Molecular Mechanisms of Cardiotoxicity Induced by ErbB Receptor Inhibitor Cancer Therapeutics

Abstract
:1. Introduction
2. ErbB Receptors and Their Ligand
3. ErbB Receptors and Cancer
4. ErbB Receptors as Targets for Cancer Therapy
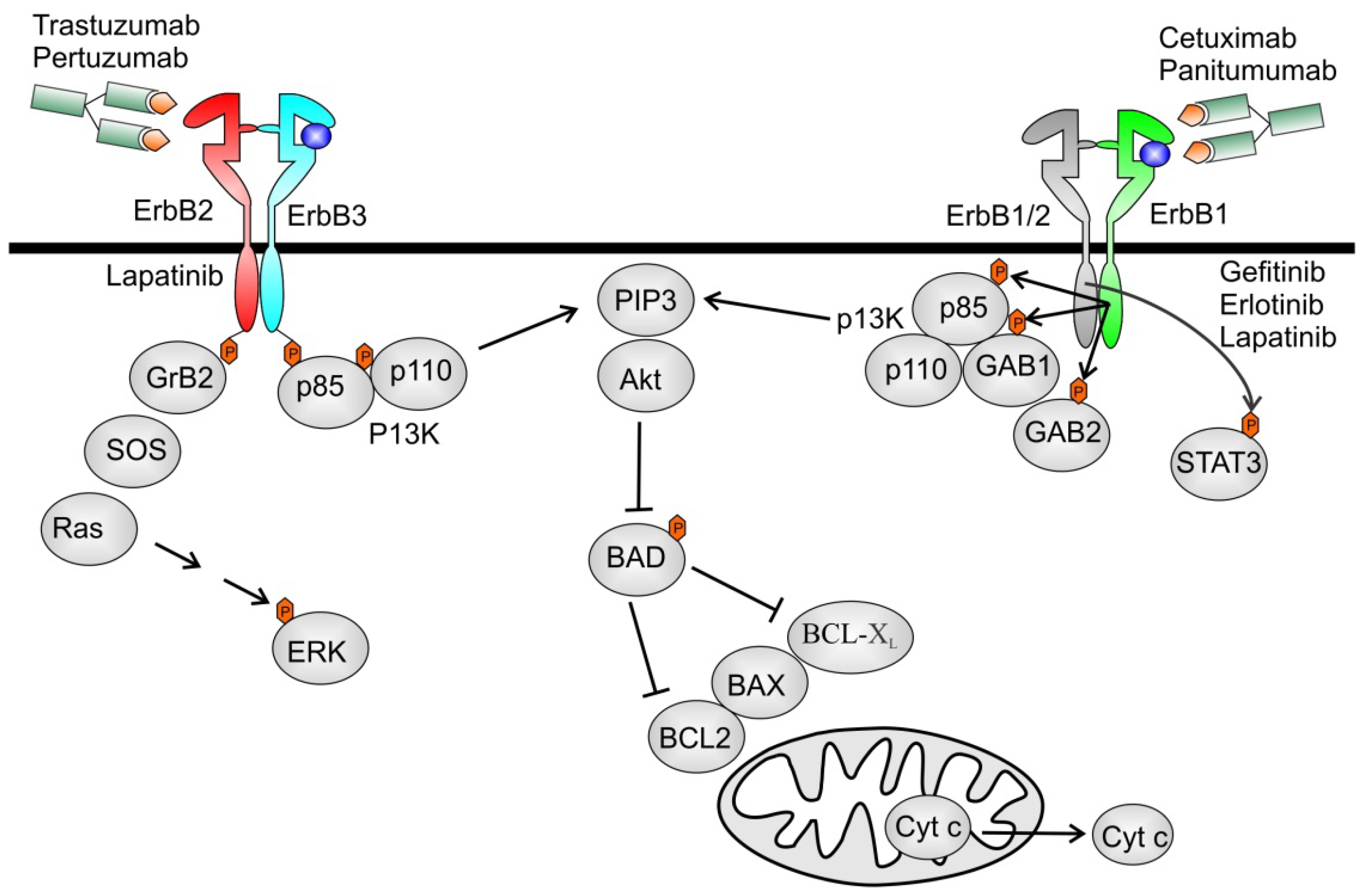
5. Cardiotoxicity
5.1. Agents Targeting ErbB1
Cardiotoxicity of ErbB1 Inhibitors
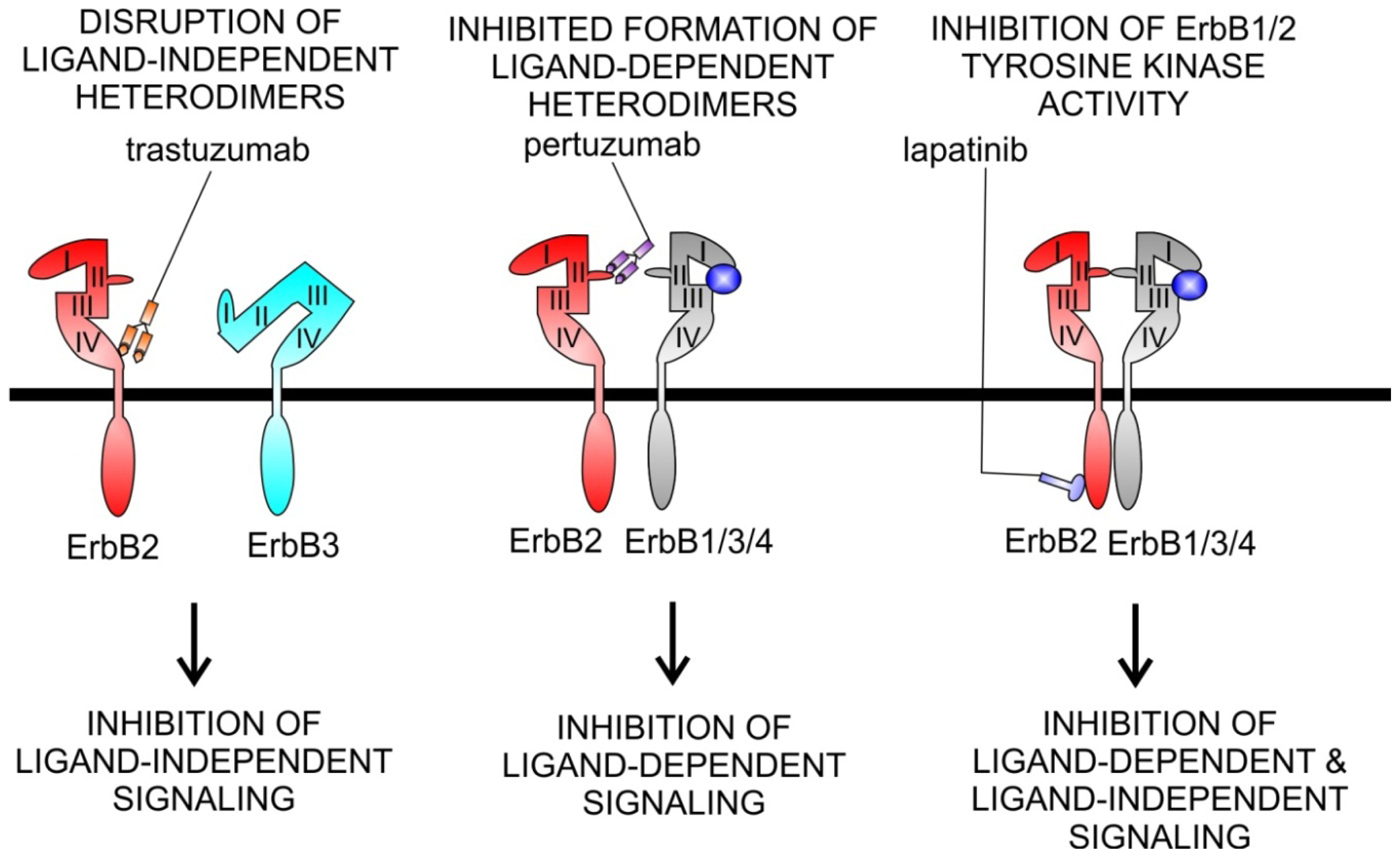
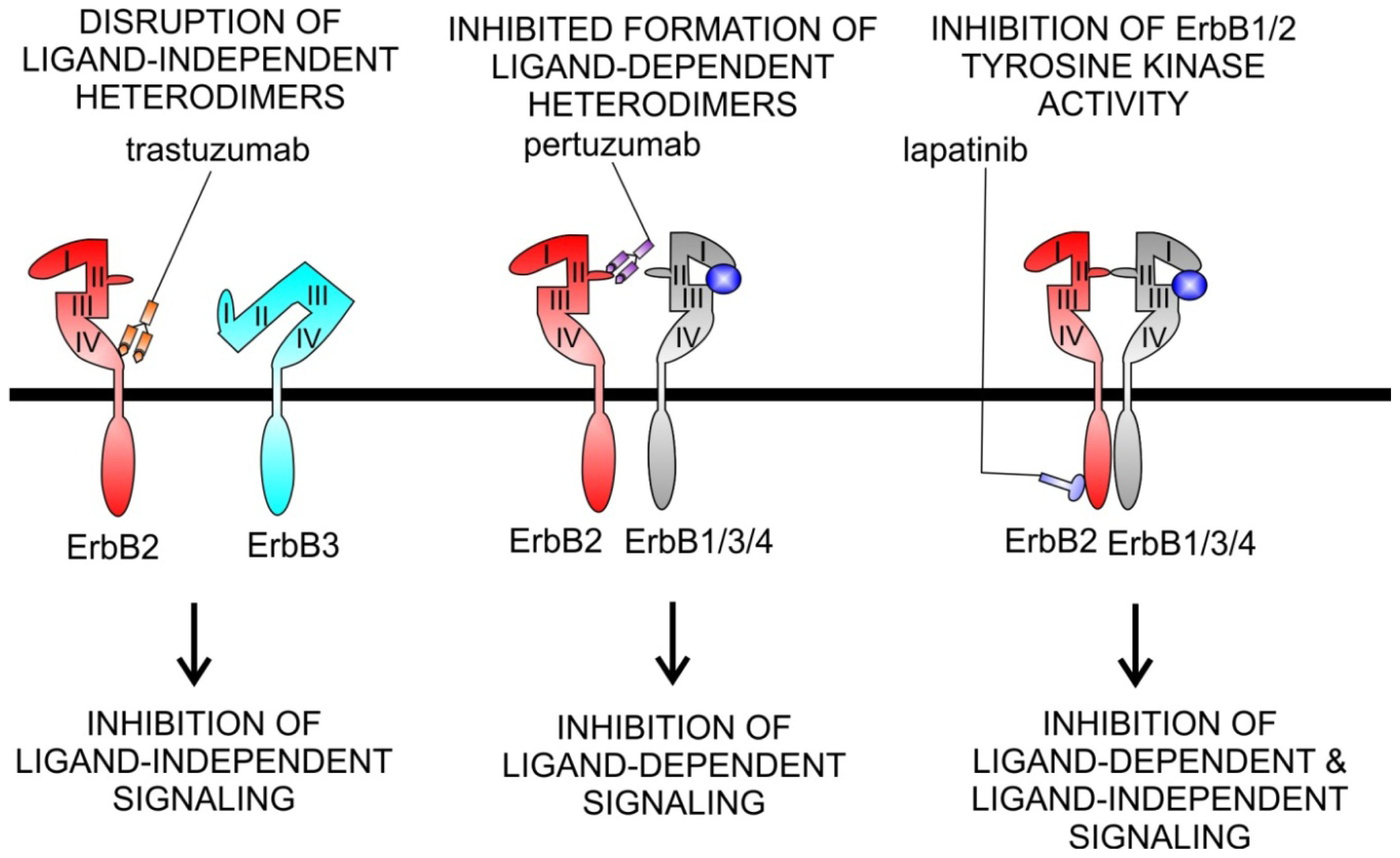
5.2. Agents Targeting ErbB2
Cardiotoxicity of ErbB2 Inhibitors
5.3. Agents Targeting ErbB3/ErbB4
6. Concluding Remarks
References
- Stupp, R.; Hegi, M.E.; van den Bent, M.J.; Mason, W.P.; Weller, M.; Mirimanoff, R.O.; Cairncross, J.G. Changing paradigms—An update on the multidisciplinary management of malignant glioma. Oncologist 2006, 11, 165–180. [Google Scholar]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against her2 for metastatic breast cancer that overexpresses her2. N. Engl. J. Med 2001, 344, 783–792. [Google Scholar]
- Yap, T.A.; Carden, C.P.; Kaye, S.B. Beyond chemotherapy: Targeted therapies in ovarian cancer. Nat. Rev. Cancer 2009, 9, 167–181. [Google Scholar]
- Agus, D.B.; Sweeney, C.J.; Morris, M.J.; Mendelson, D.S.; McNeel, D.G.; Ahmann, F.R.; Wang, J.; Derynck, M.K.; Ng, K.; Lyons, B.; et al. Efficacy and safety of single-agent pertuzumab (rhumab 2c4), a human epidermal growth factor receptor dimerization inhibitor, in castration-resistant prostate cancer after progression from taxane-based therapy. J. Clin. Oncol 2007, 25, 675–681. [Google Scholar]
- Starling, N.; Neoptolemos, J.; Cunningham, D. Role of erlotinib in the management of pancreatic cancer. Ther. Clin. Risk Manag 2006, 2, 435–445. [Google Scholar]
- Chung, K.Y.; Shia, J.; Kemeny, N.E.; Shah, M.; Schwartz, G.K.; Tse, A.; Hamilton, A.; Pan, D.; Schrag, D.; Schwartz, L.; et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J. Clin. Oncol 2005, 23, 1803–1810. [Google Scholar]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Eng. J. Med 2006, 354, 567–578. [Google Scholar]
- Holbro, T.; Hynes, N.E. Erbb receptors: Directing key signaling networks throughout life. Annu. Rev. Pharmacol. Toxicol 2004, 44, 195–217. [Google Scholar]
- Falls, D.L. Neuregulins: Functions, forms, and signaling strategies. Exp. Cell Res 2003, 284, 14–30. [Google Scholar]
- Burden, S.; Yarden, Y. Neuregulins and their receptors: A versatile signaling module in organogenesis and oncogenesis. Neuron 1997, 18, 847–855. [Google Scholar]
- Marmor, M.D.; Skaria, K.B.; Yarden, Y. Signal transduction and oncogenesis by erbb/her receptors. Int. J. Rad. Oncol. Biol. Phys 2004, 58, 903–913. [Google Scholar]
- Keefe, D.L. Trastuzumab-associated cardiotoxicity. Cancer 2002, 95, 1592–1600. [Google Scholar]
- Force, T.; Krause, D.S.; van Etten, R.A. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat. Rev. Cancer 2007, 7, 332–344. [Google Scholar]
- Stevenson, L.W.; Perloff, J.K. The limited reliability of physical signs for estimating hemodynamics in chronic heart failure. J. Am. Med. Assoc 1989, 261, 884–888. [Google Scholar]
- Riese, D.J.; Stern, D.F. Specificity within the egf family/erbb receptor family signaling network. BioEssays 1998, 20, 41–48. [Google Scholar]
- Yarden, Y.; Sliwkowski, M.X. Untangling the erbb signalling network. Nat. Rev 2001, 2, 127–137. [Google Scholar]
