Molecular Basis for Chiral Selection in RNA Aminoacylation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
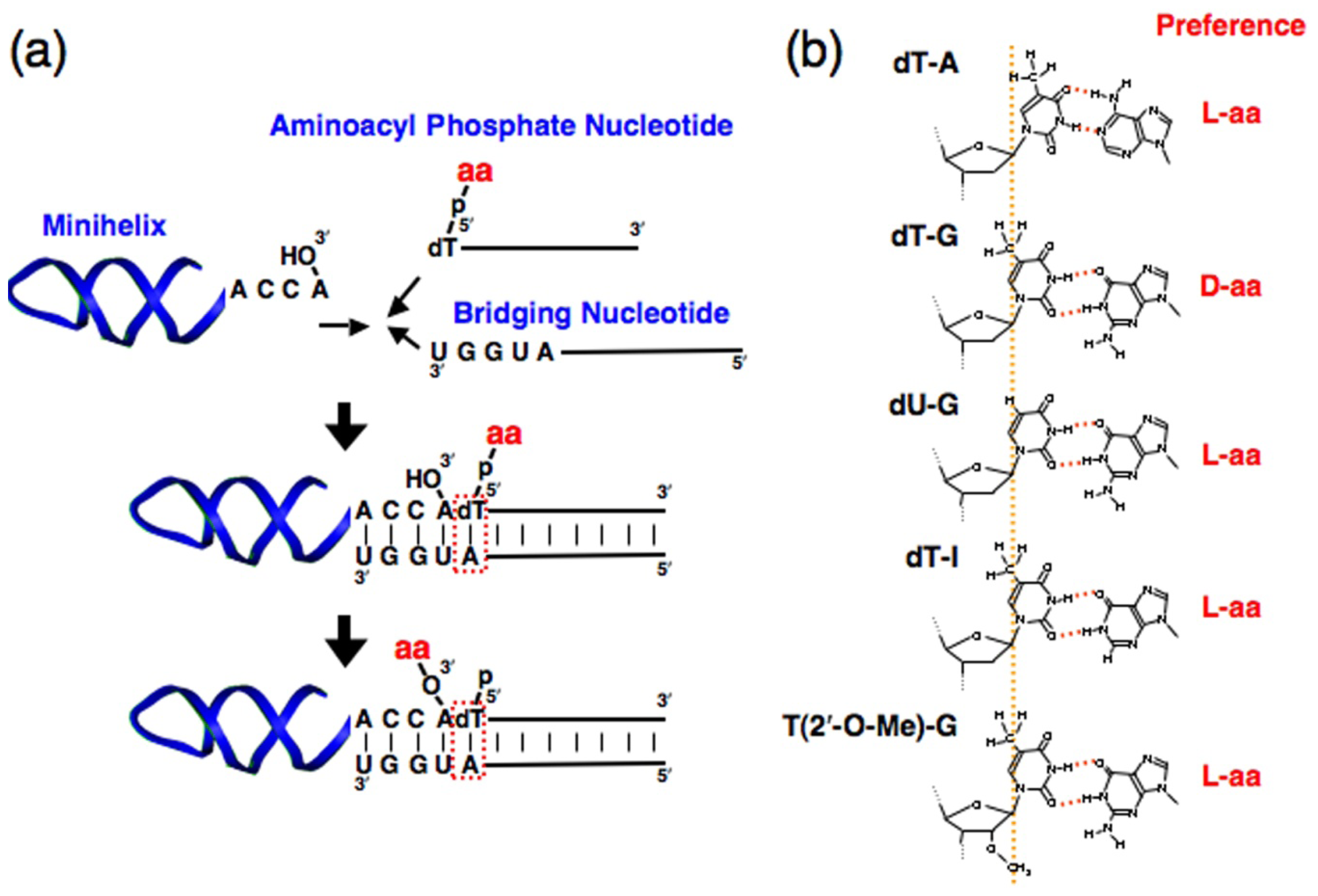
2. Chiral Selective Aminoacylation of RNA
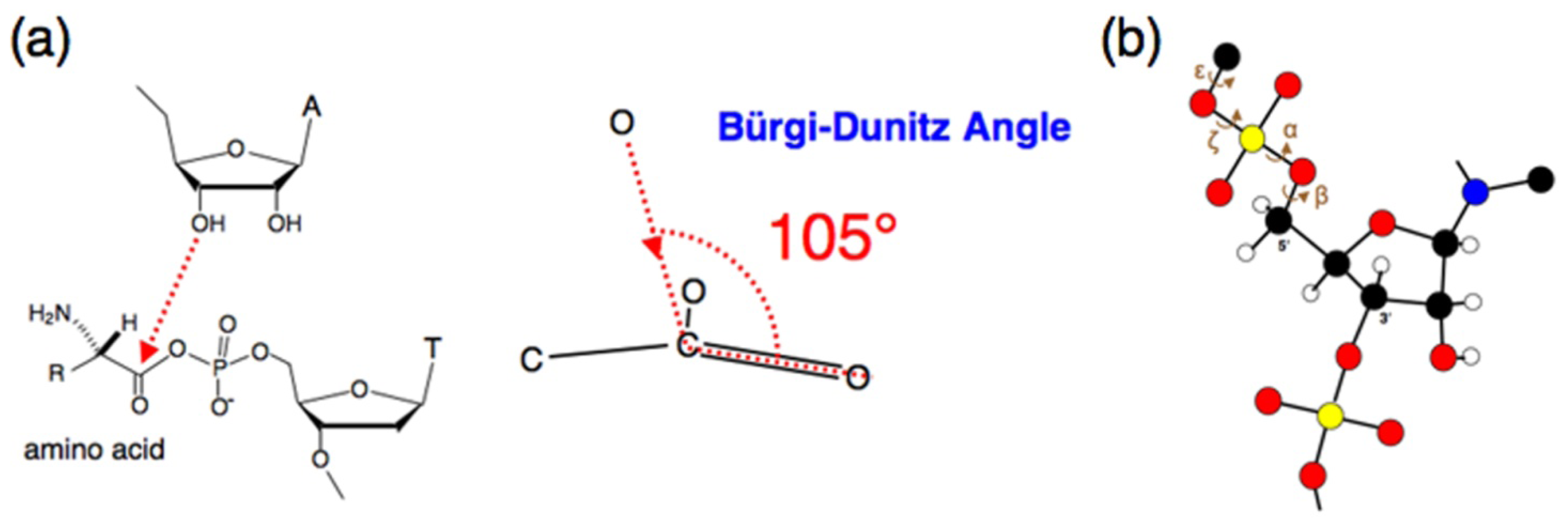
3. Chemical Features of Aminoacylation Reactions
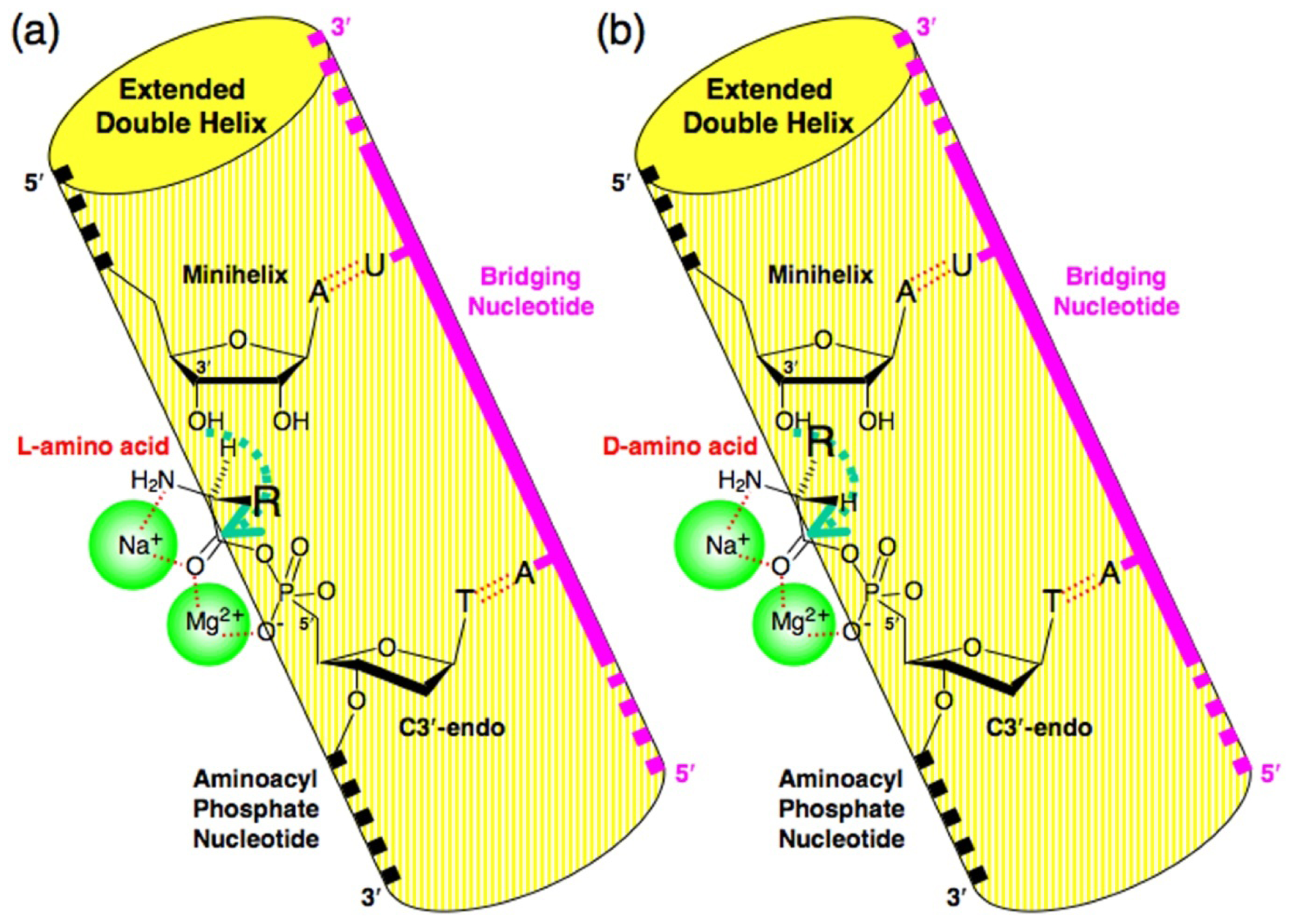
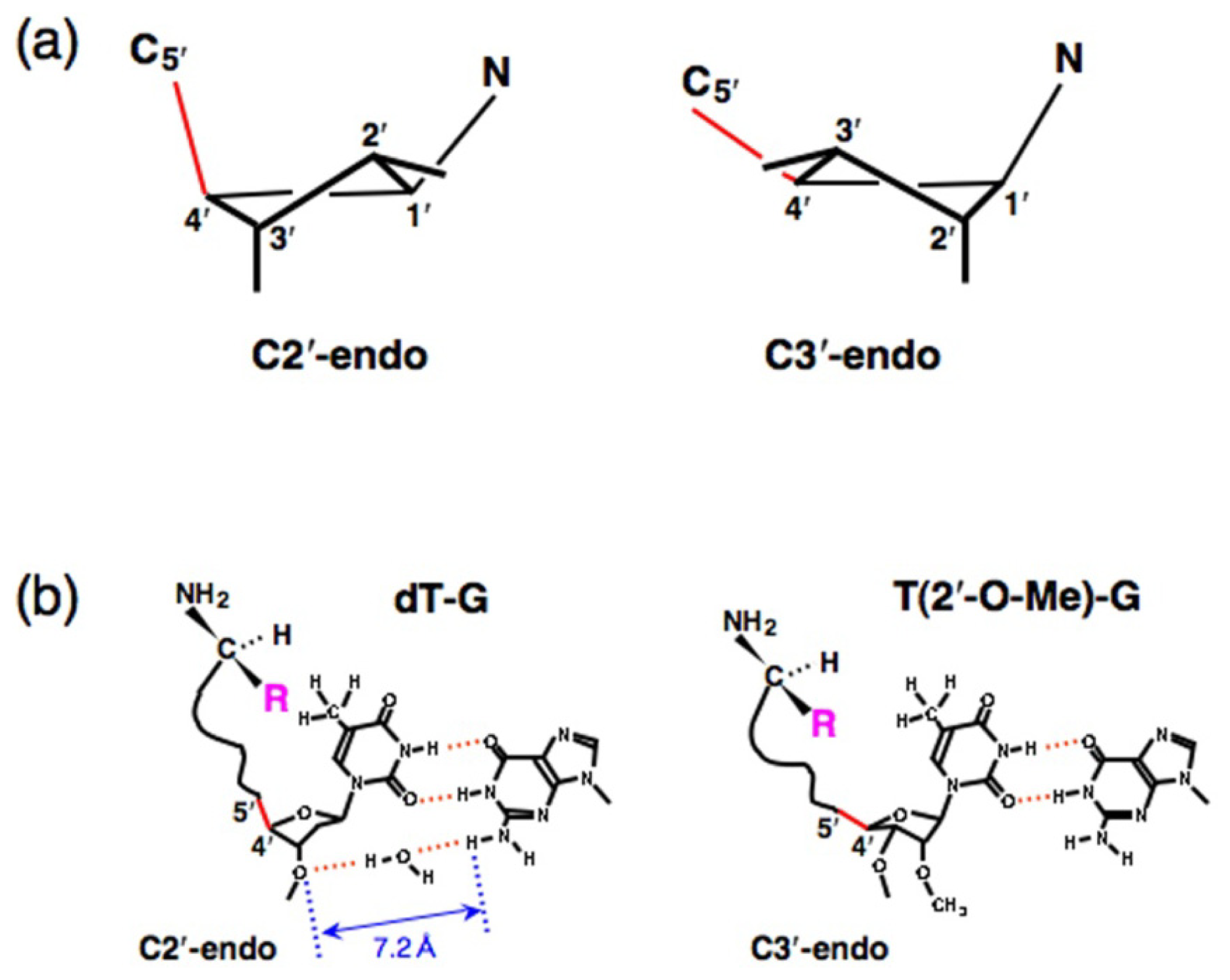
4. Proof of the Steric Effect Caused by the Positioning of Amino acid Side Chains in Aminoacylation
5. Conclusions
Acknowledgments
References
- Watson, JD; Crick, FHC. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar]
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561–563. [Google Scholar]
- Hannon, GJ. RNA interference. Nature 2002, 418, 244–251. [Google Scholar]
- Filipowicz, W. RNAi: The nuts and bolts of the RISC machine. Cell 2005, 122, 17–20. [Google Scholar]
- Siomi, H; Siomi, MC. On the road to reading the RNA-interference code. Nature 2009, 457, 396–404. [Google Scholar]
- Tamura, K; Alexander, RW. Peptide synthesis through evolution. Cell Mol Life Sci 2004, 61, 1317–1330. [Google Scholar]
