Abstract
When cellular reducing enzymes fail to shield the cell from increased amounts of reactive oxygen species (ROS), oxidative stress arises. The redox state is misbalanced, DNA and proteins are damaged and cellular transcription networks are activated. This condition can lead to the initiation and/or to the progression of atherosclerosis, tumors or pulmonary hypertension; diseases that are decisively furthered by the presence of oxidizing agents. Redox sensitive genes, like the zinc finger transcription factor early growth response 1 (Egr-1), play a pivotal role in the pathophysiology of these diseases. Apart from inducing apoptosis, signaling partners like the MEK/ERK pathway or the protein kinase C (PKC) can activate salvage programs such as cell proliferation that do not ameliorate, but rather worsen their outcome. Here, we review the currently available data on Egr-1 related signal transduction cascades in response to oxidative stress in the progression of epidemiologically significant diseases. Knowing the molecular pathways behind the pathology will greatly enhance our ability to identify possible targets for the development of new therapeutic strategies.
1. Introduction
Redox reactions contribute to countless significant biological processes and help to maintain vital cellular functions. During aerobic cellular respiration, for instance, glucose is oxidized to CO2 and oxygen is reduced to water. This key process is elementary to gain energy in the form of adenosine triphosphate (ATP). Coenzymes like nicotinamide adenine dinucleotide (NAD+) participate in these electron transfer reactions and can act as oxidizing (NAD+) or reducing agents (NADH). NADH can be further metabolized during oxidative phosphorylation, the electron transport chain across the inner mitochondrial membrane, to generate more ATP. Its related cofactor NAD phosphate (NADP+) is reduced to NADPH during the oxidative phase of the pentose phosphate pathway (PPP) in the cytosol. NADPH is an essential reducing agent, not only for anabolic reactions like nucleic acid synthesis, but also for the production and elimination of reactive oxygen species (ROS). ROS—as the name implies—are a heterogenic group of highly reactive molecules and a normal byproduct of metabolic redox reactions [1]. Among them are free radicals, which hold an unpaired valence shell electron, like superoxide (•O2−), nitric oxide (NO•) or the hydroxyl radical (•OH), and oxidizing molecules such as hydrogen peroxide (H2O2) or hypochlorus acid (HOCl). ROS are important mediators of cell signaling (called redox signaling) [2] and are indispensable for the immune defense in macrophages. The enzyme NADPH oxidase, for example, generates superoxide from molecular oxygen, which is then used to kill bacteria in the respiratory burst reaction within the cell [3]. Nonetheless, high levels of ROS contribute to cell toxicity [4], since they also damage host DNA/RNA [5], oxidize proteins [6] or cause lipid peroxidation [7]. Therefore, the proper balance (redox state) between ROS production and consumption must be maintained within the cellular compartments. Enzymes such as superoxide dismutase (SOD), catalases or peroxidases are capable of metabolizing accumulated ROS. Catalase, for example, decomposes H2O2 to H2O and O2. Glutathione uses NADPH to reduce H2O2 amounts and to regenerate itself. Other antioxidants, such as ascorbic acid (vitamin C) and tocopherol (vitamin E), are described as important radical scavengers [8]. The imbalance between the production of metabolically derived ROS and the organism’s deficiency to detoxify the cell and to repair the acquired cellular damage is called oxidative stress. This condition is related to diseases like atherosclerosis, diabetes, pulmonary hypertension (PH), cancer or Alzheimer’s disease [9–11]. Constantly increased amounts of oxidizing agents activate various signaling pathways that in turn are targeting the promoters of “redox sensitive” genes. One of these genes encodes the zinc finger transcription factor early growth response 1 (Egr-1). ROS have been shown to rapidly induce Egr-1 mRNA and protein expression [12]. Available data focusing on Egr-1 signaling after oxidative stress is limited and most of it is based on in vitro experiments [12–15]. Egr-1’s involvement in these epidemiologically relevant diseases, however, is important to understand and to develop new therapeutic strategies. Here we review the current data on Egr-1 in response to oxidative stress in the context of pathology.
