Novel Natural Inhibitors of CYP1A2 Identified by in Silico and in Vitro Screening


Abstract
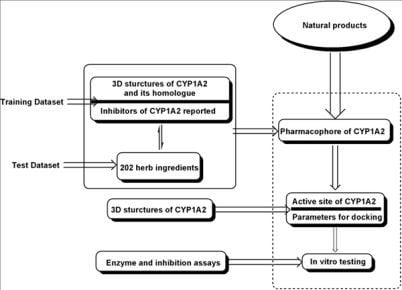
:
1. Introduction
2. Results and Discussion
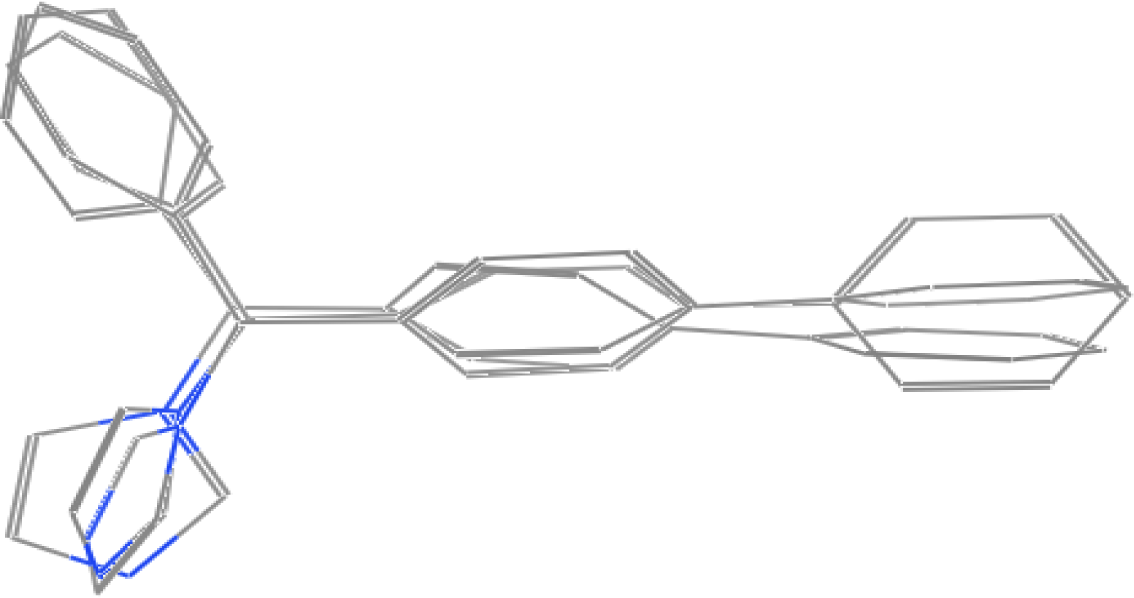
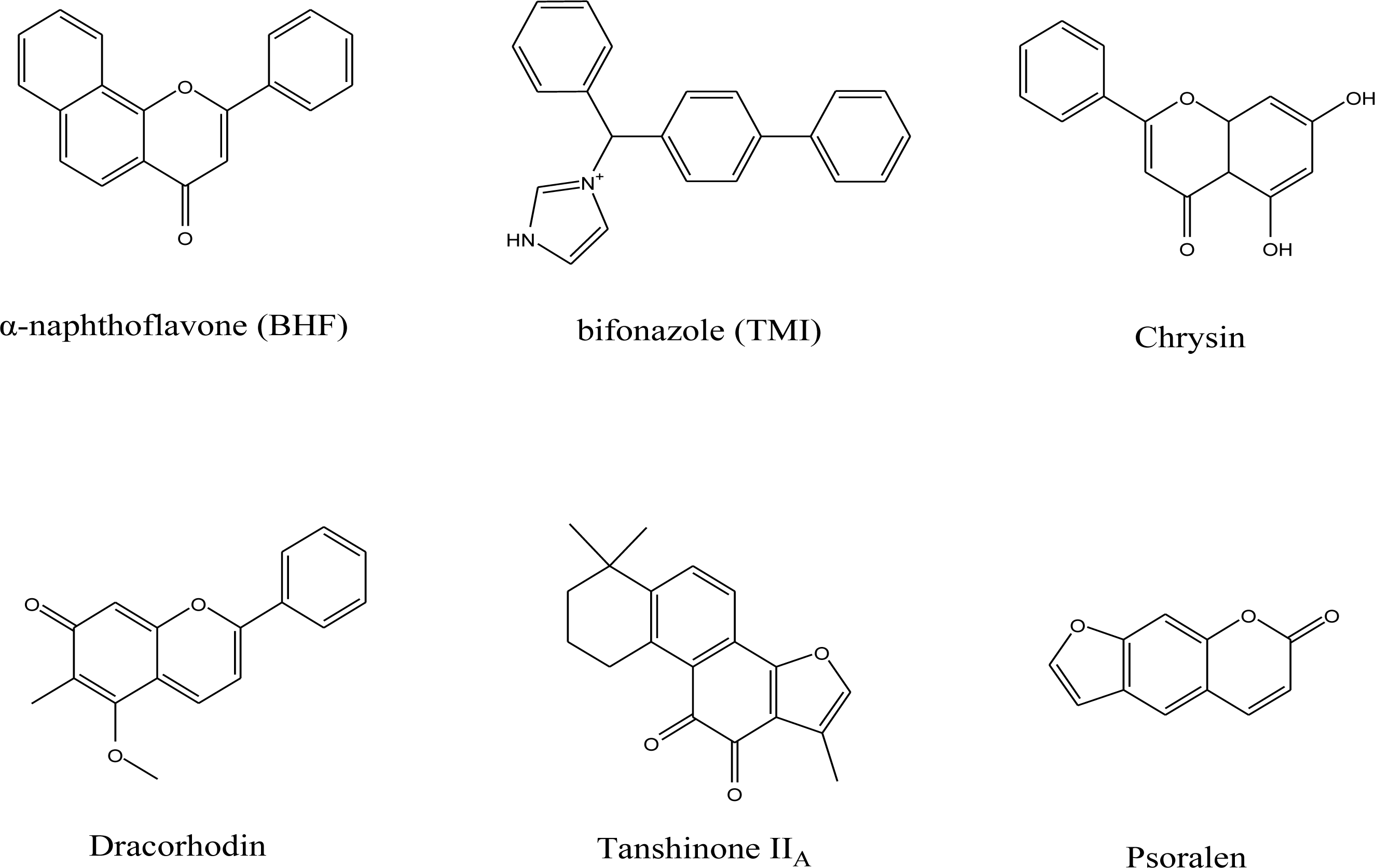
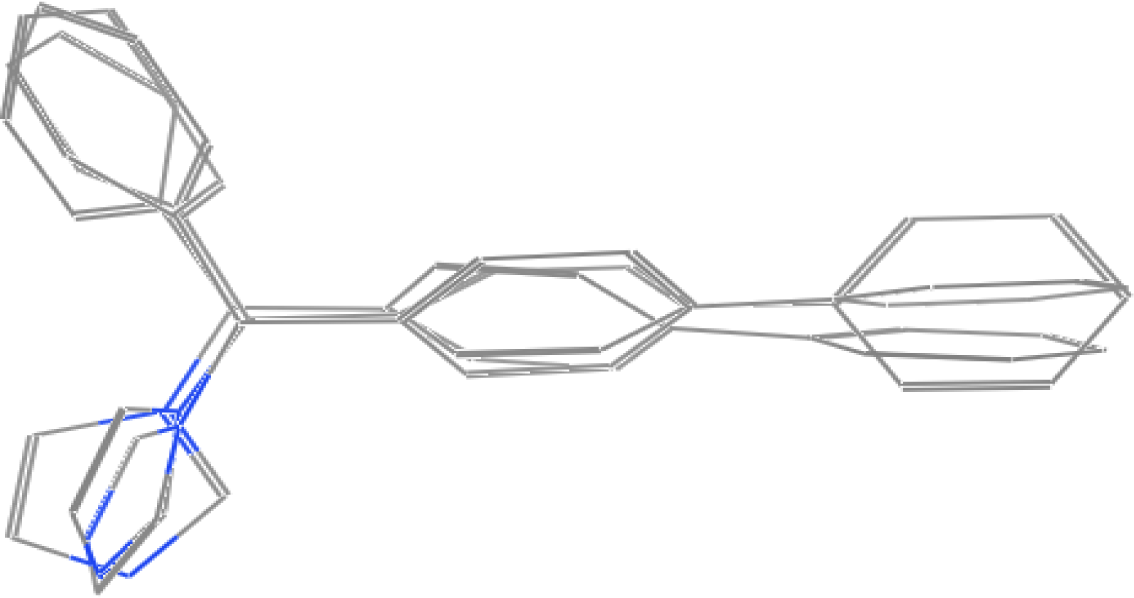
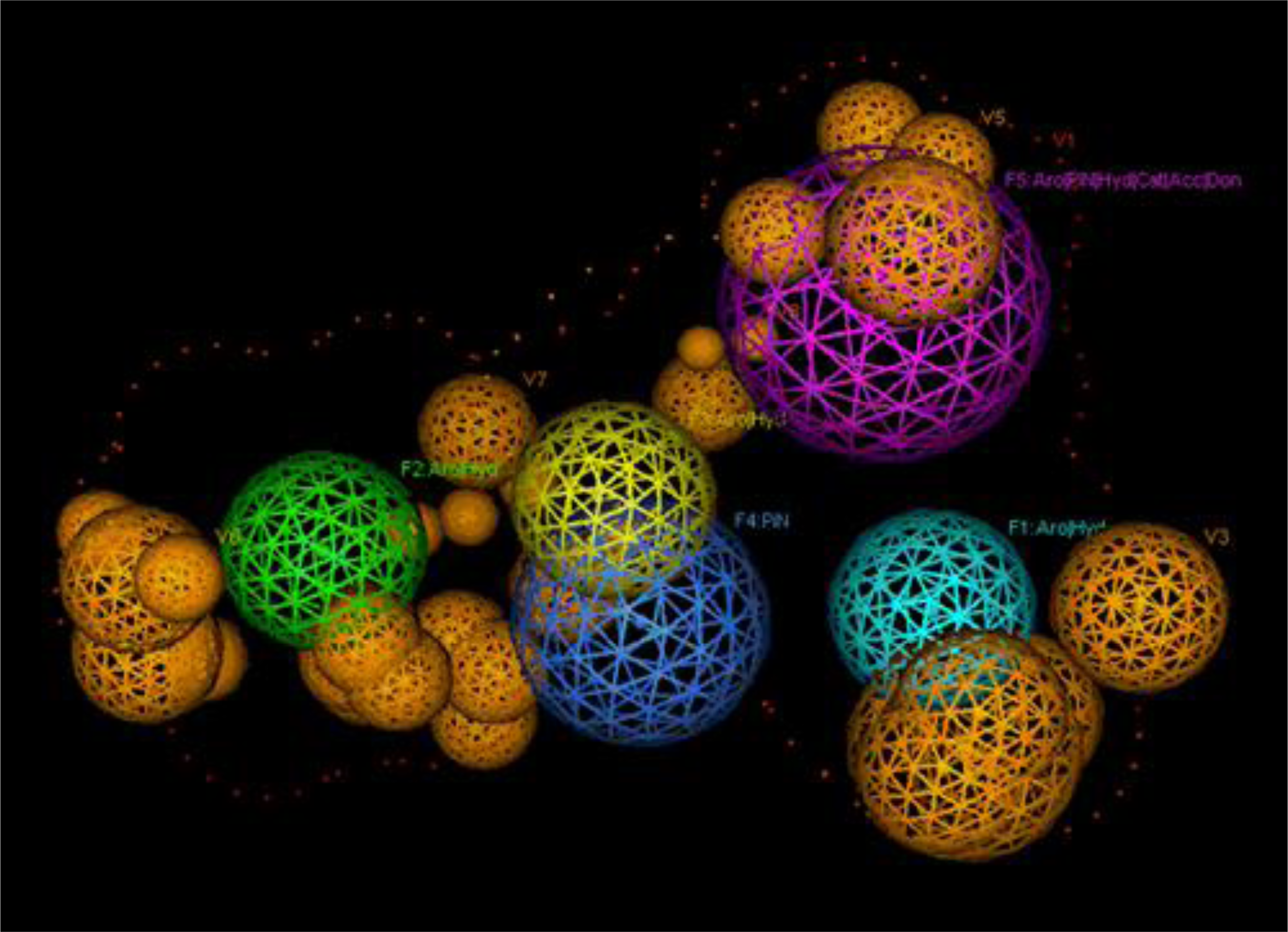
2.1. Pharmacophore Models
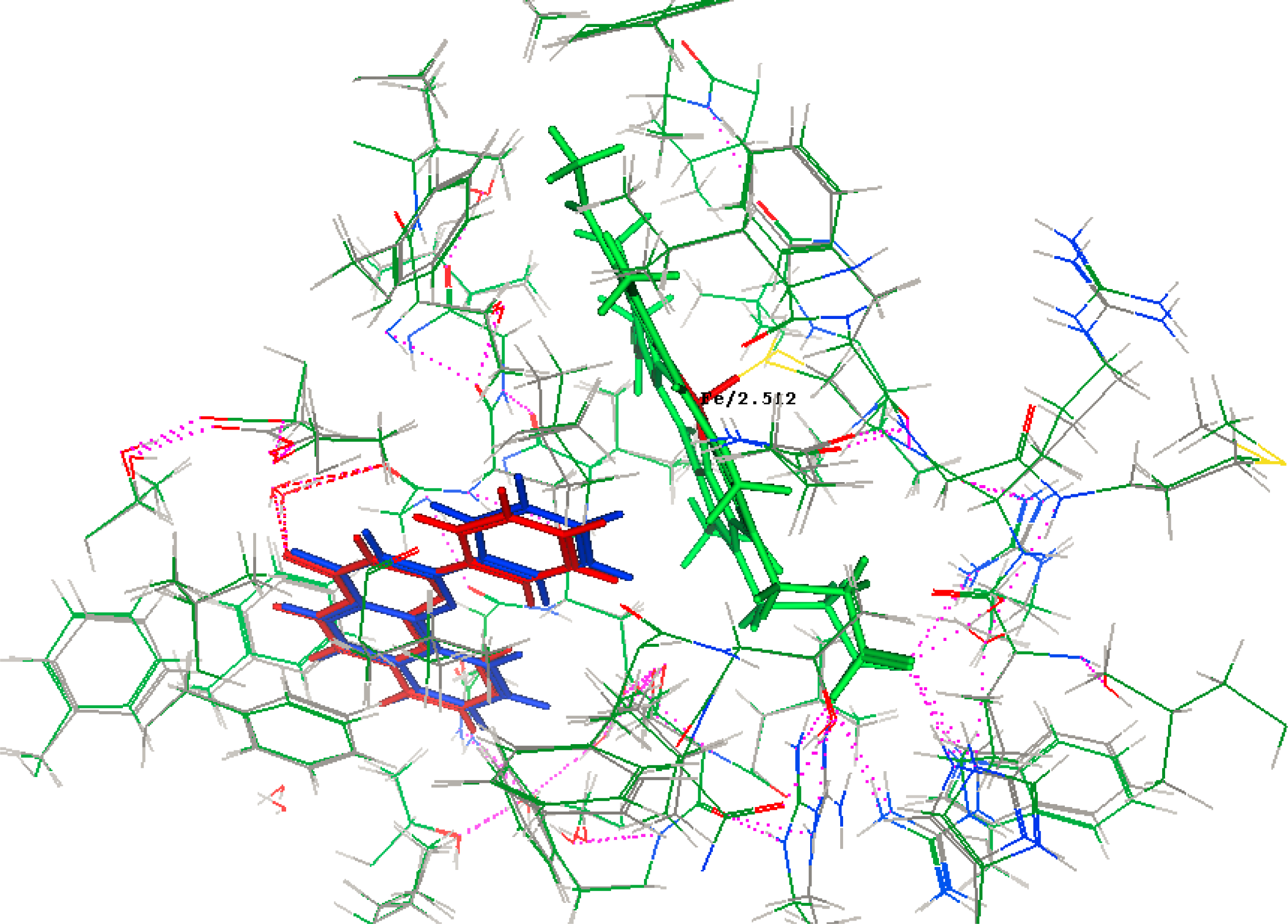
2.2. Docking Results
2.3. In Vitro Testing
3. Materials and Methods
3.1. Pharmacophore Generation
3.2. Docking Procedure
3.3. Enzyme and Inhibition Assays
4. Conclusions
Supplementary Materials
ijms-12-03250-s001.pdfAcknowledgments
References
- Kaufman, DW; Kelly, JP; Rosenberg, L; Anderson, TE; Mitchell, AA. Recent patterns of medication use in the ambulatory adult population of the United States: The Slone survey. JAMA 2002, 287, 337–344. [Google Scholar]
- Ernst, E. Are herbal medicines effective? Int. J. Clin. Pharmacol. Ther 2004, 42, 157–159. [Google Scholar]
- Klepser, TB; Doucette, WR; Horton, MR; Buys, LM; Ernst, ME; Ford, JK; Hoehns, JD; Kautzman, HA; Logemann, CD; Swegle, JM; Ritho, M; Klepser, ME. Assessment of patients’ perceptions and beliefs regarding herbal therapies. Pharmacotherapy 2000, 20, 83–87. [Google Scholar]
- Delgoda, R; Westlake, AC. Herbal interactions involving cytochrome p450 enzymes: A mini review. Toxicol. Rev 2004, 23, 239–249. [Google Scholar]
