Mercuric Compounds Induce Pancreatic Islets Dysfunction and Apoptosis in Vivo

Abstract
:1. Introduction
2. Results and Discussion
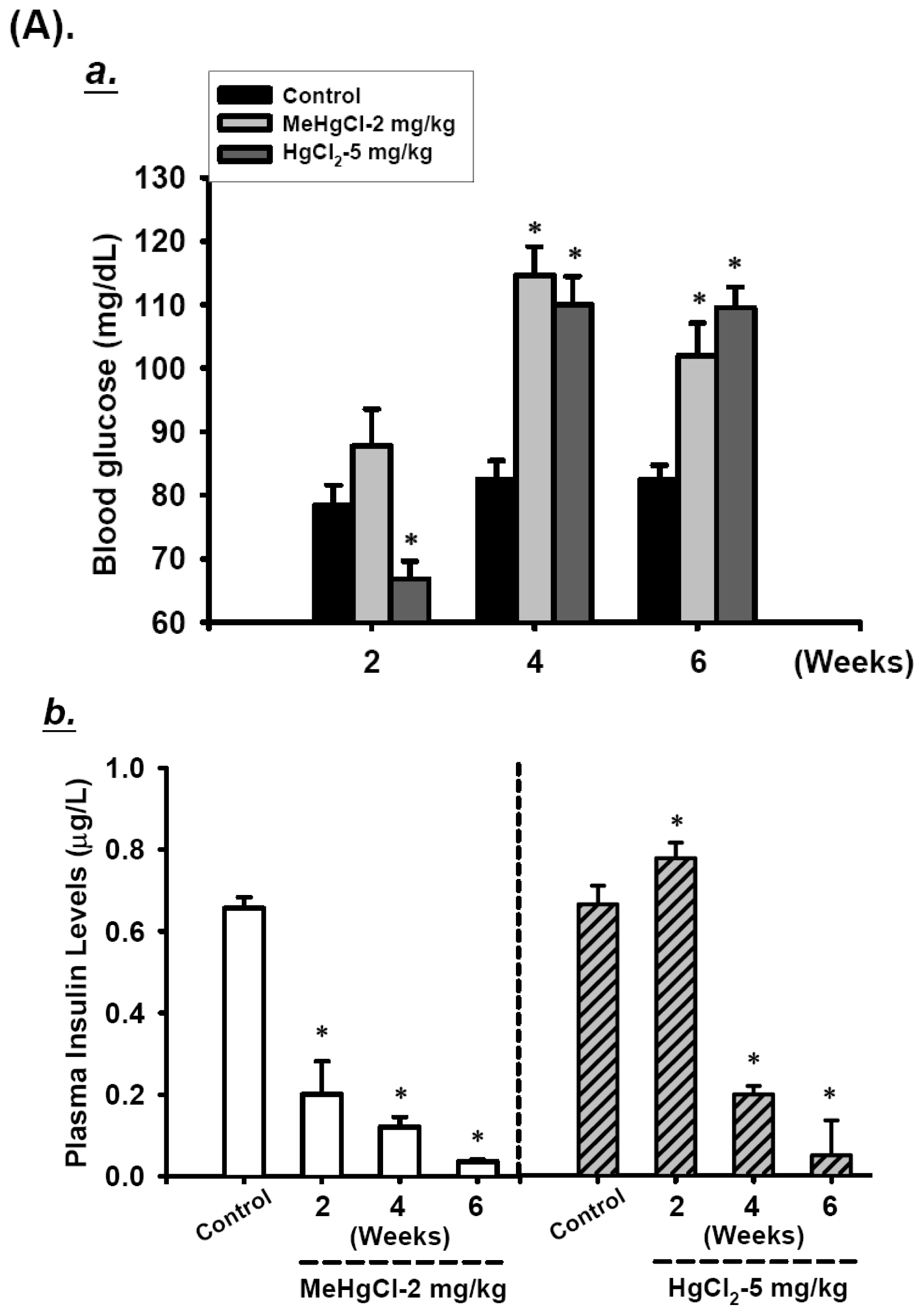
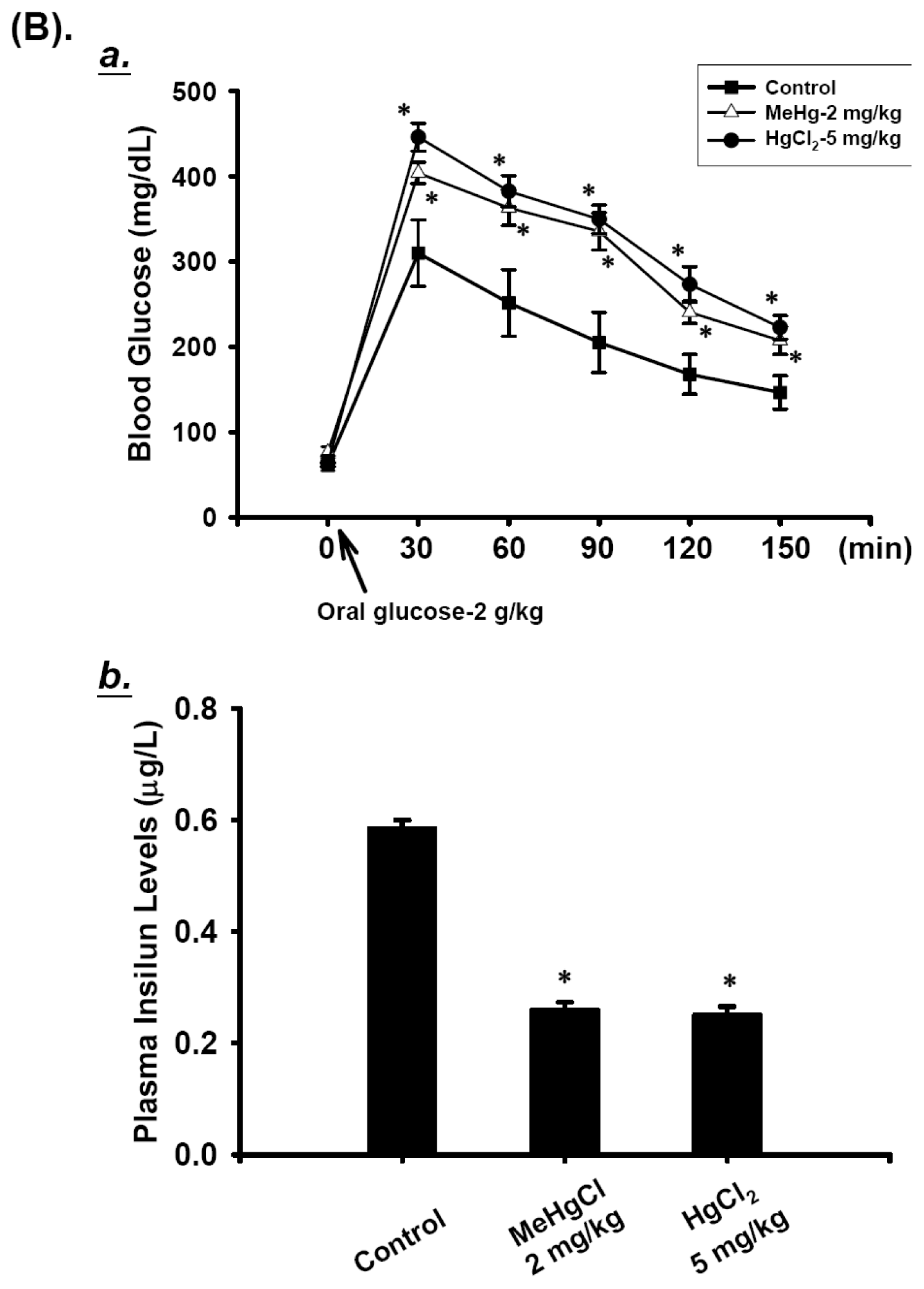
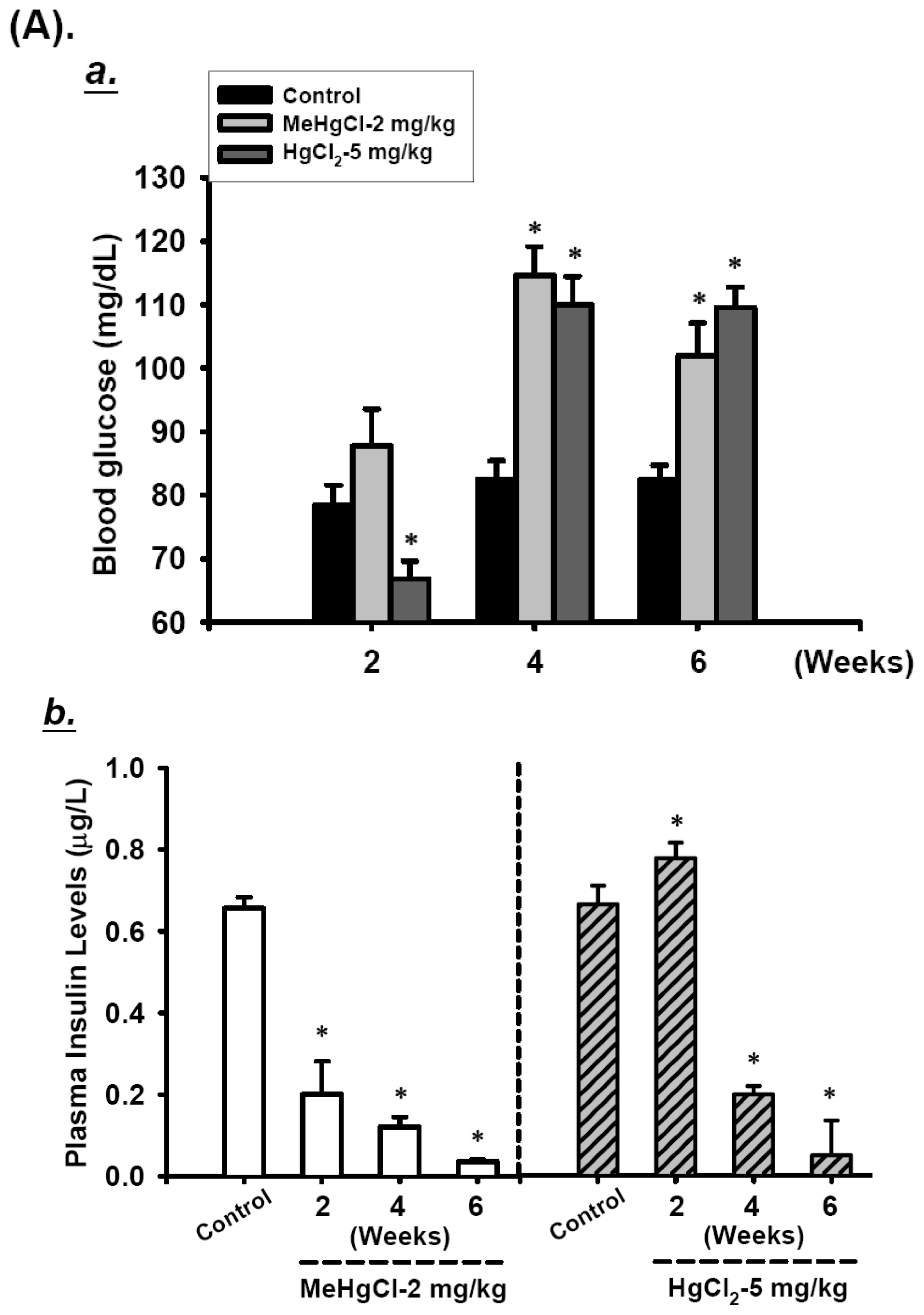
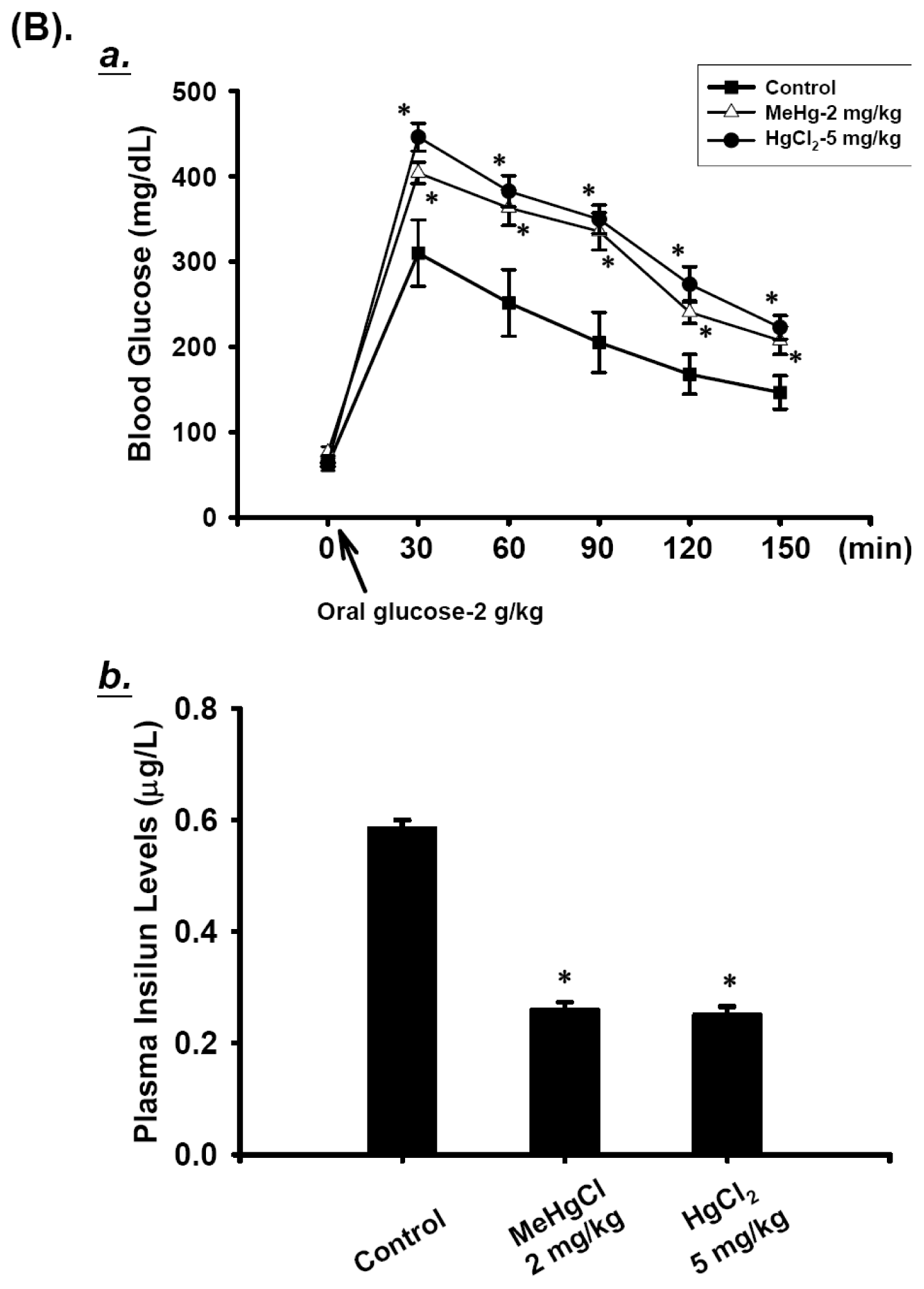
2.1. Effects of Mercuric Compounds on Blood Glucose Regulation and Plasma Insulin Levels in Mice
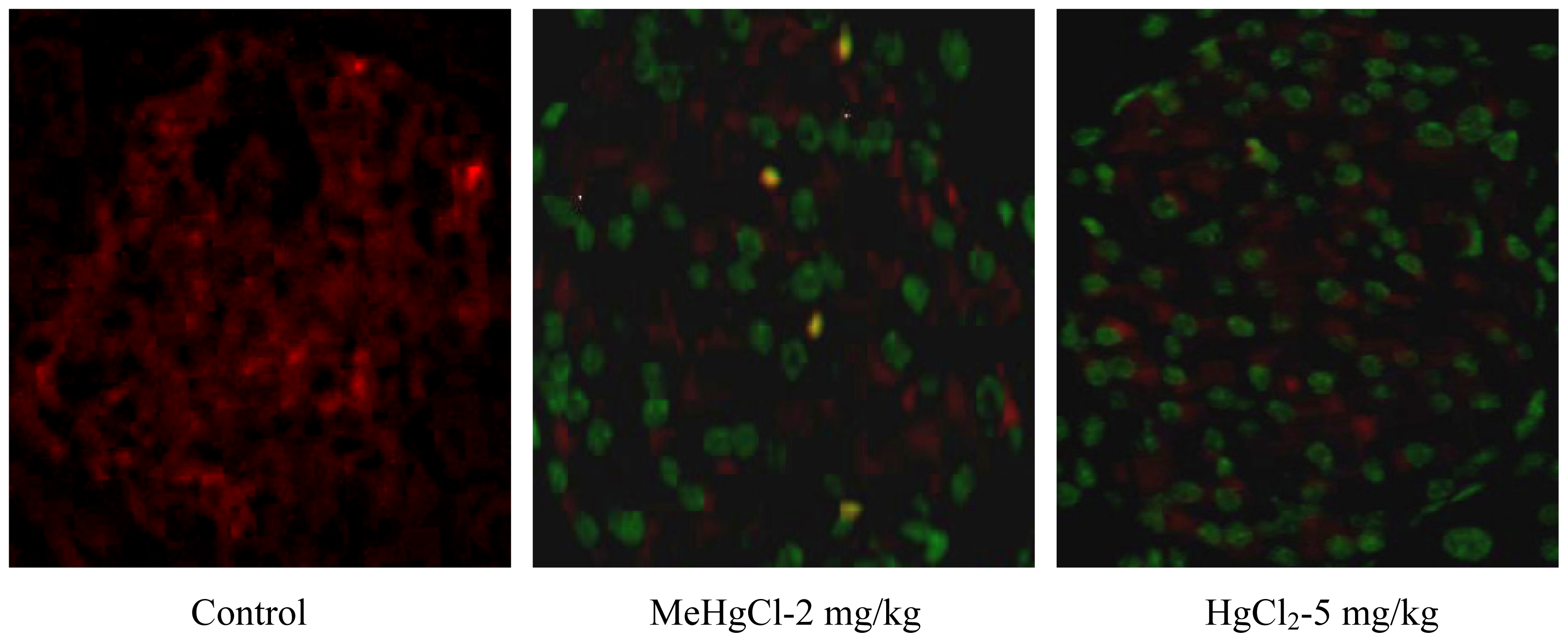
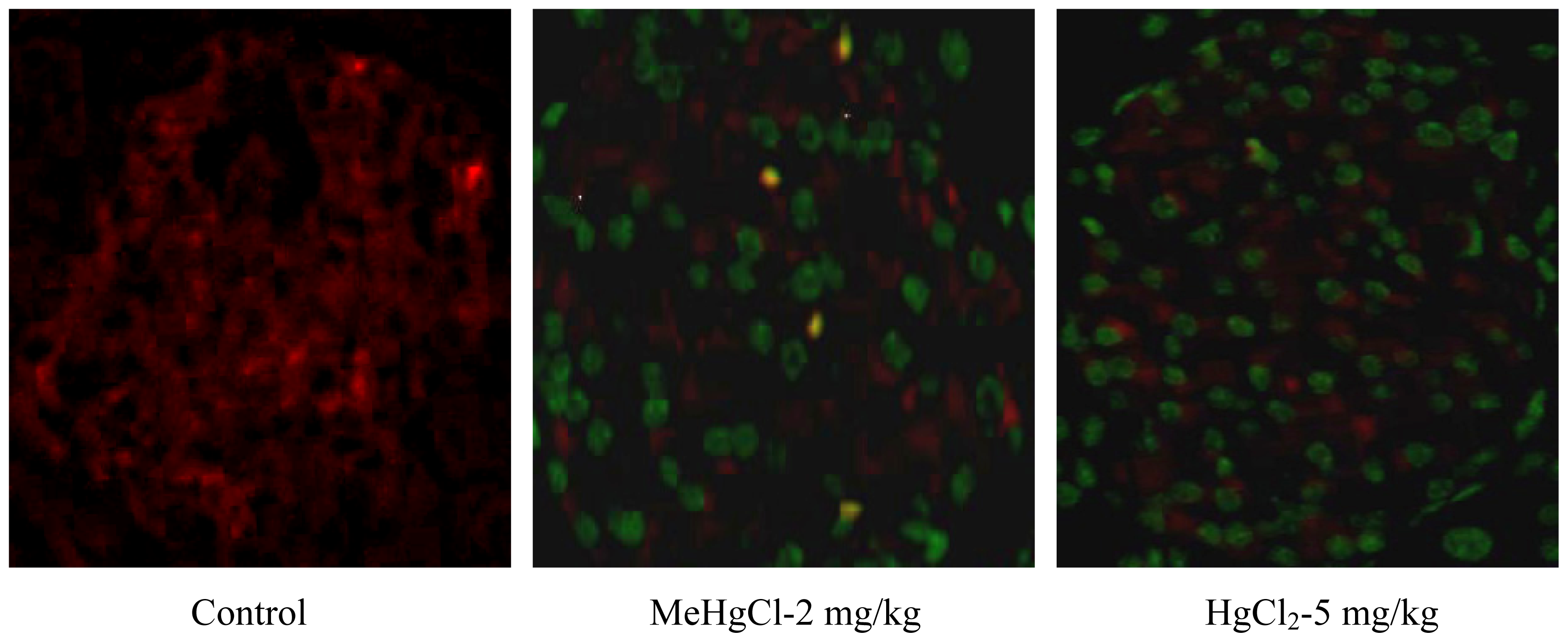
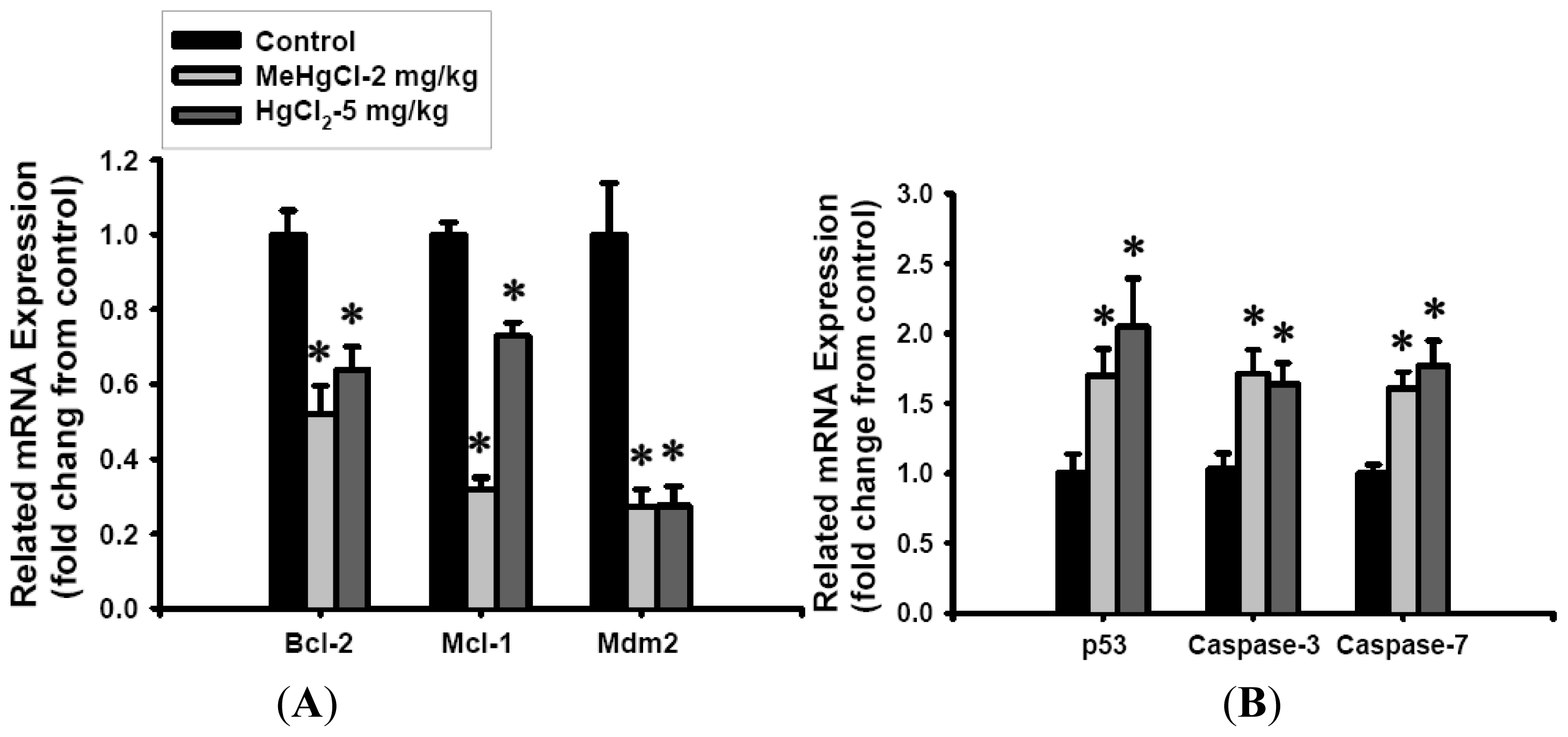
2.2. Mercuric Compounds Caused Apoptosis in the Pancreatic Islets of Exposed Mice
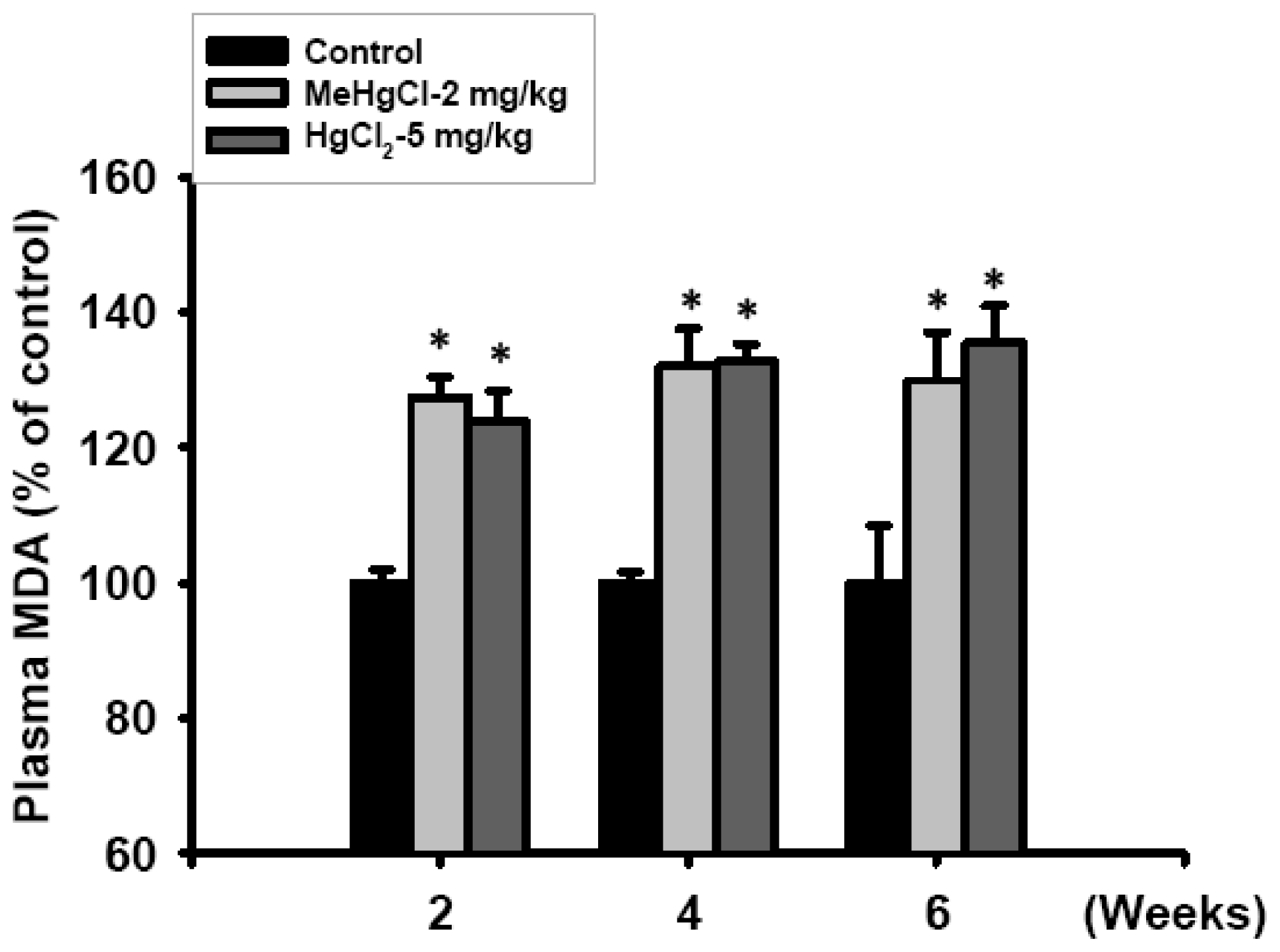
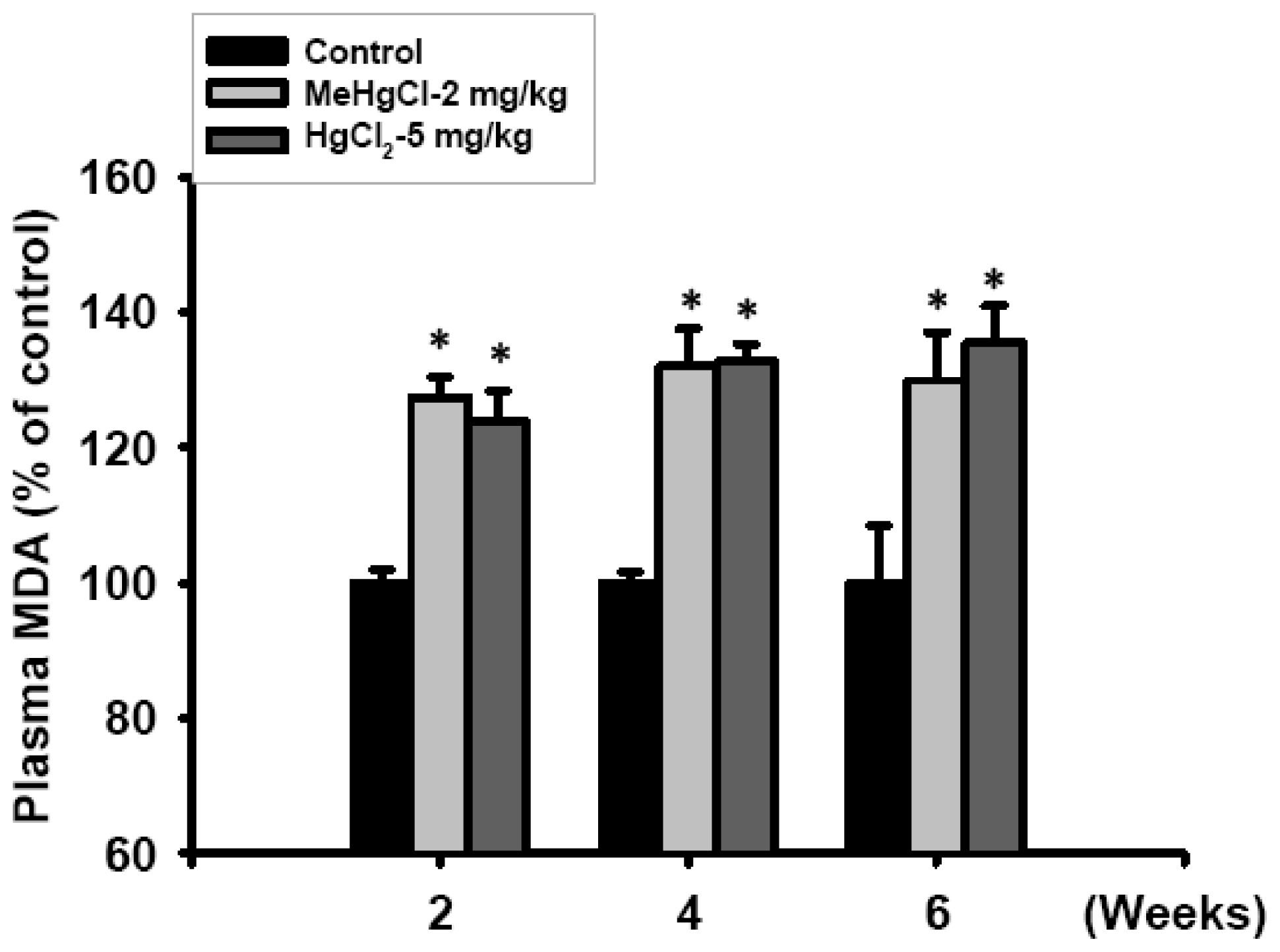
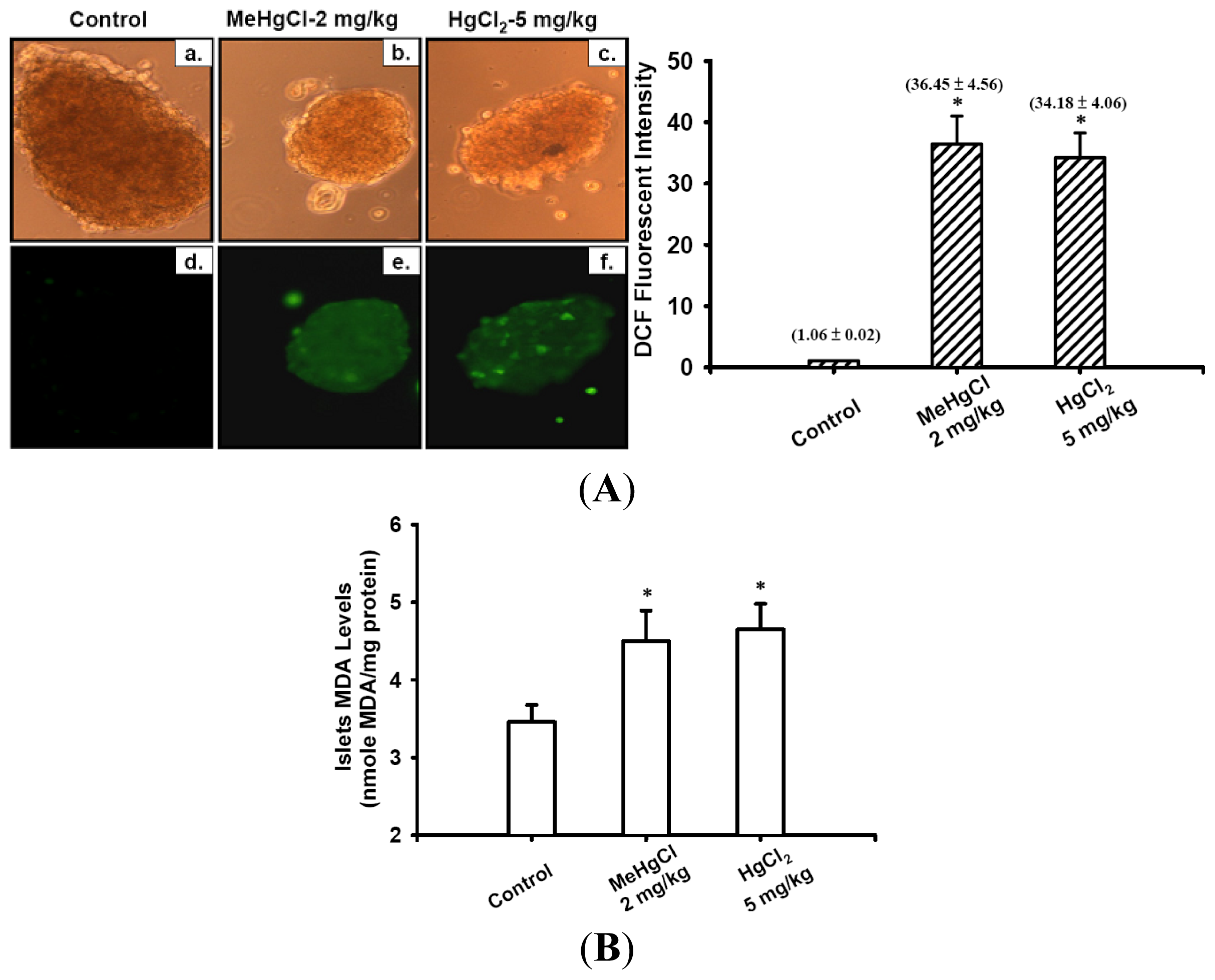
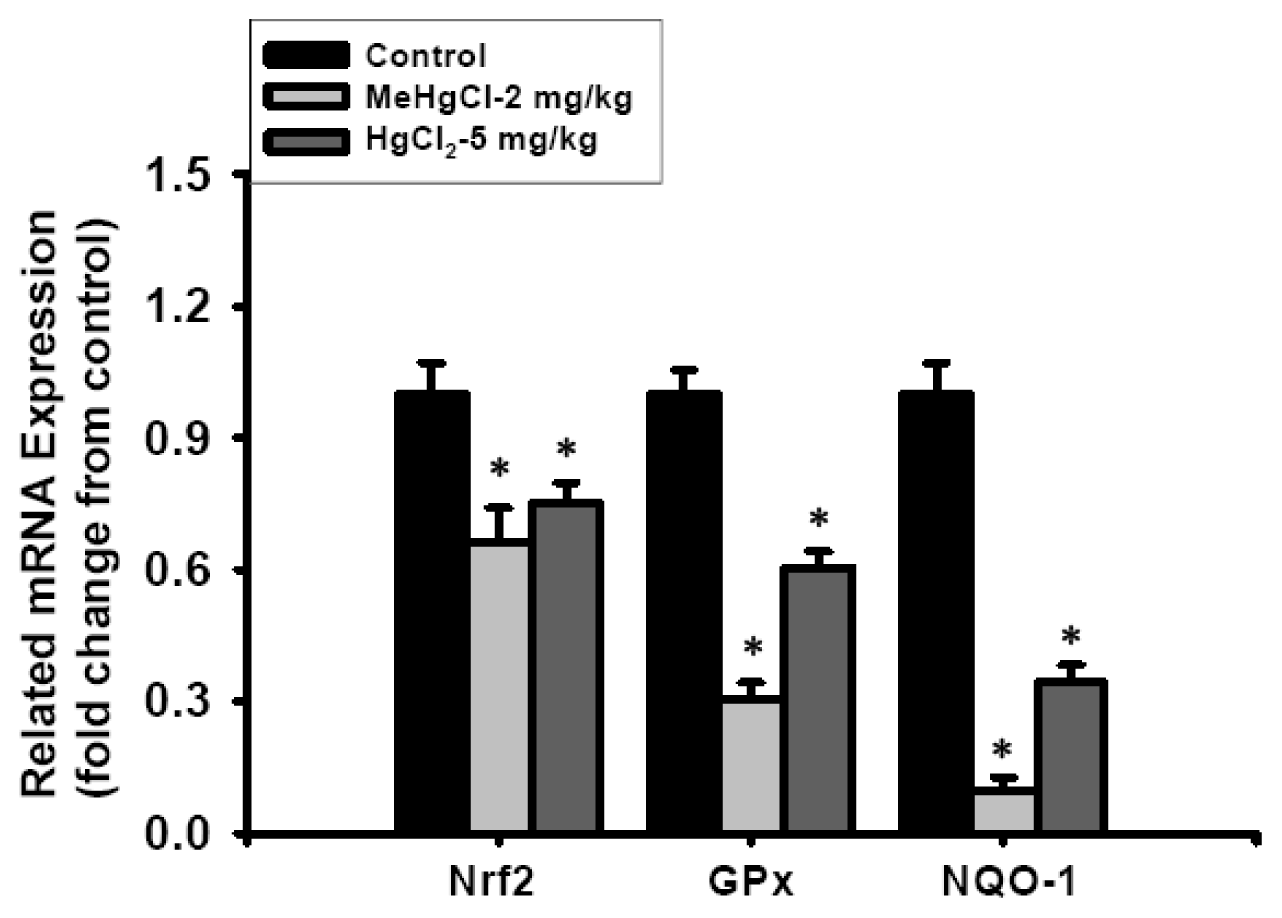
2.3. Exposure to Mercuric Compounds Induced Oxidative Stress Damage in the Pancreatic Islets
2.4. Discussion
3. Experimental Section
3.1. Animal Preparation and Study Design
3.2. Blood Glucose Measurement and Oral Glucose Tolerance Test (OGTT)
3.3. Plasma Insulin Level Detection
3.4. Determination of Mercury Concentrations
3.5. TUNEL and Insulin Double Staining
3.6. Lipid Peroxidation Detection
3.7. Pancreatic Islet Isolation and Purification Procedure
3.8. Real-Time Quantitative Reverse-Transcribed Polymerase Chain Reaction (RT-PCR) Analysis
3.9. Detection of Intracellular ROS in Islets
3.10. Statistical Analyses
4. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Karlsson, K.; Viklander, M.; Scholes, L.; Revitt, M. Heavy metal concentrations and toxicity in water and sediment from stormwater ponds and sedimentation tanks. J. Hazard Mater 2010, 178, 612–618. [Google Scholar]
- Ratcliffe, H.E.; Swanson, G.M.; Fischer, L.J. Human exposure to mercury: A critical assessment of the evidence of adverse health effects. J. Toxicol. Environ. Health 1996, 49, 221–270. [Google Scholar]
