Proposal of a New Method for Measuring Förster Resonance Energy Transfer (FRET) Rapidly, Quantitatively and Non-Destructively

Abstract
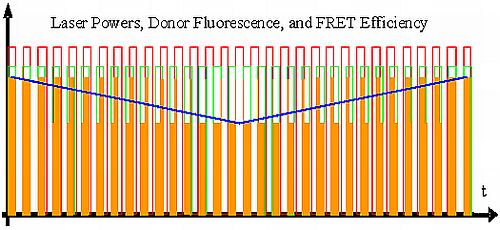
: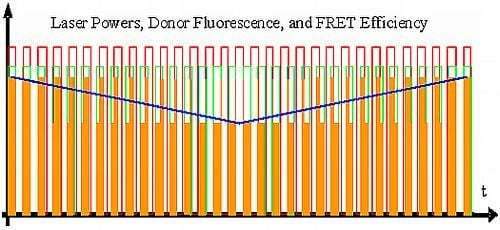
{kind=link}
{kind=link}
{kind=link}
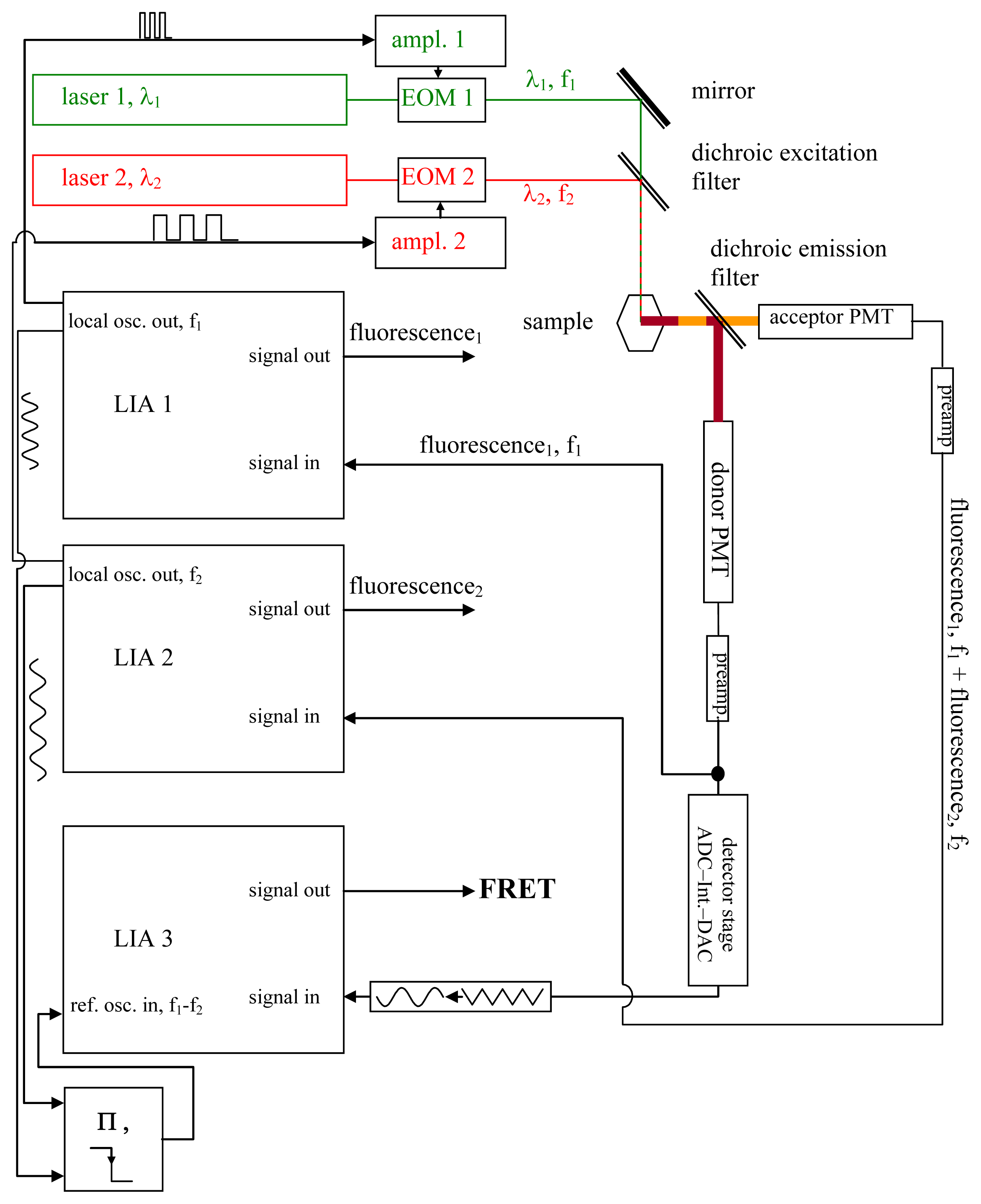
{kind=link}
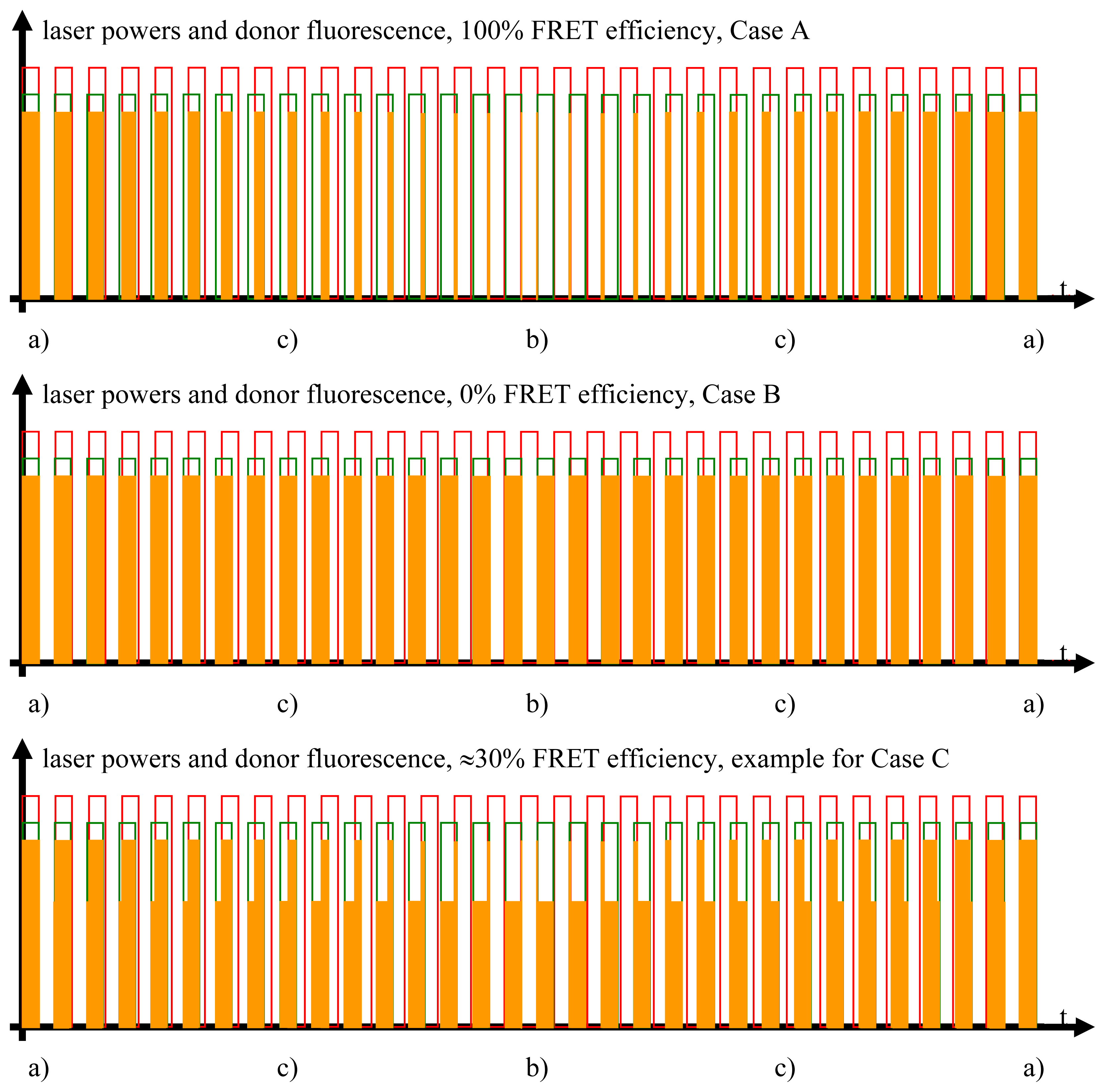
{kind=link}
{kind=link}
1. Introduction
2. Description of the System