- Saxena, A; Tripathi, KP; Roy, S; Khan, F; Sharma, A. Pharmacovigilance: Effects of herbal components on human drugs interactions involving cytochrome P450. Bioinformation 2008, 3, 198–204. [Google Scholar]
- Nebel, A; Schneider, BJ; Baker, RK; Kroll, DJ. Potential metabolic interaction between St. John’s wort and theophylline. Ann. Pharmacother 1999, 33, 502. [Google Scholar]
- Ameer, B; Weintraub, RA. Drug interactions with grapefruit juice. Clin. Pharmacokinet 1997, 33, 103–121. [Google Scholar]
- Bailey, DG; Malcolm, J; Arnold, O; Spence, JD. Grapefruit juice-drug interactions. Br. J. Clin. Pharmacol 1998, 46, 101–110. [Google Scholar]
- Kupferschmidt, HH; Fattinger, KE; Ha, HR; Follath, F; Krahenbuhl, S. Grapefruit juice enhances the bioavailability of the HIV protease inhibitor saquinavir in man. Br. J. Clin. Pharmacol 1998, 45, 355–359. [Google Scholar]
- Faber, MS; Jetter, A; Fuhr, U. Assessment of CYP1A2 activity in clinical practice: why, how, and when? Basic Clin. Pharmacol. Toxicol 2005, 97, 125–134. [Google Scholar]
- Bapiro, TE; Sayi, J; Hasler, JA; Jande, M; Rimoy, G; Masselle, A; Masimirembwa, CM. Artemisinin and thiabendazole are potent inhibitors of cytochrome P450 1A2 (CYP1A2) activity in humans. Eur. J. Clin. Pharmacol 2005, 61, 755–761. [Google Scholar]
- Qiu, F; Wang, G; Zhao, Y; Sun, H; Mao, G; A, J; Sun, J. Effect of danshen extract on pharmacokinetics of theophylline in healthy volunteers. Br. J. Clin. Pharmacol 2008, 65, 270–274. [Google Scholar]