- Yarden, Y. The egfr family and its ligands in human cancer: Signalling mechanisms and therapeutic opportunities. Eur. J. Cancer 2001, 37 Suppl 4, S3–8. [Google Scholar]
- Earp, H.S.; Calvo, B.F.; Sartor, C.I. The egf receptor family—Multiple roles in proliferation, differentiation, and neoplasia with an emphasis on her4. Trans. Am. Clin. Climatol. Assoc 2003, 114, 315–333, discussion 333–314. [Google Scholar]
- Schlessinger, J. Common and distinct elements in cellular signaling via egf and fgf receptors. Science 2004, 306, 1506–1507. [Google Scholar]
- Lee, K.F.; Simon, H.; Chen, H.; Bates, B.; Hung, M.C.; Hauser, C. Requirement for neuregulin receptor erbb2 in neural and cardiac development. Nature 1995, 378, 394–398. [Google Scholar]
- Gassmann, M.; Casagranda, F.; Orioli, D.; Simon, H.; Lai, C.; Klein, R.; Lemke, G. Aberrant neural and cardiac development in mice lacking the erbb4 neuregulin receptor. Nature 1995, 378, 390–394. [Google Scholar]
- Meyer, D.; Birchmeier, C. Multiple essential functions of neuregulin in development. Nature 1995, 378, 386–390. [Google Scholar]
- Erickson, S.L.; O’Shea, K.S.; Ghaboosi, N.; Loverro, L.; Frantz, G.; Bauer, M.; Lu, L.H.; Moore, M.W. Erbb3 is required for normal cerebellar and cardiac development: A comparison with erbb2-and heregulin-deficient mice. Development 1997, 124, 4999–5011. [Google Scholar]
- Miettinen, P.J.; Berger, J.E.; Meneses, J.; Phung, Y.; Pedersen, R.A.; Werb, Z.; Derynck, R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 1995, 376, 337–341. [Google Scholar]
- Sibilia, M.; Wagner, E.F. Strain-dependent epithelial defects in mice lacking the egf receptor. Science 1995, 269, 234–238. [Google Scholar]
- Zhao, Y.Y.; Sawyer, D.R.; Baliga, R.R.; Opel, D.J.; Han, X.; Marchionni, M.A.; Kelly, R.A. Neuregulins promote survival and growth of cardiac myocytes. Persistence of erbb2 and erbb4 expression in neonatal and adult ventricular myocytes. J. Biol. Chem 1998, 273, 10261–10269. [Google Scholar]
- Salomon, D.S.; Brandt, R.; Ciardiello, F.; Normanno, N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit. Rev. Oncol. Hematol 1995, 19, 183–232. [Google Scholar]
- Gorgoulis, V.; Aninos, D.; Mikou, P.; Kanavaros, P.; Karameris, A.; Joardanoglou, J.; Rasidakis, A.; Veslemes, M.; Ozanne, B.; Spandidos, D.A. Expression of egf, tgf-alpha and egfr in squamous cell lung carcinomas. Anticancer Res 1992, 12, 1183–1187. [Google Scholar]
- Irish, J.C.; Bernstein, A. Oncogenes in head and neck cancer. Laryngoscope 1993, 103, 42–52. [Google Scholar]
- Ekstrand, A.J.; Sugawa, N.; James, C.D.; Collins, V.P. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc. Natl. Acad. Sci. USA 1992, 89, 4309–4313. [Google Scholar]
- Moscatello, D.K.; Holgado-Madruga, M.; Godwin, A.K.; Ramirez, G.; Gunn, G.; Zoltick, P.W.; Biegel, J.A.; Hayes, R.L.; Wong, A.J. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res 1995, 55, 5536–5539. [Google Scholar]
- Benz, C.C.; Scott, G.K.; Sarup, J.C.; Johnson, R.M.; Tripathy, D.; Coronado, E.; Shepard, H.M.; Osborne, C.K. Estrogen-dependent, tamoxifen-resistant tumorigenic growth of mcf-7 cells transfected with her2/neu. Breast Cancer Res. Treat 1992, 24, 85–95. [Google Scholar]