2.1. Principle of Operation
2.2. A Suitable Test Preparation
2.3. Processes on the Molecular Level
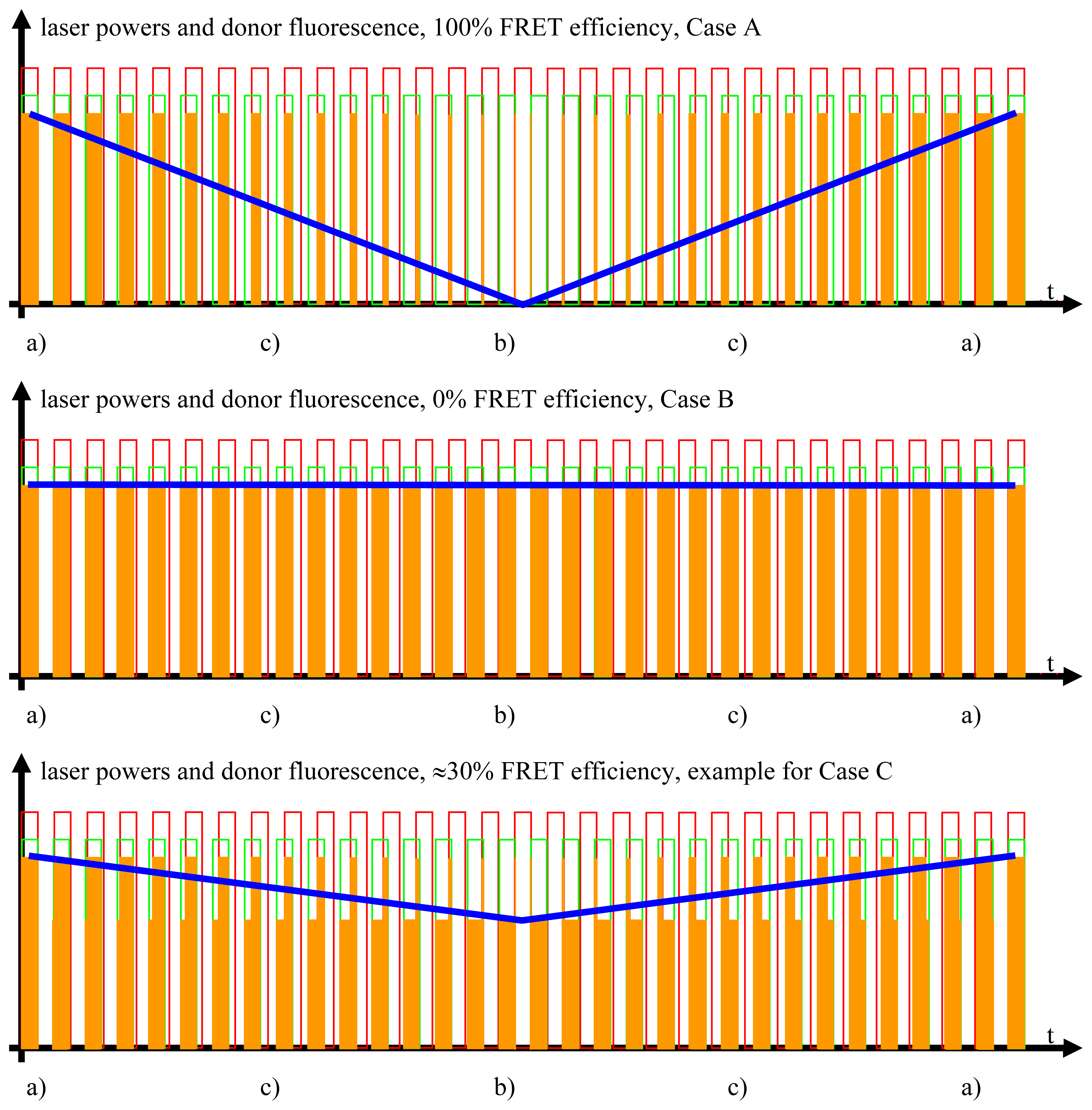
2.3.1. Case A, Ideal FRET Conditions, Donor and Acceptor Close to Each Other, Optimal FRET
- Donor excitation and acceptor excitation are close to in phase.In this case, the donor fluorescence as well as the acceptor fluorescence are modulated at either f1 or f2, at which the modulation will be quite deep. There are strong signals in LIA1 and in LIA2 before the respective amplifier stage. In this simplified analysis it is assumed that during (a) and those parts of (c) (see below) when the direct excitation of the acceptor by the laser beam at λ2 and FRET excitation of the acceptor are competing, laser excitation is dominantly strong. A quantitative analysis is provided later on in this article.