- Mozaffarian, D. Fish, mercury, selenium and cardiovascular risk: Current evidence and unanswered questions. Int. J. Environ. Res. Public. Health 2009, 6, 1894–1916. [Google Scholar]
- Iavicoli, I.; Fontana, L.; Bergamaschi, A. The effects of metals as endocrine disruptors. J. Toxicol. Environ. Health B Crit. Rev 2009, 12, 206–223. [Google Scholar]
- Michalke, B.; Halbach, S.; Nischwitz, V. JEM spotlight: Metal speciation related to neurotoxicity in humans. J. Environ. Monit 2009, 11, 939–954. [Google Scholar]
- Clarkson, T.W.; Magos, L. The toxicology of mercury and its chemical compounds. Crit. Rev. Toxicol 2006, 36, 609–662. [Google Scholar]
- Ishitobi, H.; Stern, S.; Thurston, S.W.; Zareba, G.; Langdon, M.; Gelein, R.; Weiss, B. Organic and inorganic mercury in neonatal rat brain after prenatal exposure to methylmercury and mercury vapor. Environ. Health Perspect 2010, 118, 242–248. [Google Scholar]
- Takeuchi, T.; Eto, K. Pathology and Pathogenesis of Minamata Disease. In Minamata Diseases Methyl Mercury Poisoning in Minamata and Niigata, Japan; Tsubaki, T., Irukayama, K., Eds.; Kodansya: Tokyo, Japan, 1997; pp. 103–141. [Google Scholar]
- Uchino, M.; Tanaka, Y.; Ando, Y.; Yonehara, T.; Hara, A.; Mishima, I.; Okajima, T.; Ando, M. Neurologic features of chronic minamata disease (organic mercury poisoning) and incidence of complications with aging. J. Environ. Sci. Health B 1995, 30, 699–715. [Google Scholar]
- Shigenaga, K. Pancreatic islet injury induced by methyl mercuric chloride light and electron microscopic studies. Kumamoto Med. J 1976, 29, 67–81. [Google Scholar]
- Chen, Y.W.; Huang, C.F.; Tsai, K.S.; Yang, R.S.; Yen, C.C.; Yang, C.Y.; Lin-Shiau, S.Y.; Liu, S.H. Methylmercury induces pancreatic β-cell apoptosis and dysfunction. Chem. Res. Toxicol 2006, 19, 1080–1085. [Google Scholar]