2. Egr-1—A Redox Sensitive Transcription Factor
2.1. Structural Properties of Egr-1 Protein
Two exons code for an 80–82 kDa Cys2-His2-type zinc-finger transcription factor mapping to chromosome 5 [16]. Egr-1 was found to be rapidly and transiently expressed in response of a heterogenic group of stimuli like growth factors (GFs) [17], shear stress [18], oxygen deprivation [19,20], (reperfusion) injury [21–23] and oxidative stress [12,24,25]. The central DNA binding domain (DBD) of Egr-1 consists of the three zinc-finger motives [26,27] that characteristically bind to GC-rich promoter sequences (GCG(G/T)GGCG), therefore named Egr binding sequence (EBS) [17]. Egr-1 interconnects a broad variety of cascades upstream and downstream.
2.2. Functional Motifs at the Promoter
Besides an EBS [28], several functional response elements on the Egr-1 promoter presenting targets of distinct signal transduction cascades have been investigated and characterized [29]. At the 3′ end of the promoter and next to the TATA box, five serum response elements (SRE) are located [30]. Five Ets-family transcription factor-binding sites are arranged adjacently to these SREs. Furthermore, two cyclic adenosine monophosphate (cAMP) response elements (CREs), an APETALA1 (AP1) and two gene-specific activator protein 1 (Sp1) binding sites have been described [31].
2.3. Redox Regulated Transcription Capability
Previous in vitro studies described that redox levels influence the DNA binding capacity of Egr-1 in a dose-dependent manner. Cys residues within the DNA-binding domain of the protein are oxidized and severely diminish the DNA binding capacity of Egr-1, whereas under reducing conditions, DNA binding is enhanced [15]. Under non-toxic ROS levels, Egr-1’s binding ability remains preserved by activation of an apurinic/apyrimidinic endonuclease 1 (APE1) [15,25]. APE1 is a DNA repair enzyme with nuclear redox activity [32–34]. In various cell types, ROS induce nuclear translocation of APE1 [35,36], which in turn induces DNA binding of transcriptional regulators. APE1 restores Egr-1 DNA binding by direct protein—to protein interactions without neosynthesis and subsequently enhances its transcriptional activity; most likely via posttranslational modification [25]. Evidence for a positive autoregulatory loop between APE1 and Egr-1 exists [25]. Egr-1 upregulates APE1 by protein neosynthesis and APE1 in turn preserves the DNA-binding capacity of Egr-1, therefore mutually maintaining their transcriptional activity under non-toxic redox conditions. However, to prevent a never-ending activation between APE1 [37,38] and Egr-1 [28,39], the autoregulatory loop will eventually shut down, since APE1 binding to its own promoter leads to a downregulation by its own product [37].
2.4. MAPK Signal Transduction Cascades Aiming at the SRE
Hydrogen peroxide at non-toxic doses was shown to upregulate Egr-1 mRNA in vitro [12]. Moreover, Egr-1 activation was demonstrated to be MEK/ERK and c-Jun N-terminal kinases (JNKs) dependent in H9c2 cells; a myogenic cell line derived from the embryonic rat ventricle [40]. P42/44 mitogen-activated protein (MAP) kinase (MAPK), also known as extracellular signal-regulated kinase (ERK) 1/2, is part of the classical MAPK pathway being described to activate the Egr-1 promoter [41,42]. ERK1/2 has been shown to be phosphorylated and therefore directly activated through high levels of ROS [43], such as xanthine oxidase derived H2O2 [41], though the underlying mechanism still remains to be elucidated. Raf, mitogen-activated protein kinase kinase kinase (MAPKKK), activates mitogen-activated protein kinase kinase (MAPKK = MEK) 1/2, which in turn phosphorylates the dual acceptor motif ERK1/2 (Thr-Glu-Tyr). Activated ERK translocates into the nucleus and promotes binding of Ets-family transcription factor Elk-1 to the DNA. In close proximity to Elk-1, the SREs binding sites are located, indicating mutual activation. Serum response factor (SRF) is a MADS (Mcm1 and Arg80 in yeast, Agamous and Deficiens in plants, SRF in animals) -box transcription factor targeting SRE binding sites, which contain the characteristic sequence CC(A/T)6GG also known as CArG-box [44]. SRF has been shown to be involved in the transcriptional regulation of various GF-inducible genes [45,46], among them Egr-1 [47]. SRF is classically dependent on binding of the ternary complex factors (TCFs) Elk-1, Sap1 and Sap2, to activate transcription [45]. Elk-1 and SRF form a ternary complex [48] and together activate transcription [49,50] combining the MEK/ERK pathway with SRF associated gene regulation. There are two other MAPKs, associated with mechanical stress, that have been found to interact with Egr-1 [51]. These are JNKs, referred to as stress-activated protein kinases (SAPKs) and p38 isoforms. Both of them are responsive to mechanical, oxidative or environmental stress [52]. The p38 kinase, however, was not found to be involved in H2O2 dependent upregulation of Egr-1 [42].