- Donor excitation and acceptor excitation are close to entirely counter phase.In this case, the acceptor is fluorescing all the time, since it is excited all the time, either by FRET or by the laser beam at λ2. Donor fluorescence is close to zero at all times, since the donor excitation energy is transferred to the acceptor. The modulation depths of both donor and acceptor fluorescence will be small. There will not be any considerable signal in either LIA1 or LIA2 before the respective amplifier stages.
- All intermediate states.(a) and (b) will be present, each during its respective part of the oscillation period. The average value of the donor fluorescence—integrated over many modulation periods—will oscillate at the beat frequency. The envelope of this beat oscillation is a triangular signal, which LIA3 registers if the signal by the donor fluorescence PMT has been pre-processed by a detector stage/“ADC-Integrator-DAC” circuit prior to de-modulation.
2.3.2. Case B, Ideal Non-FRET Conditions, Donor and Acceptor Far Enough from Each Other to Render Any FRET Process Impossible
2.3.3. Case C, Any Intermediate Situation
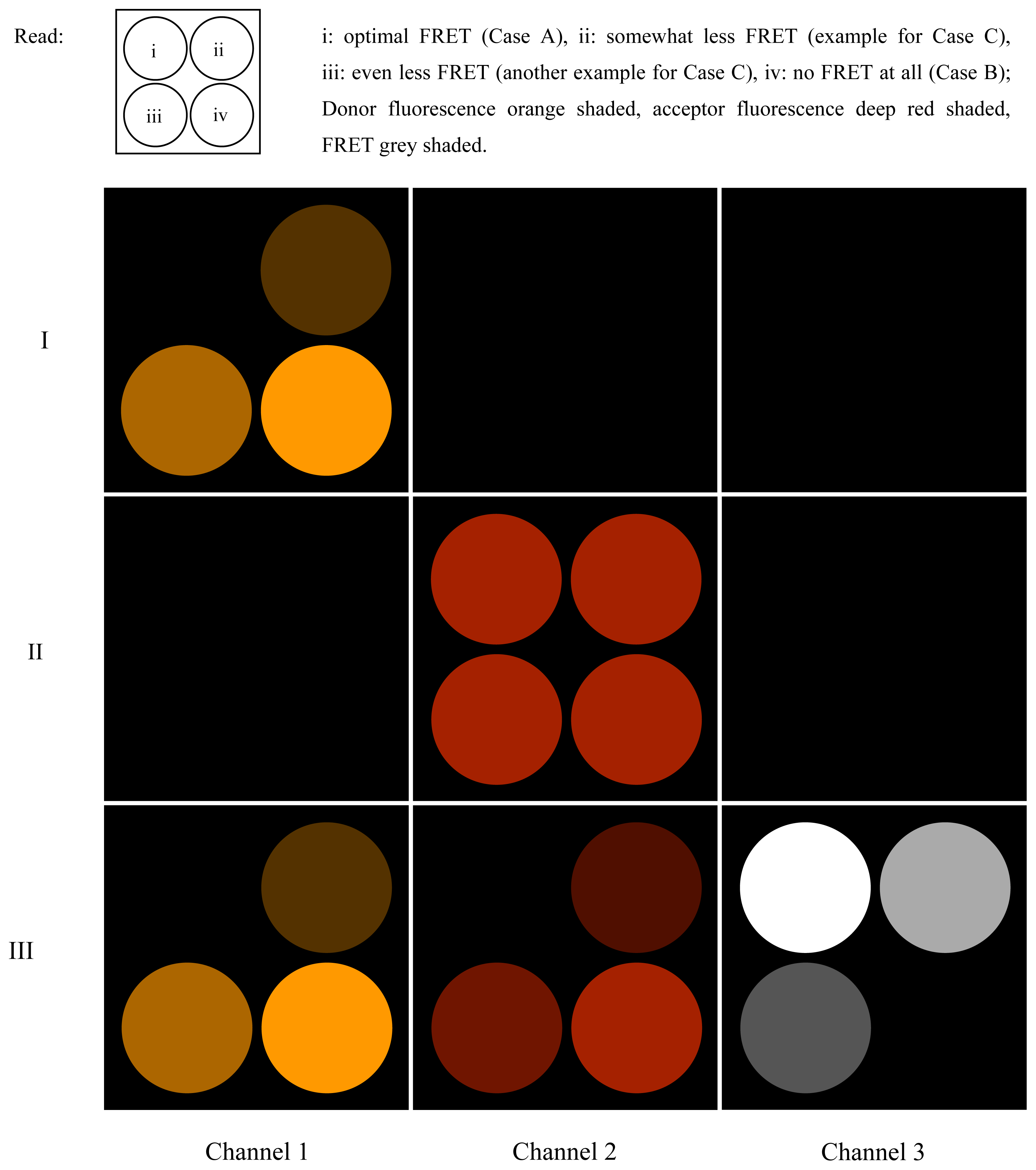
2.4. Qualitative Analysis of FRET Measurements
- At first, the preparation is illuminated solely by (λ1, f1), and the phase on LIA1 is optimized to attain the largest possible value in Channel 1 (the part of the donor fluorescence that is not “FRETed away”). Dependent on the degree of FRET, the signal will be weaker or stronger in Channel 1 (see i, ii, iii, iv). The FRET fluorescence of the acceptor cannot be seen in Channel 2 since it does not oscillate at f2 during this step, but at f1 instead. The signal is zero in Channel 3.
- Now, the preparation is illuminated solely by (λ2, f2). LIA2 is optimized for directly-excited (i.e., non-FRET) acceptor fluorescence. All the regions (i, ii, iii and iv) will shine with equal intensity, since the acceptor is being excited directly and with modulation frequency f2. Channels 1 and 3 are entirely dark.
- Finally, the preparation is illuminated simultaneously by (λ1, f1) and (λ2, f2). In region i, the donor fluorescence in Channel 1 is zero. The reason for this is that donor and acceptor are FRETing. The value of Channel 2 in i is zero since the modulation depth of the acceptor is zero.
2.5. The Quantitative Analysis of FRET Measurements
2.6. FRET and Non Linear Laser Scanning Microscopy
2.7. Multiple FRET Processes
3. Conclusions
Acknowledgments
References
- Klein, O.; Rosseland, S. Über Zusammenstöße zwischen Atomen und freien Elektronen. Z. Phys 1921, 4, 46–51. [Google Scholar]
- Cario, G.; Franck, J. Über sensibilisierte Fluoreszenz von Gasen. Z. Phys 1923, 17, 202–212. [Google Scholar]