- Yang, H.; Zhao, R.; Yang, H.Y.; Lee, M.H. Constitutively active foxo4 inhibits akt activity, regulates p27 kip1 stability, and suppresses her2-mediated tumorigenicity. Oncogene 2005, 24, 1924–1935. [Google Scholar]
- Borg, A.; Baldetorp, B.; Ferno, M.; Killander, D.; Olsson, H.; Sigurdsson, H. Erbb2 amplification in breast cancer with a high rate of proliferation. Oncogene 1991, 6, 137–143. [Google Scholar]
- Hynes, N.E.; Stern, D.F. The biology of erbb-2/neu/her-2 and its role in cancer. Biochim. Biophys. Acta 1994, 30, 2–3. [Google Scholar]
- Stephens, P.; Hunter, C.; Bignell, G.; Edkins, S.; Davies, H.; Teague, J.; Stevens, C.; O’Meara, S.; Smith, R.; Parker, A.; et al. Lung cancer: Intragenic erbb2 kinase mutations in tumours. Nature 2004, 431, 525–526. [Google Scholar]
- Holbro, T.; Beerli, R.R.; Maurer, F.; Koziczak, M.; Barbas, C.F., III; Hynes, N.E. The erbb2/erbb3 heterodimer functions as an oncogenic unit: Erbb2 requires erbb3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. USA 2003, 100, 8933–8938. [Google Scholar]
- Kew, T.Y.; Bell, J.A.; Pinder, S.E.; Denley, H.; Srinivasan, R.; Gullick, W.J.; Nicholson, R.I.; Blamey, R.W.; Ellis, I.O. C-erbb-4 protein expression in human breast cancer. Br. J. Cancer 2000, 82, 1163–1170. [Google Scholar]
- Gilbertson, R.J.; Perry, R.H.; Kelly, P.J.; Pearson, A.D.; Lunec, J. Prognostic significance of her2 and her4 coexpression in childhood medulloblastoma. Cancer Res 1997, 57, 3272–3280. [Google Scholar]
- Iannello, A.; Ahmad, A. Role of antibody-dependent cell-mediated cytotoxicity in the efficacy of therapeutic anti-cancer monoclonal antibodies. Cancer Metastasis Rev 2005, 24, 487–499. [Google Scholar]
- Imai, K.; Takaoka, A. Comparing antibody and small-molecule therapies for cancer. Nat. Rev. Cancer 2006, 6, 714–727. [Google Scholar]
- Carter, P.J. Potent antibody therapeutics by design. Nat. Rev. Immunol 2006, 6, 343–357. [Google Scholar]
- Dancey, J.; Sausville, E.A. Issues and progress with protein kinase inhibitors for cancer treatment. Nat. Rev. Drug Discov 2003, 2, 296–313. [Google Scholar]
- Huang, S.; Armstrong, E.A.; Benavente, S.; Chinnaiyan, P.; Harari, P.M. Dual-agent molecular targeting of the epidermal growth factor receptor (egfr): Combining anti-egfr antibody with tyrosine kinase inhibitor. Cancer Res 2004, 64, 5355–5362. [Google Scholar]
- Maitland, M.L.; Ratain, M.J. Terminal ballistics of kinase inhibitors: There are no magic bullets. Ann. Intern. Med 2006, 145, 702–703. [Google Scholar]
- Cheng, H.; Force, T. Why do kinase inhibitors cause cardiotoxicity and what can be done about it? Prog. Cardiovas. Dis 2010, 53, 114–120. [Google Scholar]
- Baselga, J. The egfr as a target for anticancer therapy—Focus on cetuximab. Eur. J. Cancer 2001, 37, S16–S22. [Google Scholar]
- Goldberg, R.M. Cetuximab. Nat. Rev. Drug Discov 2005, 1, S10–S11. [Google Scholar]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar]
- Herbst, R.S.; Fukuoka, M.; Baselga, J. Gefitinib [mdash] a novel targeted approach to treating cancer. Nat. Rev. Cancer 2004, 4, 979–987. [Google Scholar]
- Minna, J.D.; Dowell, J. Erlotinib hydrochloride. Nat. Rev. Drug Discov 2005, 5, S14–S15. [Google Scholar]
- Jeon, E.K.; Won, H.S.; Ko, Y.H.; Lee, I.S.; Hong, T.H.; You, Y.K.; Lee, M.A. Comparison of the efficacy and the toxicity between gemcitabine with capecitabine (gc) and gemcitabine with erlotinib (ge) in unresectable pancreatic cancer. J. Cancer Res. Clin. Oncol 2012, 138, 1625–1630. [Google Scholar]