- Gorski, JC; Huang, SM; Pinto, A; Hamman, MA; Hilligoss, JK; Zaheer, NA; Desai, M; Miller, M; Hall, SD. The effect of echinacea (Echinacea purpurea root) on cytochrome P450 activity in vivo. Clin. Pharmacol. Ther 2004, 75, 89–100. [Google Scholar]
- Appiah-Opong, R; Commandeur, JN; van Vugt-Lussenburg, B; Vermeulen, NP. Inhibition of human recombinant cytochrome P450s by curcumin and curcumin decomposition products. Toxicology 2007, 235, 83–91. [Google Scholar]
- Tang, JC; Yang, H; Song, XY; Song, XH; Yan, SL; Shao, JQ; Zhang, TL; Zhang, JN. Inhibition of cytochrome P450 enzymes by rhein in rat liver microsomes. Phytother. Res 2009, 23, 159–164. [Google Scholar]
- Peterson, S; Lampe, JW; Bammler, TK; Gross-Steinmeyer, K; Eaton, DL. Apiaceous vegetable constituents inhibit human cytochrome P-450 1A2 (hCYP1A2) activity and hCYP1A2-mediated mutagenicity of aflatoxin B1. Food Chem. Toxicol 2006, 44, 1474–1484. [Google Scholar]
- Fuhr, U; Strobl, G; Manaut, F; Anders, EM; Sorgel, F; Lopez-de-Brinas, E; Chu, DT; Pernet, AG; Mahr, G; Sanz, F; et al. Quinolone antibacterial agents: relationship between structure and in vitro inhibition of the human cytochrome P450 isoform CYP1A2. Mol. Pharmacol 1993, 43, 191–199. [Google Scholar]
- Sanz, F; López-de-Briñas, E; Rodríguez, J; Manaut, F. Theoretical Study on the Metabolism of Caffeine by Cytochrome P-450 1A2 and its Inhibition. Quant. Struct. Act. Relatsh 1994, 13, 281–284. [Google Scholar]