- Perrin, J. Fluorescence et induction moléculaire par résonance. Comptes rendus hebdomadaire des séances de l’Académie des Sciences 1927, 184, 1097–1100. [Google Scholar]
- Weber, G. Fluorescence-polarization spectrum and electronic-energy transfer in tyrosine, tryptophan and related compounds. Biochem. J 1960, 75, 335–345. [Google Scholar]
- Latt, S.A.; Cheung, H.T.; Blout, E.R. Energy transfer. A system with relatively fixed donor-acceptor separation. J. Am. Chem. Soc 1965, 87, 995–1003. [Google Scholar]
- Förster, Th. Zwischenmolekulare Energiewanderung und Fluoreszenz. Annalen der Physik 1948, 437, 55–75. [Google Scholar]
- Förster, Th. Energiewanderung und Fluoreszenz. Die Naturwissenschaften 1946, 33, 166–175. [Google Scholar]
- Arnold, W.; Oppenheimer, J.R. Internal conversion in the photosynthetic mechanism of blue-green algae. J. Gen. Physiol 1950, 33, 423–435. [Google Scholar]
- Stryer, L.; Haugland, R.P. Energy transfer: A spectroscopic ruler. Proc. Natl. Acad. Sci. USA 1967, 58, 719–726. [Google Scholar]
- Masi, A.; Cicchi, R.; Carloni, A.; Pavone, F.S.; Arcangeli, A. Optical methods in the study of protein-protein interactions. Adv. Exp. Med. Biol 2010, 674, 33–42. [Google Scholar]
- Miyawaki, A.; Llopis, J.; Heim, R.; McCaffery, J.M.; Adams, J.A.; Ikurak, M.; Tsien, R.Y. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 1997, 388, 882–887. [Google Scholar]
- Fan, G.Y.; Fujisaki, H.; Miyawaki, A.; Tsay, R.K.; Tsien, R.Y.; Ellisman, M.H. Video-rate scanning two-photon excitation fluorescence microscopy and ratio imaging with cameleons. Biophys. J 1999, 76, 2412–2420. [Google Scholar]
- Tsien, R.Y. Indicators based on fluorescence resonance energy transfer (FRET). Cold Spring Harb. Protoc 2009. [Google Scholar] [CrossRef]
- Zaccolo, M.; Pozzan, T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 2002, 295, 1711–1715. [Google Scholar]
- Gadella, T.W.J., Jr; Jovin, T.M. Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation. J. Cell Biol. 1995, 129, 1543–1558. [Google Scholar]
- Becker, W.; Benndorf, K.; Bergmann, A.; Biskup, C.; König, K.; Tirplapur, U.; Zimmer, T. FRET measurements by TCSPC laser scanning microscopy. Proc. SPIE 2001, 4431, 94–98. [Google Scholar]
- Becker, W.; Bergmann, A.; Hink, M.A.; König, K.; Benndorf, K.; Biskup, C. Fluorescence lifetime imaging by time-correlated single-photon counting. Microsc. Res. Tech 2004, 63, 58–66. [Google Scholar]
- Becker, W.; Bergmann, A.; Haustein, E.; Petrasek, Z.; Schwille, P.; Biskup, C.; Kelbauskas, L.; Benndorf, K.; Klöcker, N.; Anhut, T.; et al. Fluorescence lifetime images and correlation spectra obtained by multidimensional time-correlated single photon counting. Microsc. Res. Tech 2006, 69, 186–195. [Google Scholar]
- Pietraszewska-Bogiel, A.; Gadella, T.W.J. FRET microscopy: From principle to routine technology in cell biology. J. Microsc 2011, 241, 111–118. [Google Scholar]