- Xia, W.; Gerard, C.M.; Liu, L.; Baudson, N.M.; Ory, T.L.; Spector, N.L. Combining lapatinib (gw572016), a small molecule inhibitor of erbb1 and erbb2 tyrosine kinases, with therapeutic anti-erbb2 antibodies enhances apoptosis of erbb2-overexpressing breast cancer cells. Oncogene 2005, 24, 6213–6221. [Google Scholar]
- Silvano, G.; Lazzari, G.; Lovecchio, M.; Palazzo, C. Acute and fatal diarrhoea after erlotinib plus abdominal palliative hypofractionated radiotherapy in a metastatic non-small cell lung cancer patient: A case report. Lung Cancer 2008, 61, 270–273. [Google Scholar]
- Inoue, A.; Saijo, Y.; Maemondo, M.; Gomi, K.; Tokue, Y.; Kimura, Y.; Ebina, M.; Kikuchi, T.; Moriya, T.; Nukiwa, T. Severe acute interstitial pneumonia and gefitinib. Lancet 2003, 361, 137–139. [Google Scholar]
- Chen, M.H.; Kerkela, R.; Force, T. Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapeutics. Circulation 2008, 118, 84–95. [Google Scholar]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the her-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar]
- Romond, E.H.; Perez, E.A.; Bryant, J.; Suman, V.J.; Geyer, C.E.; Davidson, N.E.; Tan-Chiu, E.; Martino, S.; Paik, S.; Kaufman, P.A.; et al. Trastuzumab plus adjuvant chemotherapy for operable her2-positive breast cancer. N. Eng. J. Med 2005, 353, 1673–1684. [Google Scholar]
- Dahabreh, I.J.; Linardou, H.; Siannis, F.; Fountzilas, G.; Murray, S. Trastuzumab in the adjuvant treatment of early-stage breast cancer: A systematic review and meta-analysis of randomized controlled trials. Oncologist 2008, 13, 620–630. [Google Scholar]
- Joensuu, H.; Kellokumpu-Lehtinen, P.L.; Bono, P.; Alanko, T.; Kataja, V.; Asola, R.; Utriainen, T.; Kokko, R.; Hemminki, A.; Tarkkanen, M.; et al. Adjuvant docetaxel or vinorelbine with or without trastuzumab for breast cancer. N. Engl. J. Med 2006, 354, 809–820. [Google Scholar]
- Hudis, C.A. Trastuzumab—Mechanism of action and use in clinical practice. N. Engl. J. Med 2007, 357, 39–51. [Google Scholar]
- Golay, J.; Introna, M. Mechanism of action of therapeutic monoclonal antibodies: Promises and pitfalls of in vitro and in vivo assays. Arch. Biochem. Biophys 2012, 526, 146–153. [Google Scholar]
- Garrett, T.P.J.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Kofler, M.; Jorissen, R.N.; Nice, E.C.; Burgess, A.W.; et al. The crystal structure of a truncated erbb2 ectodomain reveals an active conformation, poised to interact with other erbb receptors. Mol. Cell 2003, 11, 495–505. [Google Scholar]
- Molina, M.A.; Codony-Servat, J.; Albanell, J.; Rojo, F.; Arribas, J.; Baselga, J. Trastuzumab (herceptin), a humanized anti-her2 receptor monoclonal antibody, inhibits basal and activated her2 ectodomain cleavage in breast cancer cells. Cancer Res 2001, 61, 4744–4749. [Google Scholar]
- Christianson, T.A.; Doherty, J.K.; Lin, Y.J.; Ramsey, E.E.; Holmes, R.; Keenan, E.J.; Clinton, G.M. Nh2-terminally truncated her-2/neu protein: Relationship with shedding of the extracellular domain and with prognostic factors in breast cancer. Cancer Res 1998, 58, 5123–5129. [Google Scholar]