- Lee, H; Yeom, H; Kim, YG; Yoon, CN; Jin, C; Choi, JS; Kim, BR; Kim, DH. Structure-related inhibition of human hepatic caffeine N3-demethylation by naturally occurring flavonoids. Biochem. Pharmacol 1998, 55, 1369–1375. [Google Scholar]
- de Rienzo, F; Fanelli, F; Menziani, MC; De Benedetti, PG. Theoretical investigation of substrate specificity for cytochromes P450 IA2, P450 IID6 and P450 IIIA4. J. Comput. Aided Mol. Des 2000, 14, 93–116. [Google Scholar]
- Lozano, JJ; Pastor, M; Cruciani, G; Gaedt, K; Centeno, NB; Gago, F; Sanz, F. 3D-QSAR methods on the basis of ligand-receptor complexes. Application of COMBINE and GRID/GOLPE methodologies to a series of CYP1A2 ligands. J. Comput. Aided Mol. Des 2000, 14, 341–353. [Google Scholar]
- Lewis, DF. On the recognition of mammalian microsomal cytochrome P450 substrates and their characteristics: Towards the prediction of human p450 substrate specificity and metabolism. Biochem. Pharmacol 2000, 60, 293–306. [Google Scholar]
- Moon, T; Chi, MH; Kim, D; Yoon, CN; Choi, Y. Quantitative Structure-Activity Relationships (QSAR) study of flavonoid derivatives for inhibition of cytochrome P450 1A2. Quant. Struct. Act. Relatsh 2000, 19, 257–263. [Google Scholar]
- Zhao, Y; White, MA; Muralidhara, BK; Sun, L; Halpert, JR; Stout, CD. Structure of microsomal cytochrome P450 2B4 complexed with the antifungal drug bifonazole: insight into P450 conformational plasticity and membrane interaction. J. Biol. Chem 2006, 281, 5973–5981. [Google Scholar]