3. Egr-1 Mediated Proliferation in Hypoxia Induced Pulmonary Fibrosis and Hypertension
Egr-1 is highly associated with growth, vascular cell proliferation [53], cell survival programs [18,54] and apoptosis [55]. A proliferative response to H2O2 is thought to be a protective mechanism against oxidant injury. Signal transduction of the H2O2-induced mitogenic signaling has been described to occur via the activation of MAPK and to increase the expression of Egr-1 in aortic smooth muscle cells [41]. Egr-1 regulates the expression of transforming growth factor beta 1 (TGF-β1) [56] and vice versa [57]. In an in vivo model of pulmonary fibrosis, TGF-β1 promotes epithelial apoptosis followed by mononuclear-rich inflammation, tissue fibrosis, myofibroblast and myocyte hyperplasia [55]. A null mutation of Egr-1 blocked TGF-β1 induced apoptosis in vivo, ameliorating collagen content, alveolar remodeling and parenchymal leukocyte infiltration [55]. Production of ROS has been implicated in chronic hypoxia-induced pulmonary hypertension (PH) and pulmonary vascular remodeling. Superoxide, generated under hypoxic conditions, contributed to PH through the induction of Egr-1 and its downstream gene target, tissue factor (TF) [58]. Egr-1 has been described to further the hypoxia induced autonomous proliferation of pulmonary artery adventitial fibroblasts via upregulation of the cell cycle regulator cyclin D, a key mechanism in the progression of disease [59]. Chronic hypoxia decreased lung SOD activity and SOD overexpression attenuated chronic hypoxic PH and vascular remodeling. Endothelial cell (EC) derived SOD (EC-SOD) overexpression also prevented the early hypoxia-dependent upregulation of Egr-1 and the procoagulant protein TF [58].
4. Apoptosis and Tumorigenesis
Apoptosis induced by H2O2 is thought to be a direct consequence of oxidant injury. Cellular Abelson murine leukemia viral oncogene homolog (c-Abl) is a tyrosine kinase that can act as a regulator of cell growth and apoptosis in response to oxidative stress. Significantly, H2O2-induced Egr-1 expression in vitro seems also to be induced by c-Abl kinase activity. Furthermore, c-Abl aims at the three distal SREs on the Egr-1 promoter via the MEK/ERK signaling. In addition, c-Abl-induced apoptosis is partially mitigated by Egr-1 activity, as cells, devoid of Egr-1 expression, undergo reduced rates of c-Abl-induced apoptosis [60].
When a transcription factor is participating in cell cycle control as a physiologic response to hypoxia or injury [61,62], an association with tumor growth is likely to be suspected. A number of tumor supressor genes are regulated directly by Egr-1, among them p53 [56], and the already mentioned relation betweeen GFs and Egr-1 has also been described for tumor dependent angiogenesis [63]. Cells expressing the breakpoint cluster region-abelson (bcr-Abl) oncogene demonstrate increased levels of intracellular ROS [64] and signaling initiated by the bcr-Abl kinase causes chronic myelogenous leukemia (CML). A recent publication reported that transcriptional upregulation of Fyn, a ROS sensitive src-family member, was strongly dependent on Egr-1 in an in vitro model [65], indicating participation of Egr-1 in the pathogenesis of CML.