- Cooley, S.; Burns, L.J.; Repka, T.; Miller, J.S. Natural killer cell cytotoxicity of breast cancer targets is enhanced by two distinct mechanisms of antibody-dependent cellular cytotoxicity against lfa-3 and her2/neu. Exp. Hematol 1999, 27, 1533–1541. [Google Scholar]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med 2000, 6, 443–446. [Google Scholar]
- Cameron, D.A.; Stein, S. Drug insight: Intracellular inhibitors of her2[mdash]clinical development of lapatinib in breast cancer. Nat. Clin. Prac. Oncol 2008, 5, 512–520. [Google Scholar]
- Valachis, A.; Nearchou, A.; Lind, P.; Mauri, D. Lapatinib, trastuzumab or the combination added to preoperative chemotherapy for breast cancer: A meta-analysis of randomized evidence. Breast Cancer Res. Treat 2012, 135, 655–662. [Google Scholar]
- Baselga, J.; Bradbury, I.; Eidtmann, H.; Di Cosimo, S.; de Azambuja, E.; Aura, C.; Gomez, H.; Dinh, P.; Fauria, K.; van Dooren, V.; et al. Lapatinib with trastuzumab for her2-positive early breast cancer (neoaltto): A randomised, open-label, multicentre, phase 3 trial. Lancet 2012, 379, 633–640. [Google Scholar]
- Rusnak, D.W.; Lackey, K.; Affleck, K.; Wood, E.R.; Alligood, K.J.; Rhodes, N.; Keith, B.R.; Murray, D.M.; Knight, W.B.; Mullin, R.J.; et al. The effects of the novel, reversible epidermal growth factor receptor/erbb-2 tyrosine kinase inhibitor, gw2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol. Cancer Ther 2001, 1, 85–94. [Google Scholar]
- Taskar, K.S.; Rudraraju, V.; Mittapalli, R.K.; Samala, R.; Thorsheim, H.R.; Lockman, J.; Gril, B.; Hua, E.; Palmieri, D.; Polli, J.W.; et al. Lapatinib distribution in her2 overexpressing experimental brain metastases of breast cancer. Pharm. Res 2012, 29, 770–781. [Google Scholar]
- Lim, E.; Lin, N.U. New insights and emerging therapies for breast cancer brain metastases. Oncology 2012, 26. [Google Scholar]
- Agus, D.B.; Akita, R.W.; Fox, W.D.; Lewis, G.D.; Higgins, B.; Pisacane, P.I.; Lofgren, J.A.; Tindell, C.; Evans, D.P.; Maiese, K.; et al. Targeting ligand-activated erbb2 signaling inhibits breast and prostate tumor growth. Cancer Cell 2002, 2, 127–137. [Google Scholar]
- Gordon, M.S.; Matei, D.; Aghajanian, C.; Matulonis, U.A.; Brewer, M.; Fleming, G.F.; Hainsworth, J.D.; Garcia, A.A.; Pegram, M.D.; Schilder, R.J.; et al. Clinical activity of pertuzumab (rhumab 2c4), a her dimerization inhibitor, in advanced ovarian cancer: Potential predictive relationship with tumor her2 activation status. J. Clin. Oncol 2006, 24, 4324–4332. [Google Scholar]
- Gianni, L.; Llado, A.; Bianchi, G.; Cortes, J.; Kellokumpu-Lehtinen, P.L.; Cameron, D.A.; Miles, D.; Salvagni, S.; Wardley, A.; Goeminne, J.C.; et al. Open-label, phase ii, multicenter, randomized study of the efficacy and safety of two dose levels of pertuzumab, a human epidermal growth factor receptor 2 dimerization inhibitor, in patients with human epidermal growth factor receptor 2-negative metastatic breast cancer. J. Clin. Oncol 2010, 28, 1131–1137. [Google Scholar]
- De Bono, J.S.; Bellmunt, J.; Attard, G.; Droz, J.P.; Miller, K.; Flechon, A.; Sternberg, C.; Parker, C.; Zugmaier, G.; Hersberger-Gimenez, V.; et al. Open-label phase ii study evaluating the efficacy and safety of two doses of pertuzumab in castrate chemotherapy-naive patients with hormone-refractory prostate cancer. J. Clin. Oncol 2007, 25, 257–262. [Google Scholar]