- Raychaudhuri, S; Jain, V; Dongre, M. Identification of a constitutively active variant of LuxO that affects production of HA/protease and biofilm development in a non-O1, non-O139 Vibrio cholerae O110. Gene 2006, 369, 126–133. [Google Scholar]
- Vieth, M; Hirst, JD; Brooks, CR. Do active site conformations of small ligands correspond to low free-energy solution structures? J. Comput. Aided Mol. Des 1998, 12, 563–572. [Google Scholar]
- Chen, IJ; Foloppe, N. Conformational sampling of druglike molecules with MOE and catalyst: implications for pharmacophore modeling and virtual screening. J. Chem. Inf. Model 2008, 48, 1773–1791. [Google Scholar]
- Butler, KT; Luque, FJ; Barril, X. Toward accurate relative energy predictions of the bioactive conformation of drugs. J. Comput. Chem 2009, 30, 601–610. [Google Scholar]
- Zhu, RX; Zhang, XL; Dong, XC; Chen, MB. Searching Inhibitors of Adenosine Kinase by Simulation Methods. Chin. J. Chem 2006, 24, 1493–1497. [Google Scholar]
- Shen, J; Xu, X; Cheng, F; Liu, H; Luo, X; Shen, J; Chen, K; Zhao, W; Shen, X; Jiang, H. Virtual screening of natural products for discovering active compounds and target information. Curr. Med. Chem 2003, 10, 2327–2342. [Google Scholar]
- Krovat, EM; Steindl, T; Langer, T. Recent Advances in Docking and Scoring. Curr. Comput. Aided Drug Des 2005, 1, 93–102. [Google Scholar]
- Sousa, SF; Fernandes, PA; Ramos, MJ. Protein-ligand docking: current status and future challenges. Proteins 2006, 65, 15–26. [Google Scholar]