In a majority of human prostate carcinoma specimens Egr-1 protein expression control was lost, suggesting that high levels of Egr-1 plays a central role in the initiation of human prostate cancers [66]. Indeed it was evidenced that Egr-1 deficient mice demonstrated impaired prostate tumor growth [67]. Alterations in the androgen receptor signaling were found to be a major cause of the disease and it has been shown that Egr-1 promotes the translocation of the androgen receptor into the nucleus [68]. Anti-hormonal therapy of prostate cancer becomes limited in the state of androgen-independent disease [69] and Egr-1 seems also capable to govern prostate cancer progression under androgen resistance [70,71]. Therefore, Egr-1 might be a new and interesting target of anti tumor therapy especially when anti-hormonal drugs are no longer effective.
5. Involvement of Egr-1 in Viral Pathogenesis
Several viral infections lead to the activation of Egr-1 [72–74]. Infections with the Herpesviridae family are not only characterized by a high prevalence in the human population, such as herpes simplex virus 1 and 2 (HHV1/2) [75], but also have specifically been described to promote tumors such as the Eppstein- Barr Virus (HHV4) or the Kaposi’s sarcoma-associated herpesvirus (KSHV) also known as HHV8. High stress levels can trigger and reactivate viral infections that have been latent for a long time or even can promote virus-associated malignancies on the long term [76,77]. Egr-1 has also been shown to critically participate in the KSHV reactivation process directly by mediating transcription of the gene encoding for replication and transcription activator (RTA) [78], a viral component known to control the switch from latent to lytic infection [79,80]. This seems also to be true for the reactivation of EBV, where Egr-1 has also been shown to positively regulate RTA via a positive feedback mechanism [81].
6. Signaling Involved in the Pathogenesis of Atherosclerosis
6.1. NADPH-Oxidase, Hemin and Egr-1
As mentioned above, NADPH-oxidase is a ROS generating enzyme. Many different stimuli including angiotensin II, glucose, and oxidized LDL activate NADPH-oxidases in the vascular wall [82]. Furthermore, NADPH-oxidase was found in coronary specimens and a relationship between plaque formation and NADPH-oxidase expression could be detected [83]. Extracorpuscular heme (ferroprotoporphyrin IX) released from hemoglobin is a potent proinflammatory ROS inducer. Heme catalyzes the oxidation of LDL, thus promoting foam cell formation and vascular smooth muscle cell (vSMC) proliferation. Hemin, oxidized heme, has been found to mediate redox-sensitive gene expression and to contribute to atherosclerotic plaque progression. Via MEK/ERK, hemin upregulated Egr-1 in vSMCs. This was directly dependent on NADPH-oxidase activity. The novel NADPH-oxidase inhibitors apocynin and diphenyleneiodonium chloride were also tested and could block hemin induced Egr-1 expression [84].
6.2. PKC and Egr-1
The role of Egr-1 for the pathogenesis of atherosclerosis is quite well described [85]. The protein kinase C (PKC) isoforms have been shown to lie upstream of Egr-1 [86]. In human aortic smooth muscle cells, PKCβII and PKCΦ activation and MEK/ERK mediated Egr-1 expression were essential for low dense lipoprotein (LDL)-induced cell proliferation [87], furthering the progression of the disease. It is well established, that H2O2 also leads to PKC activation [88] and in vitro, the autoregulatory loop between APE1 and Egr-1 was shown to be PKC dependent [25]. In an in vivo study, atherosclerosis was markedly impaired in mice deficient for both PKCβ and ApoE when compared to ApoE null mice [89]. Finally, recent reports have shown that gastrin effects on Egr-1 expression were dependent on activation of PKC family kinases, but do not require Ras (as involved in GF mediated MEK/ERK activation), phosphoinositol-3-kinase (PI3K) or intracellular calcium signals and are therefore arguing for a PKC/Raf/MEK/ERK/Egr-1 pathway [90].