- Herbst, R.S.; Davies, A.M.; Natale, R.B.; Dang, T.P.; Schiller, J.H.; Garland, L.L.; Miller, V.A.; Mendelson, D.; van den Abbeele, A.D.; Melenevsky, Y.; et al. Efficacy and safety of single-agent pertuzumab, a human epidermal receptor dimerization inhibitor, in patients with non small cell lung cancer. Clin. Cancer Res 2007, 13, 6175–6181. [Google Scholar]
- Cho, H.S.; Mason, K.; Ramyar, K.X.; Stanley, A.M.; Gabelli, S.B.; Denney, D.W., Jr; Leahy, D.J. Structure of the extracellular region of her2 alone and in complex with the herceptin fab. Nature 2003, 421, 756–760. [Google Scholar]
- Franklin, M.C.; Carey, K.D.; Vajdos, F.F.; Leahy, D.J.; de Vos, A.M.; Sliwkowski, M.X. Insights into erbb signaling from the structure of the erbb2-pertuzumab complex. Cancer Cell 2004, 5, 317–328. [Google Scholar]
- Gianni, L.; Pienkowski, T.; Im, Y.H.; Roman, L.; Tseng, L.M.; Liu, M.C.; Lluch, A.; Staroslawska, E.; de la Haba-Rodriguez, J.; Im, S.A.; et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early her2-positive breast cancer (neosphere): A randomised multicentre, open-label, phase 2 trial. Lancet Oncol 2012, 13, 25–32. [Google Scholar]
- Baselga, J.; Cortes, J.; Kim, S.B.; Im, S.A.; Hegg, R.; Im, Y.H.; Roman, L.; Pedrini, J.L.; Pienkowski, T.; Knott, A.; et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N. Engl. J. Med 2012, 366, 109–119. [Google Scholar]
- Lemmens, K.; Segers, V.F.; Demolder, M.; de Keulenaer, G.W. Role of neuregulin-1/erbb2 signaling in endothelium-cardiomyocyte cross-talk. J. Biol. Chem 2006, 281, 19469–19477. [Google Scholar]
- Bersell, K.; Arab, S.; Haring, B.; Kühn, B. Neuregulin1/erbb4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 2009, 138, 257–270. [Google Scholar]
- Kuramochi, Y.; Guo, X.; Sawyer, D.B. Neuregulin activates erbb2-dependent src/fak signaling and cytoskeletal remodeling in isolated adult rat cardiac myocytes. J. Mol. Cell. Cardiol 2006, 41, 228–235. [Google Scholar]
- Russell, K.S.; Stern, D.F.; Polverini, P.J.; Bender, J.R. Neuregulin activation of erbb receptors in vascular endothelium leads to angiogenesis. Am. J. Physiol 1999, 277, H2205–H2211. [Google Scholar]
- Lemmens, K.; Fransen, P.; Sys, S.U.; Brutsaert, D.L.; de Keulenaer, G.W. Neuregulin-1 induces a negative inotropic effect in cardiac muscle: Role of nitric oxide synthase. Circulation 2004, 109, 324–326. [Google Scholar]
- De Keulenaer, G.W.; Doggen, K.; Lemmens, K. The vulnerability of the heart as a pluricellular paracrine organ: Lessons from unexpected triggers of heart failure in targeted erbb2 anticancer therapy. Circ. Res 2010, 106, 35–46. [Google Scholar]
- Seidman, A.; Hudis, C.; Pierri, M.K.; Shak, S.; Paton, V.; Ashby, M.; Murphy, M.; Stewart, S.J.; Keefe, D. Cardiac dysfunction in the trastuzumab clinical trials experience. J. Clin. Oncol 2002, 20, 1215–1221. [Google Scholar]
- Ewer, M.S.; Vooletich, M.T.; Durand, J.B.; Woods, M.L.; Davis, J.R.; Valero, V.; Lenihan, D.J. Reversibility of trastuzumab-related cardiotoxicity: New insights based on clinical course and response to medical treatment. J. Clin. Oncol 2005, 23, 7820–7826. [Google Scholar]
- Sawyer, D.B.; Peng, X.; Chen, B.; Pentassuglia, L.; Lim, C.C. Mechanisms of anthracycline cardiac injury: Can we identify strategies for cardioprotection? Prog. Cardiovasc. Dis 2010, 53, 105–113. [Google Scholar]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol 2012, 52, 1213–1225. [Google Scholar]