- Chen, Y; Shoichet, BK. Molecular docking and ligand specificity in fragment-based inhibitor discovery. Nat. Chem. Biol 2009, 5, 358–364. [Google Scholar]
- Cermak, R; Wolffram, S. The potential of flavonoids to influence drug metabolism and pharmacokinetics by local gastrointestinal mechanisms. Curr. Drug Metab 2006, 7, 729–744. [Google Scholar]
- Hertog, MGL; Hollman, PCH; van de Putte, B. Content of potentially anticarcinogenic flavonoids of tea infusions wines, and fruit juices. J. Agric. Food Chem 1993, 41, 1242–1246. [Google Scholar]
- Peterson, J; Dwyer, J. Taxonomic classification helps identify flavonoid-containing foods on a semiquantitative food frequency questionnaire. J Am Diet Assoc 1998, 98. [Google Scholar]
- Bravo, L. Polyphenols: chemistry, dietary sources, metabolism, and nutritional significance. Nutr. Rev 1998, 56, 317–333. [Google Scholar]
- State Food and Drug Administration. Catalog of the Drugs in China; State Food and Drug Administration: Beijing, China, 2008. [Google Scholar]
- Choi, JS; Choi, BC; Choi, KE. Effect of quercetin on the pharmacokinetics of oral cyclosporine. Am. J. Health Syst. Pharm 2004, 61, 2406–2409. [Google Scholar]
- Peng, WX; Li, HD; Zhou, HH. Effect of daidzein on CYP1A2 activity and pharmacokinetics of theophylline in healthy volunteers. Eur. J. Clin. Pharmacol 2003, 59, 237–241. [Google Scholar]
- Rajnarayana, K; Reddy, MS; Krishna, DR. Diosmin pretreatment affects bioavailability of metronidazole. Eur. J. Clin. Pharmacol 2003, 58, 803–807. [Google Scholar]