- Chen, Y.W.; Huang, C.F.; Yang, C.Y.; Yen, C.C.; Tsai, K.S.; Liu, S.H. Inorganic mercury causes pancreatic β-cell death via the oxidative stress-induced apoptotic and necrotic pathways. Toxicol. Appl. Pharmacol 2010, 243, 323–331. [Google Scholar]
- Chatenoud, L.; You, S.; Okada, H.; Kuhn, C.; Michaud, B.; Bach, J.F. 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: Immune therapies of type 1 diabetes: New opportunities based on the hygiene hypothesis. Clin. Exp. Immunol 2010, 160, 106–112. [Google Scholar]
- Johnson, J.D.; Luciani, D.S. Mechanisms of pancreatic β-cell apoptosis in diabetes and its therapies. Adv. Exp. Med. Biol 2010, 654, 447–462. [Google Scholar]
- Ma, K.; Nunemaker, C.S.; Wu, R.; Chakrabarti, S.K.; Taylor-Fishwick, D.A.; Nadler, J.L. 12-lipoxygenase products reduce insulin secretion and β-cell viability in human islets. J. Clin. Endocrinol. Metab 2010, 95, 887–893. [Google Scholar]
- Hou, N.; Torii, S.; Saito, N.; Hosaka, M.; Takeuchi, T. Reactive oxygen species-mediated pancreatic β-cell death is regulated by interactions between stress-activated protein kinases, p38 and c-Jun N-terminal kinase, and mitogen-activated protein kinase phosphatases. Endocrinology 2008, 149, 1654–1665. [Google Scholar]
- Kaneto, H.; Kawamori, D.; Matsuoka, T.A.; Kajimoto, Y.; Yamasaki, Y. Oxidative stress and pancreatic β-cell dysfunction. Am. J. Ther 2005, 12, 529–533. [Google Scholar]
- Chen, Y.W.; Yang, C.Y.; Huang, C.F.; Hung, D.Z.; Leung, Y.M.; Liu, S.H. Heavy metals, islet function and diabetes development. Islets 2009, 1, 169–176. [Google Scholar]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem 2010, 345, 91–104. [Google Scholar]
- Lu, T.H.; Hsieh, S.Y.; Yen, C.C.; Wu, H.C.; Chen, K.L.; Hung, D.Z.; Chen, C.H.; Wu, C.C.; Su, Y.C.; Chen, Y.W.; et al. Involvement of oxidative stress-mediated ERK1/2 and p38 activation regulated mitochondria-dependent apoptotic signals in methylmercury-induced neuronal cell injury. Toxicol. Lett 2011, 204, 71–80. [Google Scholar]