- Tamura, K. Peptide bond formation: RNA’s big bang. J Cosmol 2011, 13, 3800–3810. [Google Scholar]
- Bonner, W. Physical Origin of Homochirality in Life; Cline, DB, Ed.; American Institute of Physics: New York, NY, USA, 1996. [Google Scholar]
- Hegstrom, RA. Parity violation and symmetry breaking of a racemic mixture. Biosystems 1987, 20, 49–56. [Google Scholar]
- Fukue, T; Tamura, M; Kandori, R; Kusakabe, N; Hough, JH; Bailey, J; Whittet, DC; Lucas, PW; Nakajima, Y; Hashimoto, J. Extended high circular polarization in the Orion massive star forming region: Implications for the origin of homochirality in the solar system. Orig Life Evol Biosph 2010, 40, 335–346. [Google Scholar]
- Soai, K; Shibata, T; Morioka, H; Choji, K. Asymmetric autocatalysis and amplification of enantiomeric excess of a chiral molecule. Nature 1995, 378, 767–768. [Google Scholar]
- Blackmond, DG. Asymmetric autocatalysis and its implications for the origin of homochirality. Proc Natl Acad Sci USA 2004, 101, 5732–5736. [Google Scholar]
- Kawasaki, T; Matsumura, Y; Tsutsumi, T; Suzuki, K; Ito, M; Soai, K. Asymmetric autocatalysis triggered by carbon isotope (13C/12C) chirality. Science 2009, 324, 492–495. [Google Scholar]
- Schimmel, P. Aminoacyl tRNA synthetases: General scheme of structure-function relationships in the polypeptides and recognition of transfer RNAs. Annu Rev Biochem 1987, 56, 125–158. [Google Scholar]
- Root-Bernstein, R. Simultaneous origin of homochirality, the genetic code and its directionality. BioEssays 2007, 29, 689–698. [Google Scholar]
- Paecht-Horowitz, M; Katchalsky, A. Synthesis of amino acyl-adenylates under prebiotic conditions. J Mol Evol 1973, 2, 91–98. [Google Scholar]
- Lohrmann, R; Bridson, PK; Orgel, LE. Efficient metal-ion catalyzed template-directed oligonucleotide synthesis. Science 1980, 208, 1464–1465. [Google Scholar]
- Schimmel, P; Giegé, R; Moras, D; Yokoyama, S. An operational RNA code for amino acids and possible relationship to genetic code. Proc Natl Acad Sci USA 1993, 90, 8763–8768. [Google Scholar]
- Tamura, K; Schimmel, P. Chiral-selective aminoacylation of an RNA minihelix. Science 2004, 305, 1253. [Google Scholar]
