0103 | Class I | Phase I/II | |||
| Histone methyltrans ferase inhibitors (HMTi) | S-adenosylhomocysteine hydrolase inhibitor | 3-Deazaneplanocin A (DZNep) | Polycomb group proteins | [116,117,120] | ||
| Fungal mycotoxin | Chaetocin | SU(VAR)3-9 | [121] | |||
| Small molecule inhibitor | BIX-01294 | G9a histone methyl transferase | [122] | |||
| Histone demethylase inhibitor (HDMi) | Polyamine analog | Polyamine analog | Histone demethylase LSD1 | [123,124] | ||
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kwon, M.J.; Shin, Y.K. Epigenetic Regulation of Cancer-Associated Genes in Ovarian Cancer. Int. J. Mol. Sci. 2011, 12, 983-1008. https://doi.org/10.3390/ijms12020983
Kwon MJ, Shin YK. Epigenetic Regulation of Cancer-Associated Genes in Ovarian Cancer. International Journal of Molecular Sciences. 2011; 12(2):983-1008. https://doi.org/10.3390/ijms12020983
Chicago/Turabian StyleKwon, Mi Jeong, and Young Kee Shin. 2011. "Epigenetic Regulation of Cancer-Associated Genes in Ovarian Cancer" International Journal of Molecular Sciences 12, no. 2: 983-1008. https://doi.org/10.3390/ijms12020983
APA StyleKwon, M. J., & Shin, Y. K. (2011). Epigenetic Regulation of Cancer-Associated Genes in Ovarian Cancer. International Journal of Molecular Sciences, 12(2), 983-1008. https://doi.org/10.3390/ijms12020983