- Carvalho, M.C.; Franco, J.L.; Ghizoni, H.; Kobus, K.; Nazari, E.M.; Rocha, J.B.; Nogueira, C.W.; Dafre, A.L.; Muller, Y.M.; Farina, M. Effects of 2,3-dimercapto-1-propanesulfonic acid (DMPS) on methylmercury-induced locomotor deficits and cerebellar toxicity in mice. Toxicology 2007, 239, 195–203. [Google Scholar]
- Goulet, S.; Dore, F.Y.; Mirault, M.E. Neurobehavioral changes in mice chronically exposed to methylmercury during fetal and early postnatal development. Neurotoxicol. Teratol 2003, 25, 335–347. [Google Scholar]
- De Freitas, A.S.; Funck, V.R.; Rotta Mdos, S.; Bohrer, D.; Morschbacher, V.; Puntel, R.L.; Nogueira, C.W.; Farina, M.; Aschner, M.; Rocha, J.B. Diphenyl diselenide, a simple organoselenium compound, decreases methylmercury-induced cerebral, hepatic and renal oxidative stress and mercury deposition in adult mice. Brain Res. Bull 2009, 79, 77–84. [Google Scholar]
- Glaser, V.; Nazari, E.M.; Muller, Y.M.; Feksa, L.; Wannmacher, C.M.; Rocha, J.B.; de Bem, A.F.; Farina, M.; Latini, A. Effects of inorganic selenium administration in methylmercury-induced neurotoxicity in mouse cerebral cortex. Int. J. Dev. Neurosci 2010, 28, 631–637. [Google Scholar]
- Lu, T.H.; Chen, C.H.; Lee, M.J.; Ho, T.J.; Leung, Y.M.; Hung, D.Z.; Yen, C.C.; He, T.Y.; Chen, Y.W. Methylmercury chloride induces alveolar type II epithelial cell damage through an oxidative stress-related mitochondrial cell death pathway. Toxicol. Lett 2010, 194, 70–78. [Google Scholar]
- Zhao, J.Q.; Wen, Y.F.; Bhadauria, M.; Nirala, S.K.; Sharma, A.; Shrivastava, S.; Shukla, S.; Agrawal, O.P.; Mathur, R. Protective effects of propolis on inorganic mercury induced oxidative stress in mice. Indian J. Exp. Biol 2009, 47, 264–269. [Google Scholar]
- Zhang, Q.; Pi, J.; Woods, C.G.; Andersen, M.E. A systems biology perspective on Nrf2-mediated antioxidant response. Toxicol. Appl. Pharmacol 2010, 244, 84–97. [Google Scholar]