- Gianni, L.; Herman, E.H.; Lipshultz, S.E.; Minotti, G.; Sarvazyan, N.; Sawyer, D.B. Anthracycline cardiotoxicity: From bench to bedside. J. Clin. Oncol 2008, 26, 3777–3784. [Google Scholar]
- Burris, H.A., III; Hurwitz, H.I.; Dees, E.C.; Dowlati, A.; Blackwell, K.L.; O’Neil, B.; Marcom, P.K.; Ellis, M.J.; Overmoyer, B.; Jones, S.F.; et al. Phase i safety, pharmacokinetics, and clinical activity study of lapatinib (gw572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J. Clin. Oncol 2005, 23, 5305–5313. [Google Scholar]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus capecitabine for her2-positive advanced breast cancer. N. Engl. J. Med 2006, 355, 2733–2743. [Google Scholar]
- De Azambuja, E.; Bedard, P.; Suter, T.; Piccart-Gebhart, M. Cardiac toxicity with anti-her-2 therapies-what have we learned so far? Target. Oncol 2009, 4, 77–88. [Google Scholar]
- Sliwkowski, M.X.; Lofgren, J.A.; Lewis, G.D.; Hotaling, T.E.; Fendly, B.M.; Fox, J.A. Nonclinical studies addressing the mechanism of action of trastuzumab (herceptin). Semin. Oncol 1999, 26, 60–70. [Google Scholar]
- Lenihan, D.; Suter, T.; Brammer, M.; Neate, C.; Ross, G.; Baselga, J. Pooled analysis of cardiac safety in patients with cancer treated with pertuzumab. Ann. Oncol 2012, 23, 791–800. [Google Scholar]
- Shell, S.A.; Lyass, L.; Trusk, P.B.; Pry, K.J.; Wappel, R.L.; Bacus, S.S. Activation of ampk is necessary for killing cancer cells and sparing cardiac cells. Cell Cycle 2008, 7, 1769–1775. [Google Scholar]
- Spector, N.L.; Yarden, Y.; Smith, B.; Lyass, L.; Trusk, P.; Pry, K.; Hill, J.E.; Xia, W.; Seger, R.; Bacus, S.S. Activation of amp-activated protein kinase by human egf receptor 2/egf receptor tyrosine kinase inhibitor protects cardiac cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10607–10612. [Google Scholar]
- Jathal, M.K.; Chen, L.; Mudryj, M.; Ghosh, P.M. Targeting erbb3: The new rtk(id) on the prostate cancer block. Immun. Endocr. Metab. Agents Med. Chem 2011, 11, 131–149. [Google Scholar]
- McDonagh, C.F.; Huhalov, A.; Harms, B.D.; Adams, S.; Paragas, V.; Oyama, S.; Zhang, B.; Luus, L.; Overland, R.; Nguyen, S.; et al. Antitumor activity of a novel bispecific antibody that targets the erbb2/erbb3 oncogenic unit and inhibits heregulin-induced activation of erbb3. Mol. Cancer Ther 2012. [Google Scholar] [CrossRef]
- Langdon, S.P.; Faratian, D.; Nagumo, Y.; Mullen, P.; Harrison, D.J. Pertuzumab for the treatment of ovarian cancer. Exp. Opin. Biol. Ther 2010, 10, 1113–1120. [Google Scholar]
 ). All four members of the ErbB receptor family share high homology in the extracellular domain and the kinase domain. However, ErbB3 lacks tyrosine kinase activity. So far no ligand has been found for ErbB2, which has been found to be the preferred dimerization partner for other receptors. EGFR (ErbB1), ErbB2, ErbB3 and ErbB4 have different ligand binding and signaling. EGF, epidermal growth factor; TGF α, transforming growth factor alpha; AR, amphiregulin; EPR, epiregulin; BTC, betacellulin; HB-EGF, heparin binding EGF; NRG, neuregulin. Ligand binding causes homo/heterodimerization by ErbB family members enhancing complexity of signal transduction.