- Tamura, K; Schimmel, PR. Chiral-selective aminoacylation of an RNA minihelix: Mechanistic features and chiral suppression. Proc Natl Acad Sci USA 2006, 103, 13750–13752. [Google Scholar]
- Tamura, K. Origin of amino acid homochirality: Relationship with the RNA world and origin of tRNA aminoacylation. Biosystems 2008, 92, 91–98. [Google Scholar]
- Gilbert, W. The RNA world. Nature 1986, 319, 618. [Google Scholar]
- Kruger, K; Grabowski, PJ; Zaug, AJ; Sands, J; Gottschling, DE; Cech, TR. Self-splicing RNA: Autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 1982, 31, 147–157. [Google Scholar]
- Guerrier-Takada, C; Gardiner, K; Marsh, T; Pace, N; Altman, S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983, 35, 849–857. [Google Scholar]
- Tamura, K. Molecular handedness of life: Significance of RNA aminoacylation. J Biosci 2009, 34, 991–994. [Google Scholar]
- Tamura, K. Amino acid homochirality and the RNA world: Necessities for life on earth. J Cosmol 2010, 5, 883–889. [Google Scholar]
- Tamura, K. Origin of asymmetry of amino acids: Relationship to the RNA world. Biophysics 2010, 50, 180–181. [Google Scholar]
- Tamura, K. RNA-directed molecular asymmetry of amino acids. Viva Origino 2010, 38, 18–22. [Google Scholar]
- Eriani, G; Delarue, M; Poch, O; Gangloff, J; Moras, D. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature 1990, 347, 203–206. [Google Scholar]
- Carpenter, FH. The free energy in hydrolytic reactions: The non-ionized compound convention. J Am Chem Soc 1960, 82, 1111–1122. [Google Scholar]
- Bürgi, HB; Dunitz, JD; Lehn, JM; Wipff, G. Stereochemistry of reaction paths at carbonyl centre. Tetrahedron 1974, 30, 1563–1572. [Google Scholar]
- Tamura, K. Tokyo University of Science, Noda, Chiba, Japan; Unpublished work; 2006. [Google Scholar]
- Saenger, W. Principles of Nucleic Acid Structure; Springer: New York, NY, USA, 1984. [Google Scholar]
- IUPAC-IUB Joint Commission on Biochemical Nomenclature. Abbreviations and symbols for the description of conformations of polynucleotide chains. Eur J Biochem 1983, 131, 9–15.