- Drews, G.; Krippeit-Drews, P.; Dufer, M. Oxidative stress and β-cell dysfunction. Pflugers Arch 2010, 460, 703–718. [Google Scholar]
- Rains, J.L.; Jain, S.K. Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med 2011, 50, 567–575. [Google Scholar]
- Bazuine, M.; Ouwens, D.M.; Gomes de Mesquita, D.S.; Maassen, J.A. Arsenite stimulated glucose transport in 3T3-L1 adipocytes involves both Glut4 translocation and p38 MAPK activity. Eur. J. Biochem 2003, 270, 3891–3903. [Google Scholar]
- Xue, P.; Hou, Y.; Zhang, Q.; Woods, C.G.; Yarborough, K.; Liu, H.; Sun, G.; Andersen, M.E.; Pi, J. Prolonged inorganic arsenite exposure suppresses insulin-stimulated AKT S473 phosphorylation and glucose uptake in 3T3-L1 adipocytes: Involvement of the adaptive antioxidant response. Biochem. Biophys. Res. Commun 2011, 407, 360–365. [Google Scholar]
- Walton, F.S.; Harmon, A.W.; Paul, D.S.; Drobna, Z.; Patel, Y.M.; Styblo, M. Inhibition of insulin-dependent glucose uptake by trivalent arsenicals: possible mechanism of arsenic-induced diabetes. Toxicol. Appl. Pharmacol 2004, 198, 424–433. [Google Scholar]
- Barnes, D.M.; Hanlon, P.R.; Kircher, E.A. Effects of inorganic HgCl2 on adipogenesis. Toxicol. Sci 2003, 75, 368–377. [Google Scholar]
- Barnes, D.M.; Kircher, E.A. Effects of mercuric chloride on glucose transport in 3T3-L1 adipocytes. Toxicol. In Vitro 2005, 19, 207–214. [Google Scholar]
- Kasahara, T.; Kasahara, M. Characterization of rat Glut4 glucose transporter expressed in the yeast Saccharomyces cerevisiae: Comparison with Glut1 glucose transporter. Biochim. Biophys. Acta 1997, 1324, 111–119. [Google Scholar]
- Miller, D.S.; Shehata, A.T.; Lerner, J. HgCl2 inhibition of D-glucose transport in jejunal tissue from 2 day and 21 day chicks. J. Pharmacol. Exp. Ther 1980, 214, 101–105. [Google Scholar]