). All four members of the ErbB receptor family share high homology in the extracellular domain and the kinase domain. However, ErbB3 lacks tyrosine kinase activity. So far no ligand has been found for ErbB2, which has been found to be the preferred dimerization partner for other receptors. EGFR (ErbB1), ErbB2, ErbB3 and ErbB4 have different ligand binding and signaling. EGF, epidermal growth factor; TGF α, transforming growth factor alpha; AR, amphiregulin; EPR, epiregulin; BTC, betacellulin; HB-EGF, heparin binding EGF; NRG, neuregulin. Ligand binding causes homo/heterodimerization by ErbB family members enhancing complexity of signal transduction.
). All four members of the ErbB receptor family share high homology in the extracellular domain and the kinase domain. However, ErbB3 lacks tyrosine kinase activity. So far no ligand has been found for ErbB2, which has been found to be the preferred dimerization partner for other receptors. EGFR (ErbB1), ErbB2, ErbB3 and ErbB4 have different ligand binding and signaling. EGF, epidermal growth factor; TGF α, transforming growth factor alpha; AR, amphiregulin; EPR, epiregulin; BTC, betacellulin; HB-EGF, heparin binding EGF; NRG, neuregulin. Ligand binding causes homo/heterodimerization by ErbB family members enhancing complexity of signal transduction.
). All four members of the ErbB receptor family share high homology in the extracellular domain and the kinase domain. However, ErbB3 lacks tyrosine kinase activity. So far no ligand has been found for ErbB2, which has been found to be the preferred dimerization partner for other receptors. EGFR (ErbB1), ErbB2, ErbB3 and ErbB4 have different ligand binding and signaling. EGF, epidermal growth factor; TGF α, transforming growth factor alpha; AR, amphiregulin; EPR, epiregulin; BTC, betacellulin; HB-EGF, heparin binding EGF; NRG, neuregulin. Ligand binding causes homo/heterodimerization by ErbB family members enhancing complexity of signal transduction.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | Class | Target | Malignancies | Other toxicity |
|---|---|---|---|---|
| Drugs with known or likely cardiotoxicity | ||||
| Trastuzumab (Herceptin®) | Humanized mAb | ErbB2 | ErbB2+ breast cancer | Infusion reactions, neutropaenia |
| Drugs with low cardiotoxicity * | ||||
| Lapatinib (Tykerb®) | TKI | EGFR/ErbB1; ErbB2 | ErbB2+ breast cancer, ovarian cancer, gliomas, NSCLC | Skin rash, diarrhoea |
| Cetuximab (Erbitux®) | Chimeric mAb | EGFR/ErbB1 | CRC, squamous cell carcinoma of head/neck | Skin rash, infusion reactions, interstitial lung disease, hypmagnesaemia |
| Panitumumab (Vectibix®) | Human mAb | EGFR/ErbB1 | CRC | Skin rash |
| Gefitinib (Iressa®) | TKI | EGFR/ErbB1 | NSCLC, gliomas | Skin rash, nausea, diarrhoea, interstitial lung disease |
| Erlotinib (Tarceva®) | TKI | EGFR/ErbB1 | NSCLC, pancreatic cancer, gliomas | Skin rash, nausea, diarrhoea, interstitial lung disease |
| Pertuzumab (Omnitarg®) | Humanized mAb | ErbB2 | Breast, ovarian, prostate cancer, NSCLC | Skin rash |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hervent, A.-S.; De Keulenaer, G.W. Molecular Mechanisms of Cardiotoxicity Induced by ErbB Receptor Inhibitor Cancer Therapeutics. Int. J. Mol. Sci. 2012, 13, 12268-12286. https://doi.org/10.3390/ijms131012268
Hervent A-S, De Keulenaer GW. Molecular Mechanisms of Cardiotoxicity Induced by ErbB Receptor Inhibitor Cancer Therapeutics. International Journal of Molecular Sciences. 2012; 13(10):12268-12286. https://doi.org/10.3390/ijms131012268
Chicago/Turabian StyleHervent, Anne-Sophie, and Gilles W. De Keulenaer. 2012. "Molecular Mechanisms of Cardiotoxicity Induced by ErbB Receptor Inhibitor Cancer Therapeutics" International Journal of Molecular Sciences 13, no. 10: 12268-12286. https://doi.org/10.3390/ijms131012268