- Eyring, H; Grant, DM; Hecht, H. The rotational barrier in ethane. J Chem Educ 1962, 39, 466–468. [Google Scholar]
- Lavelle, L; Fresco, JR. UV spectroscopic identification and thermodynamic analysis of protonated third strand deoxycytidine residues at neutrality in the triplex d(C+-T)6:[d(A-G)6d(C-T)6]; evidence for a proton switch. Nucleic Acids Res 1995, 23, 2692–2705. [Google Scholar]
- Grilley, D; Soto, AM; Draper, DE. Mg2+-RNA interaction free energies and their relationship to the folding of RNA tertiary structures. Proc Natl Acad Sci USA 2006, 103, 14003–14008. [Google Scholar]
- Cerda, BA; Hoyau, S; Ohanessian, G; Wesdemiotis, C. Na+ binding to cyclic and linear dipeptides. Bond energies, entropies of Na+ complexation, and attachment sites from the dissociation of Na+-bound heterodimers and ab initio calculations. J Am Chem Soc 1998, 120, 2437–2448. [Google Scholar]
- Bouchonnet, S; Hoppilliard, Y. Proton and sodium ion of affinities of glycine and its sodium salt in the gas phase. Ab initio calculations. J Mass Spectrom 1992, 27, 71–76. [Google Scholar]
- Jensen, F. Structure and stability of complexes of glycine and glycine methyl analogs with H+, Li+, and Na+. J Am Chem Soc 1992, 114, 9533–9537. [Google Scholar]
- Meksuriyen, D; Fukuchi-Shimogori, T; Tomitori, H; Kashiwagi, K; Toida, T; Imanari, T; Kawai, G; Igarashi, K. Formation of a complex containing ATP, Mg2+, and spermine. Structural evidence and biological significance. J Biol Chem 1998, 273, 30939–30944. [Google Scholar]
- Joyce, CM; Steitz, TA. Polymerase structures and function: Variations on a theme. J Bacteriol 1995, 177, 6321–6329. [Google Scholar]
- Steitz, TA. DNA polymerases: Structural diversity and common mechanisms. J Biol Chem 1999, 274, 17395–17398. [Google Scholar]
- Song, Z; Sims, A; Eady, J; Zhao, H; Olubajo, O. Determining nucleotide acidity and cation binding constants by 31P NMR. Can J Anal Sci Spec 2008, 53, 45–51. [Google Scholar]
- Gorenstein, DG. Phosphorus-31NMR, Principles and Applications; Academic Press: Orlando, FL, USA, 1984. [Google Scholar]
- Crick, FHC. Codon-anticodon pairing: The wobble hypothesis. J Mol Biol 1966, 19, 548–555. [Google Scholar]
- Yokoyama, S; Watanabe, T; Murao, K; Ishikura, H; Yamaizumi, Z; Nishimura, S; Miyazawa, T. Molecular mechanism of codon recognition by tRNA species with modified uridine in the first position of anticodon. Proc Natl Acad Sci USA 1985, 82, 4905–4909. [Google Scholar]
- Osawa, S; Jukes, TH; Watanabe, K; Muto, A. Recent evidence for evolution of the genetic code. Microbiol Rev 1992, 56, 229–264. [Google Scholar]
- Venkateswarlu, D; Lind, KE; Mohan, V; Manoharan, M; Ferguson, DM. Structural properties of DNA:RNA duplexes containing 2′-O-methyl and 2′-S-methyl substitutions: A molecular dynamics investigation. Nucleic Acids Res 1999, 27, 2189–2195. [Google Scholar]
- Isaacs, RJ; Rayens, WS; Spielmann, HP. Structural differences in the NOE-derived structure of G-T mismatched DNA relative to normal DNA are correlated with differences in 13C relaxation-based internal dynamics. J Mol Biol 2002, 319, 191–207. [Google Scholar]