- Loh, K.P.; Huang, S.H.; De Silva, R.; Tan, B.K.; Zhu, Y.Z. Oxidative stress: Apoptosis in neuronal injury. Curr. Alzheimer Res 2006, 3, 327–337. [Google Scholar]
- Willis, S.N.; Fletcher, J.I.; Kaufmann, T.; van Delft, M.F.; Chen, L.; Czabotar, P.E.; Ierino, H.; Lee, E.F.; Fairlie, W.D.; Bouillet, P.; et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007, 315, 856–859. [Google Scholar]
- McKenzie, M.D.; Jamieson, E.; Jansen, E.S.; Scott, C.L.; Huang, D.C.; Bouillet, P.; Allison, J.; Kay, T.W.; Strasser, A.; Thomas, H.E. Glucose induces pancreatic islet cell apoptosis that requires the BH3-only proteins Bim and Puma and multi-BH domain protein Bax. Diabetes 2010, 59, 644–652. [Google Scholar]
- Hu, C.; Smith, S.D.; Pang, L.; Sadovsky, Y.; Nelson, D.M. Enhanced basal apoptosis in cultured term human cytotrophoblasts is associated with a higher expression and physical interaction of p53 and Bak. Placenta 2006, 27, 978–983. [Google Scholar]
- Iwakuma, T.; Lozano, G. MDM2, an introduction. Mol. Cancer Res 2003, 1, 993–1000. [Google Scholar]
- Lu, T.H.; Su, C.C.; Chen, Y.W.; Yang, C.Y.; Wu, C.C.; Hung, D.Z.; Chen, C.H.; Cheng, P.W.; Liu, S.H.; Huang, C.F. Arsenic induces pancreatic β-cell apoptosis via the oxidative stress-regulated mitochondria-dependent and endoplasmic reticulum stress-triggered signaling pathways. Toxicol. Lett 2011, 201, 15–26. [Google Scholar]
- Yen, C.C.; Ho, T.J.; Wu, C.C.; Chang, C.F.; Su, C.C.; Chen, Y.W.; Jinn, T.R.; Lu, T.H.; Cheng, P.W.; et al. Inorganic arsenic causes cell apoptosis in mouse cerebrum through an oxidative stress-regulated signaling pathway. Arch. Toxicol 2011, 85, 565–575. [Google Scholar]
- Jaiswal, A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med 2004, 36, 1199–1207. [Google Scholar]
- Ade, N.; Leon, F.; Pallardy, M.; Peiffer, J.L.; Kerdine-Romer, S.; Tissier, M.H.; Bonnet, P.A.; Fabre, I.; Ourlin, J.C. HMOX1 and NQO1 genes are upregulated in response to contact sensitizers in dendritic cells and THP-1 cell line: role of the Keap1/Nrf2 pathway. Toxicol. Sci 2009, 107, 451–460. [Google Scholar]