- Ishikawa-Ankerhold, H.C.; Ankerhold, R.; Drummen, G.P.C. Advanced fluorescence microscopy techniques—FRAP, FLIP, FLAP, FRET and FLIM. Molecules 2012, 17, 4047–4132. [Google Scholar]
- Gordon, G.W.; Berry, G.; Liang, X.H.; Levine, B.; Herman, B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J 1998, 74, 2702–2713. [Google Scholar]
- Jalink, K.; van Rheenen, J. Chapter 7 FilterFRET: Quantitative imaging of sensitized emission. Lab. Tech. Biochem. Mol. Biol 2009, 33, 289–349. [Google Scholar]
- Zimmermann, T.; Rietdorf, J.; Girod, A.; Georget, V.; Pepperkok, R. Spectral imaging and linear un-mixing enables improved FRET efficiency with a novel GFP2-YFP FRET pair. FEBS Lett 2002, 531, 245–249. [Google Scholar]
- Thaler, C.; Koushik, S.V.; Blank, P.S.; Vogel, S.S. Quantitative multiphoton spectral imaging and its use for measuring resonance energy transfer. Biophys. J 2005, 89, 2736–2749. [Google Scholar]
- Bastiaens, P.I.H.; Majoul, I.V.; Verveer, P.J.; Söling, H.D.; Jovin, T.M. Imaging the intracellular trafficking and state of the AB 5 quaternary structure of cholera toxin. EMBO J 1996, 15, 4246–4253. [Google Scholar]
- Bastiaens, P.I.H.; Jovin, T.M. Fluorescence Resonance Energy Transfer Microscopy. In Cell Biology: A Laboratory Handbook; Celis, J.E., Ed.; Academic Press: New York, NY, USA, 1998; Volume 3, pp. 136–146. [Google Scholar]
- Berney, C.; Danuser, G. FRET or no FRET: A quantitative comparison. Biophys. J 2003, 84, 3992–4010. [Google Scholar]
- Van Munster, E.B.; Kremers, G.J.; Adjobo-Hermans, M.J.W.; Gadella, T.W.J., Jr. Fluorescence resonance energy transfer (FRET) measurement by gradual acceptor photobleaching. J. Microsc. 2005, 218, 253–262. [Google Scholar]
- Jovin, T.M.; Arndt-Jovin, D.J. FRET Microscopy: Digital Imaging of Fluorescence Resonance Energy Transfer. Application in Cell Biology. In Cell Structure and Function by Microspectrofluorometry; Kohen, E., Hirschberg, J.G., Eds.; Academic Press: San Diego, CA, USA, 1989; pp. 99–117. [Google Scholar]
- Young, R.M.; Arnette, J.K.; Roess, D.A.; Barisas, B.G. Quantitation of fluorescence energy transfer between cell surface proteins via fluorescence donor photobleaching kinetics. Biophys. J 1994, 67, 881–888. [Google Scholar]
- Weigert, F. Bericht über polarisiertes Fluoreszenzlicht. Verh. Dtsch. Physik. Ges 1920, 23, 100–102. [Google Scholar]
- Gaviola, E.; Pringsheim, P. Über den Einfluß der Konzentration auf die Polarisation der Fluoreszenz von Farbstofflösungen. Z. Phys 1924, 24, 24–36. [Google Scholar]
- Clayton, H.A.; Hanley, Q.S.; Arndt-Jovin, D.J.; Subramanian, V.; Jovin, T.M. Dynamic fluorescence anisotropy imaging microscopy in the frequency domain (rFLIM). Biophys. J 2002, 83, 1631–1649. [Google Scholar]