- Sansen, S; Yano, JK; Reynald, RL; Schoch, GA; Griffin, KJ; Stout, CD; Johnson, EF. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P4501A2. J. Biol. Chem 2007, 282, 14348–14355. [Google Scholar]
- Molecular Operation Environment (MOE), version 200810; Chemical Computing Group Inc: Montreal, Canada, 2008.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds Used as a Template | True Positive Rate | True Negative Rate |
|---|---|---|
| BHF, TMI | 61.5% | 85.2% |
| BHF, Chrysin and Tanshinone IIA | 69.2% | 96.8% |
| Chrysin, Psoralen and Dracorhodin | 61.5% | 89.9% |
| Three TMI in different conformations without optimization | 84.6% | 77.2% |
| Three TMI in different conformations with sophisticated optimization | 84.6% | 86.8% |
| Number | Active Ingredients | % of Control Indication Activity |
|---|---|---|
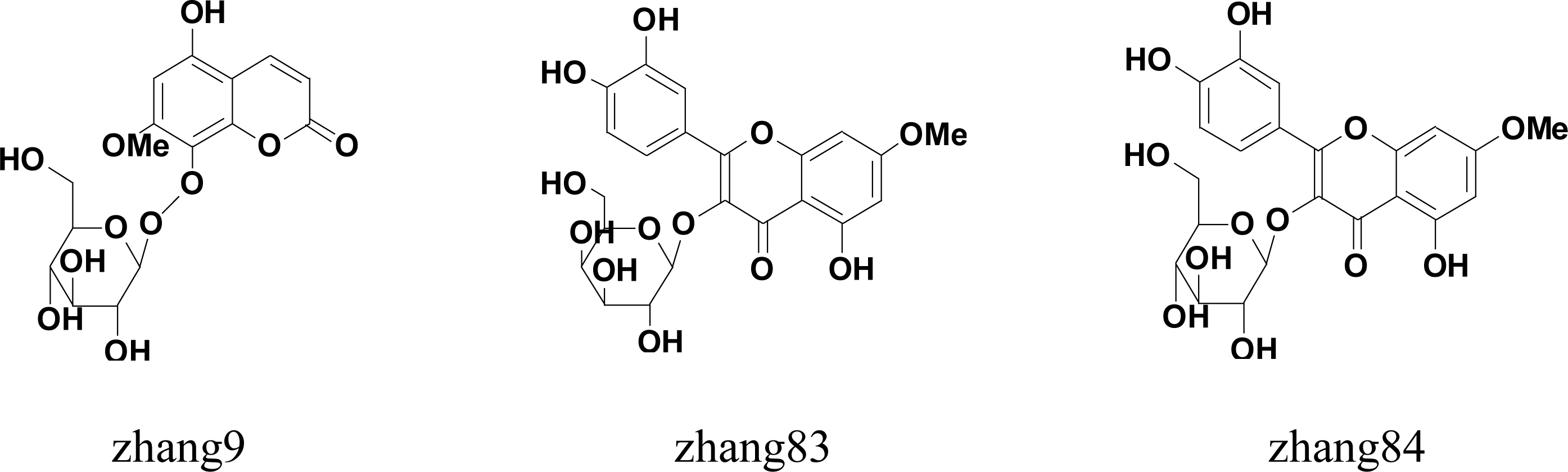
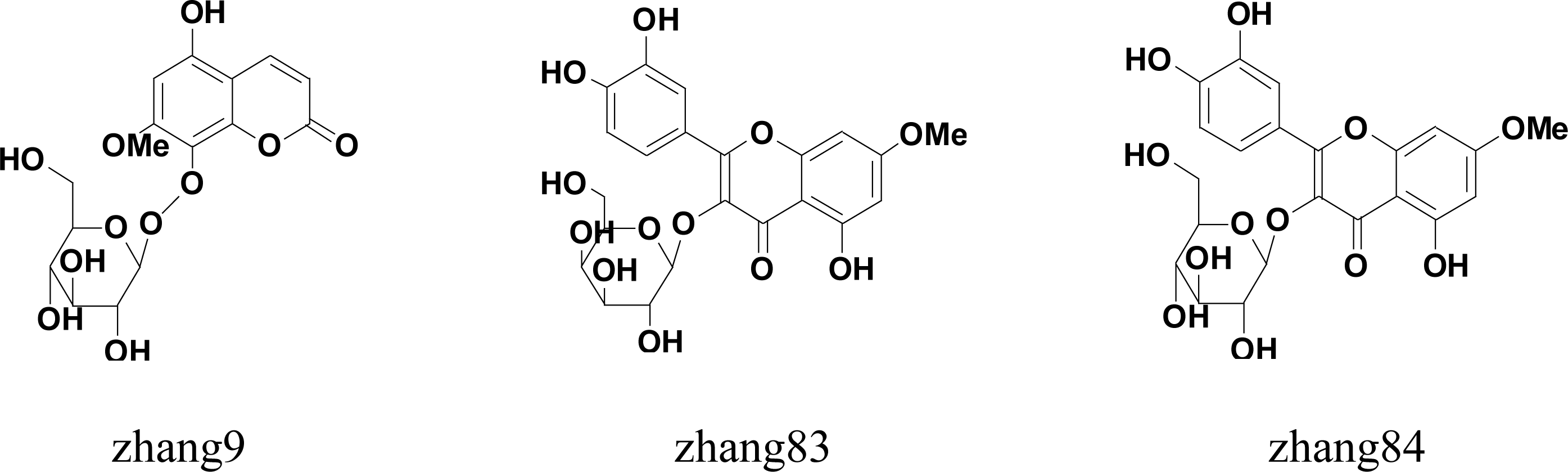
| zhang 9 | 5-hydroxy-7-methoxycoumarin-8-O-d-glucopyranoside | >90% |
| zhang 83 | rhamnetin 3-O-β-d-galactopyranoside | >90% |
| zhang 84 | rhamnetin 3-O-β-d-glucopyranoside | >90% |
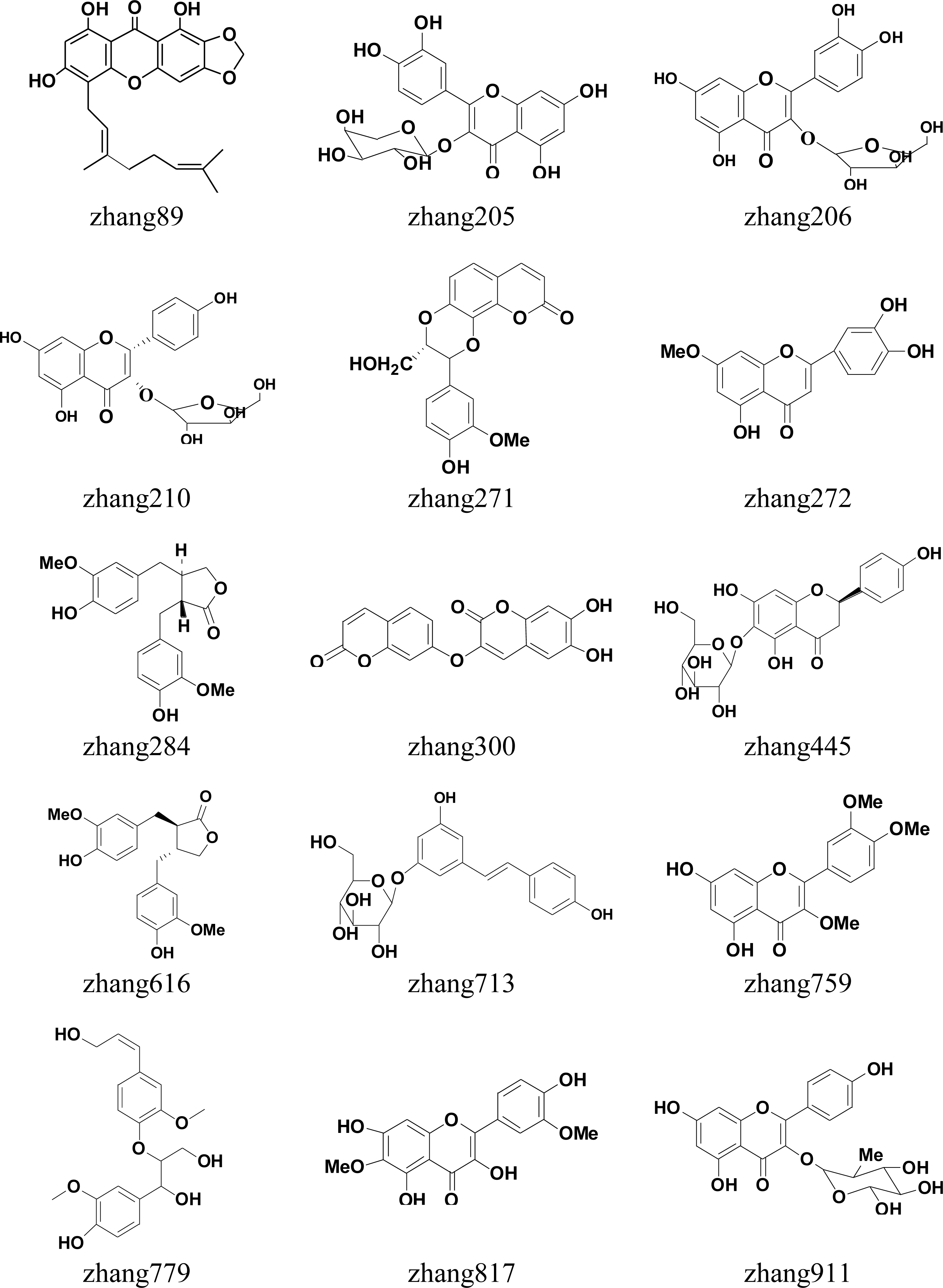
| zhang 89 | 1,6,8-trihydroxy-2,3-methylenedioxy-5-geranylxanthone | >90% |
| zhang 205 | quercetin-3-O-β-d-arabinopyranoside | >90% |
| zhang 206 | quercetin-3-O-α-d-arabinofuranose | >90% |
| zhang 210 | dihydrokaempferol-3-O-α-d-arabinofuranose | >90% |
| zhang 271 | 5′-demethoxy Daphneticin | >90% |
| zhang 272 | 3′-hydroxy-Genkwanin | <90% |