6.3. Egr-1 and Accelerated Atherosclerosis in Diabetic Disease
It has been shown that insulin stimulates Egr-1 protein expression in endothelial cells (ECs) [91] and vSMCs [92] via the MEK/ERK pathway. Moreover, oxidative stress combined with insulin as initial stimulus further enhanced Egr-1 activation. Insulin resistance is characterized by compensatory hyperinsulinemia, with a functional MEK/ERK signaling but with selective impairment of PI3K [93]. Glucose can also induce Egr-1 expression, but PKC dependent in EC [91]. Egr-1 influences on the insulin gene itself could be mediated via pancreas duodenum homeobox-1 (PDX-1) [94,95], important for glucose homeostasis. Egr-1 signaling, PI3K or PKC might be interesting targets for drug therapy in atherosclerosis and diabetic disease in the future.
7. Conclusion—Daily Antioxidants as Dietary Supplements for the Prevention of Disease?
Oxidizing agents such as ROS or free radicals can be initiators and mediators of disease. When natural detoxifying enzymes fail, oxidative stress occurs and may cause aging, atherosclerosis or tumors. Egr-1 is rapidly induced after exposure to oxidants and interacts with various signaling partners mostly in the direction of disease progression (Figure 1).
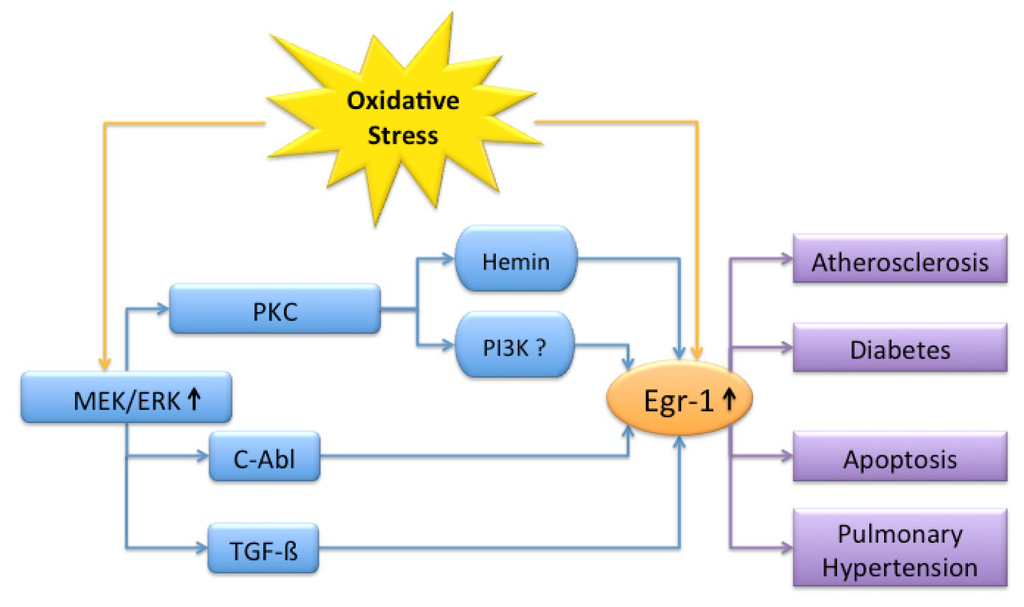
Figure 1.
Overview of signaling partners involved in oxidative stress mediated Egr-1 signaling. Oxidative stress leads to Egr-1 activation (↑) and promotes atherosclerosis, diabetes, apoptosis and pulmonary hypertension. The MEK/ERK pathway is the main signal transduction cascade involved. Depending on the targeted cell type and in vitro or in vivo data, different elements are involved.