- Arnott, S; Chandrasekaran, R; Millane, RP; Park, H-S. DNA-RNA hybrid secondary structures. J Mol Biol 1986, 188, 631–640. [Google Scholar]
- Tinoco, I, Jr; Borer, PN; Dengler, B; Levine, MD. Improved estimation of secondary structure in ribonucleic acids. Nat New Biol 1973, 246, 40–41. [Google Scholar]
- Bailey, JM. RNA-directed amino acid homochirality. FASEB J 1998, 12, 503–507. [Google Scholar]
- Profy, AT; Usher, DA. Stereoselective aminoacylation of a dinucleotide monophosphate by the imidazolides of dl-alanine and N-(tert-butoxycarbonyl)-dl-alanine. J Mol Evol 1984, 20, 147–156. [Google Scholar]
- Saxinger, C; Ponnamperuma, C; Woese, C. Evidence for the interaction of nucleotides with immobilized amino-acids and its significance for the origin of the genetic code. Nat New Biol 1971, 234, 172–174. [Google Scholar]
- Walker, GW. Nucleotide-binding site data and the origin of the genetic code. Biosystems 1977, 9, 139–150. [Google Scholar]
- Shimizu, M. Molecular basis for the genetic code. J Mol Evol 1982, 18, 297–303. [Google Scholar]
- Yarus, M. A specific amino acid binding site composed of RNA. Science 1988, 240, 1751–1758. [Google Scholar]
- Hobish, MK; Wickramasinghe, NS; Ponnamperuma, C. Direct interaction between amino acids and nucleotides as a possible physicochemical basis for the origin of the genetic code. Adv Space Res 1995, 13, 365–382. [Google Scholar]
- Yarus, M. RNA-ligand chemistry: A testable source for the genetic code. RNA 2000, 6, 475–484. [Google Scholar]
- Legiewicz, M; Yarus, M. A more complex isoleucine aptamer with a cognate triplet. J Biol Chem 2005, 280, 19815–19822. [Google Scholar]
- Majerfield, I; Puthenvedu, D; Yarus, M. RNA affinity for molecular l-histidine; genetic code origins. J Mol Evol 2005, 61, 226–235. [Google Scholar]
- Majerfield, I; Yarus, M. A diminutive and specific RNA binding site for l-tryptophan. Nucleic Acids Res 2005, 33, 5482–5493. [Google Scholar]
- Brack, A; Spach, G. Structures of polypeptides with l- and d-residues, part III: Experimental evidence for enrichment in enantiomers. J Mol Evol 1980, 15, 231–238. [Google Scholar]
- Brack, A; Spach, G. Enantiomer enrichment in early peptides. Orig Life 1981, 11, 135–142. [Google Scholar]




© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tamura, K. Molecular Basis for Chiral Selection in RNA Aminoacylation. Int. J. Mol. Sci. 2011, 12, 4745-4757. https://doi.org/10.3390/ijms12074745
Tamura K. Molecular Basis for Chiral Selection in RNA Aminoacylation. International Journal of Molecular Sciences. 2011; 12(7):4745-4757. https://doi.org/10.3390/ijms12074745
Chicago/Turabian StyleTamura, Koji. 2011. "Molecular Basis for Chiral Selection in RNA Aminoacylation" International Journal of Molecular Sciences 12, no. 7: 4745-4757. https://doi.org/10.3390/ijms12074745
APA StyleTamura, K. (2011). Molecular Basis for Chiral Selection in RNA Aminoacylation. International Journal of Molecular Sciences, 12(7), 4745-4757. https://doi.org/10.3390/ijms12074745