| zhang 284 | (−)-Matairesinol | <90% |
| zhang 300 | Edgeworthin | <90% |
| zhang 445 | Hemiphloin | >90% |
| zhang 616 | (+)-Matairesinol | <90% |
| zhang 713 | Piceid | >90% |
| zhang 759 | 3,3′,4′-Tri-Me ether-3,3′,4′,5,7-Pentahydroxyflavone | >90% |
| zhang 779 | (−)-Threo-guaiacylglycerol-8-O-4′-(coniferyl alcohol) ether | >90% |
| zhang 817 | Spinacetin | <90% |
| zhang 911 | Kaempferol-3-O-α-l-rhamnopyranoside | >90% |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhu, R.; Hu, L.; Li, H.; Su, J.; Cao, Z.; Zhang, W. Novel Natural Inhibitors of CYP1A2 Identified by in Silico and in Vitro Screening. Int. J. Mol. Sci. 2011, 12, 3250-3262. https://doi.org/10.3390/ijms12053250
Zhu R, Hu L, Li H, Su J, Cao Z, Zhang W. Novel Natural Inhibitors of CYP1A2 Identified by in Silico and in Vitro Screening. International Journal of Molecular Sciences. 2011; 12(5):3250-3262. https://doi.org/10.3390/ijms12053250
Chicago/Turabian StyleZhu, Ruixin, Liwei Hu, Haiyun Li, Juan Su, Zhiwei Cao, and Weidong Zhang. 2011. "Novel Natural Inhibitors of CYP1A2 Identified by in Silico and in Vitro Screening" International Journal of Molecular Sciences 12, no. 5: 3250-3262. https://doi.org/10.3390/ijms12053250
APA StyleZhu, R., Hu, L., Li, H., Su, J., Cao, Z., & Zhang, W. (2011). Novel Natural Inhibitors of CYP1A2 Identified by in Silico and in Vitro Screening. International Journal of Molecular Sciences, 12(5), 3250-3262. https://doi.org/10.3390/ijms12053250