Since Harman postulated the free radical theory of aging (FRTA) in 1956 [96], extensive research has been conducted to discover the key to a longer lifespan. Prevention of the formation of oxide radicals became an important target of the pharmaceutical industry and today a large group of over-the-counter drugs flood the drug market. But can the largely propagated daily supply of antioxidants, like vitamin C, E or selenium, in form of pills in fact reduce the incidence or outcome of a disease? In recent meta-analyses, the authors analyzed clinical trials investigating the oral supply of vitamins in tablet form on quality of life, mortality or the incidence of cardiovascular diseases and colon cancer. Unfortunately, they came to the conclusion that these high doses of antioxidants had no proven positive effect or even led to an increased mortality [97–100]. By scavenging ROS, fine-tuned feed back mechanisms such as the APE1/Egr-1 relation may become deranged. In turn, reducing ROS levels might also increase Egr-1 binding activity and even promote tumor progression or atherosclerosis. Therefore, the approach of disease prevention by scavenging radicals with high-dose supplements is neither reasonable nor safe. Probably we underestimate the complexity of fine regulated cellular signals in oxygen metabolism and in diseases. NADPH-oxidase inhibitors are currently in the very early stages of development and serious side effects regarding immune competence might be expected. The best guideline for a longer lifespan and the prevention of disease is a healthy lifestyle. This involves a balanced diet with high intake of fiber and natural antioxidants found in fruit and vegetables as well as daily physical activity and the cessation of smoking. Further studies at the molecular level are necessary to dissect the pathophysiological mechanisms behind ROS-induced signaling. Not until then, will we be able to design a distinct and individually matched therapy that will help to improve the outcome of diseases induced by oxidative stress.
Acknowledgements
This work was supported in part by grants from the “Förderprogramm für Forschung und Lehre—(FöFoLe)” and the Walter-Brendel-Centre of Experimental Medicine, Ludwig-Maximilians-Universität (Munich, Germany), as well as from the von Behring-Röntgen-Foundation (Marburg, Germany).
- Conflicts of InterestThe authors declare no conflict of interest.
References
- Sies, H. Role of reactive oxygen species in biological processes. Klin. Wochenschr 1991, 69, 965–968. [Google Scholar]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic signaling mediated by oxidants in ras-transformed fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar]
- Peskin, A.V.; Khramtsov, A.V.; Morozov, I.A.; Zemskov, V.M.; Zbarsky, I.B. Visualization of reactive oxygen species formation by phagocytizing macrophages. Exp. Cell. Res 1984, 151, 247–251. [Google Scholar]
- Sies, H.; de Groot, H. Role of reactive oxygen species in cell toxicity. Toxicol. Lett 1992, 64–65, 547–551. [Google Scholar]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate mmp-3-induced emt and genomic instability. Nature 2005, 436, 123–127. [Google Scholar]
- Dixit, R.; Mukhtar, H.; Bickers, D.R. Destruction of microsomal cytochrome p-450 by reactive oxygen species generated during photosensitization of hematoporphyrin derivative. Photochem. Photobiol 1983, 37, 173–176. [Google Scholar]
- Dixit, R.; Mukhtar, H.; Bickers, D.R. Studies on the role of reactive oxygen species in mediating lipid peroxide formation in epidermal microsomes of rat skin. J. Invest. Dermatol 1983, 81, 369–375. [Google Scholar]
- Förstermann, U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch 2010, 459, 923–939. [Google Scholar]
- Halliwell, B.; Cross, C.E. Oxygen-derived species: Their relation to human disease and environmental stress. Environ. Health Perspect 1994, 102, 5–12. [Google Scholar]