- Xie, F.; Zhu, J.; Deng, C.; Huang, G.; Mitchelson, K.; Cheng, J. General and reliable quantitative measurements of fluorescence resonance energy transfer using three fluorescence channels. Analyst 2012, 137, 1013–1019. [Google Scholar]
- Åslund, N.; Carlsson, K. Confocal scanning microfluorometry of dual-labelled specimens using two excitation wavelengths and lock-in detection technique. Micron 1993, 24, 603–609. [Google Scholar]
- Carlsson, K.; Åslund, N.; Mossberg, K.; Philip, J. Simultaneous confocal recording of multiple fluorescent labels with improved channel separation. J. Microsc 1994, 176, 287–299. [Google Scholar]
- Carlsson, K. Signal-to-noise ratio for confocal microscopy when using the IMS technique. Micron 1995, 26, 317–322. [Google Scholar]
- Helm, P.J.; Franksson, O.; Carlsson, K. A confocal scanning laser microscope for quantitative ratiometric 3D measurements of [Ca2+] and Ca2+ diffusions in living cells stained with Fura-2. Pflügers Arch. Eur. J. Physiol 1995, 429, 672–681. [Google Scholar]
- Carlsson, K.; Liljeborg, A. Confocal fluorescence microscopy using spectral and lifetime information to simultaneously record four fluorophores with high channel separation. J. Microsc 1997, 185, 37–46. [Google Scholar]
- Carlsson, K.; Liljeborg, A. Simultaneous confocal lifetime imaging of multiple fluorophores using the intensity-modulated multiple-wavelength scanning (IMS) technique. J. Microsc 1998, 191, 119–127. [Google Scholar]
- Åslund, N.; Carlsson, K.S. Apparatus for Quantitative Imaging of Multiple Fluorophores. U.S. Patent 5,294,799, 15 March 1994. [Google Scholar]
- Åslund, N.; Carlsson, K.S. Apparatus for Quantitative Imaging of Multiple Fluorophores Using Dual Detectors. U.S. Patent 5,418,371, 23 May 1995. [Google Scholar]




© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Helm, P.J. Proposal of a New Method for Measuring Förster Resonance Energy Transfer (FRET) Rapidly, Quantitatively and Non-Destructively. Int. J. Mol. Sci. 2012, 13, 12367-12382. https://doi.org/10.3390/ijms131012367
Helm PJ. Proposal of a New Method for Measuring Förster Resonance Energy Transfer (FRET) Rapidly, Quantitatively and Non-Destructively. International Journal of Molecular Sciences. 2012; 13(10):12367-12382. https://doi.org/10.3390/ijms131012367
Chicago/Turabian StyleHelm, Paul Johannes. 2012. "Proposal of a New Method for Measuring Förster Resonance Energy Transfer (FRET) Rapidly, Quantitatively and Non-Destructively" International Journal of Molecular Sciences 13, no. 10: 12367-12382. https://doi.org/10.3390/ijms131012367
APA StyleHelm, P. J. (2012). Proposal of a New Method for Measuring Förster Resonance Energy Transfer (FRET) Rapidly, Quantitatively and Non-Destructively. International Journal of Molecular Sciences, 13(10), 12367-12382. https://doi.org/10.3390/ijms131012367