- Simmons, S.O.; Fan, C.Y.; Yeoman, K.; Wakefield, J.; Ramabhadran, R. NRF2 oxidative stress induced by heavy metals is cell type dependent. Curr. Chem. Genomics 2011, 5, 1–12. [Google Scholar]
- Pi, J.; Freeman, M.L.; Yamamoto, M. Nrf2 in toxicology and pharmacology: The good, the bad and the ugly? Toxicol. Appl. Pharmacol 2010, 244, 1–3. [Google Scholar]
- Ni, M.; Li, X.; Yin, Z.; Jiang, H.; Sidoryk-Wegrzynowicz, M.; Milatovic, D.; Cai, J.; Aschner, M. Methylmercury induces acute oxidative stress, altering Nrf2 protein level in primary microglial cells. Toxicol. Sci 2010, 116, 590–603. [Google Scholar]
- Pi, J.; Zhang, Q.; Woods, C.G.; Wong, V.; Collins, S.; Andersen, M.E. Activation of Nrf2-mediated oxidative stress response in macrophages by hypochlorous acid. Toxicol. Appl. Pharmacol 2008, 226, 236–243. [Google Scholar]
- Siegel, D.; Gustafson, D.L.; Dehn, D.L.; Han, J.Y.; Boonchoong, P.; Berliner, L.J.; Ross, D. NAD(P)H:quinone oxidoreductase 1: Role as a superoxide scavenger. Mol. Pharmacol 2004, 65, 1238–1247. [Google Scholar]
- Kudin, A.P.; Augustynek, B.; Lehmann, A.K.; Kovacs, R.; Kunz, W.S. The contribution of thioredoxin-2 reductase and glutathione peroxidase to H(2)O(2) detoxification of rat brain mitochondria. Biochim. Biophys. Acta 2012, 1817, 1901–1906. [Google Scholar]
- Huang, C.F.; Chen, Y.W.; Yang, C.Y.; Lin, H.Y.; Way, T.D.; Chiang, W.; Liu, S.H. Extract of lotus leaf (Nelumbo nucifera) and its active constituent catechin with insulin secretagogue activity. J. Agric. Food Chem 2011, 59, 1087–1094. [Google Scholar]
- Huang, G.C.; Zhao, M.; Jones, P.; Persaud, S.; Ramracheya, R.; Lobner, K.; Christie, M.R.; Banga, J.P.; Peakman, M.; Sirinivsan, P.; et al. The development of new density gradient media for purifying human islets and islet-quality assessments. Transplantation 2004, 77, 143–145. [Google Scholar]
- Valenzuela, M.T.; Guerrero, R.; Nunez, M.I.; Ruiz de Almodovar, J.M.; Sarker, M.; de Murcia, G.; Oliver, F.J. PARP-1 modifies the effectiveness of p53-mediated DNA damage response. Oncogene 2002, 21, 1108–1116. [Google Scholar]
- Barve, A.; Khor, T.O.; Nair, S.; Reuhl, K.; Suh, N.; Reddy, B.; Newmark, H.; Kong, A.N. γ-tocopherol-enriched mixed tocopherol diet inhibits prostate carcinogenesis in TRAMP mice. Int. J. Cancer 2009, 124, 1693–1699. [Google Scholar]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 2002, 30, e36–e45. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Weeks | Group | ||
|---|---|---|---|
| Control | MeHgCl-2 (mg/kg) | HgCl2-5 (mg/kg) | |
| 2 | 2.4 ± 0.3 | 4970.8 ± 38.8 * | 432.0 ± 111.2 * |
| 4 | 2.6 ± 0.4 | 14827.5 ± 1938.7 * | 683.4 ± 47.9 * |
| 6 | 3.0 ± 0.5 | 27741.4 ± 6747.1 * | 865.8 ± 222.5 * |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, K.-L.; Liu, S.-H.; Su, C.-C.; Yen, C.-C.; Yang, C.-Y.; Lee, K.-I.; Tang, F.-C.; Chen, Y.-W.; Lu, T.-H.; Su, Y.-C.; et al. Mercuric Compounds Induce Pancreatic Islets Dysfunction and Apoptosis in Vivo. Int. J. Mol. Sci. 2012, 13, 12349-12366. https://doi.org/10.3390/ijms131012349
Chen K-L, Liu S-H, Su C-C, Yen C-C, Yang C-Y, Lee K-I, Tang F-C, Chen Y-W, Lu T-H, Su Y-C, et al. Mercuric Compounds Induce Pancreatic Islets Dysfunction and Apoptosis in Vivo. International Journal of Molecular Sciences. 2012; 13(10):12349-12366. https://doi.org/10.3390/ijms131012349
Chicago/Turabian StyleChen, Kuo-Liang, Shing-Hwa Liu, Chin-Chuan Su, Cheng-Chieh Yen, Ching-Yao Yang, Kuan-I Lee, Feng-Cheng Tang, Ya-Wen Chen, Tien-Hui Lu, Yi-Chang Su, and et al. 2012. "Mercuric Compounds Induce Pancreatic Islets Dysfunction and Apoptosis in Vivo" International Journal of Molecular Sciences 13, no. 10: 12349-12366. https://doi.org/10.3390/ijms131012349