- Bray, T.M. Antioxidants and oxidative stress in health and disease: Introduction. Proc. Soc. Exp. Biol. Med 1999, 222. [Google Scholar] [CrossRef]
- Forsberg, L.; de Faire, U.; Morgenstern, R. Oxidative stress, human genetic variation, and disease. Arch. Biochem. Biophys 2001, 389, 84–93. [Google Scholar]
- Nose, K.; Ohba, M. Functional activation of the Egr-1 (early growth response-1) gene by hydrogen peroxide. Biochem. J 1996, 316, 381–383. [Google Scholar]
- Paron, I.; D’Elia, A.; D’Ambrosio, C.; Scaloni, A.; D’Aurizio, F.; Prescott, A.; Damante, G.; Tell, G. A proteomic approach to identify early molecular targets of oxidative stress in human epithelial lens cells. Biochem. J 2004, 378, 929–937. [Google Scholar]
- Nose, K.; Shibanuma, M.; Kikuchi, K.; Kageyama, H.; Sakiyama, S.; Kuroki, T. Transcriptional activation of early-response genes by hydrogen peroxide in a mouse osteoblastic cell line. Eur. J. Biochem 1991, 201, 99–106. [Google Scholar]
- Huang, R.P.; Adamson, E.D. Characterization of the DNA-binding properties of the early growth response-1 (Egr-1) transcription factor: Evidence for modulation by a redox mechanism. DNA Cell Biol 1993, 12, 265–273. [Google Scholar]
- Gashler, A.; Sukhatme, V.P. Early growth response protein 1 (Egr-1): Prototype of a zinc-finger family of transcription factors. Prog. Nucleic Acid Res. Mol. Biol 1995, 50, 191–224. [Google Scholar]
- Cao, X.M.; Koski, R.A.; Gashler, A.; McKiernan, M.; Morris, C.F.; Gaffney, R.; Hay, R.V.; Sukhatme, V.P. Identification and characterization of the Egr-1 gene product, a DNA-binding zinc finger protein induced by differentiation and growth signals. Mol. Cell. Biol 1990, 10, 1931–1939. [Google Scholar]
- Khachigian, L.M.; Anderson, K.R.; Halnon, N.J.; Gimbrone, M.A., Jr; Resnick, N.; Collins, T. Egr-1 is activated in endothelial cells exposed to fluid shear stress and interacts with a novel shear-stress-response element in the pdgf a-chain promoter. Arterioscler. Thromb. Vasc. Biol 1997, 17, 2280–2286. [Google Scholar]
- Yan, S.F.; Mackman, N.; Kisiel, W.; Stern, D.M.; Pinsky, D.J. Hypoxia/hypoxemia-induced activation of the procoagulant pathways and the pathogenesis of ischemia-associated thrombosis. Arterioscler. Thromb. Vasc. Biol 1999, 19, 2029–2035. [Google Scholar]
- Nishi, H.; Nishi, K.H.; Johnson, A.C. Early growth response-1 gene mediates up-regulation of epidermal growth factor receptor expression during hypoxia. Cancer Res 2002, 62, 827–834. [Google Scholar]
- Ouellette, A.J.; Malt, R.A.; Sukhatme, V.P.; Bonventre, J.V. Expression of two “immediate early” genes, Egr-1 and c-fos, in response to renal ischemia and during compensatory renal hypertrophy in mice. J. Clin. Invest 1990, 85, 766–771. [Google Scholar]
- Bonventre, J.V.; Sukhatme, V.P.; Bamberger, M.; Ouellette, A.J.; Brown, D. Localization of the protein product of the immediate early growth response gene, Egr-1, in the kidney after ischemia and reperfusion. Cell. Regul 1991, 2, 251–260. [Google Scholar]
- Brand, T.; Sharma, H.S.; Fleischmann, K.E.; Duncker, D.J.; McFalls, E.O.; Verdouw, P.D.; Schaper, W. Proto-oncogene expression in porcine myocardium subjected to ischemia and reperfusion. Circ. Res 1992, 71, 1351–1360. [Google Scholar]
- Hirata, H.; Asanuma, M.; Cadet, J.L. Superoxide radicals are mediators of the effects of methamphetamine on zif268 (Egr-1, ngfi-a) in the brain: Evidence from using cuzn superoxide dismutase transgenic mice. Brain Res. Mol. Brain Res 1998, 58, 209–216. [Google Scholar]
- Pines, A.; Bivi, N.; Romanello, M.; Damante, G.; Kelley, M.R.; Adamson, E.D.; D’Andrea, P.; Quadrifoglio, F.; Moro, L.; Tell, G. Cross-regulation between Egr-1 and ape/ref-1 during early response to oxidative stress in the human osteoblastic hobit cell line: Evidence for an autoregulatory loop. Free Radic Res 2005, 39, 269–281. [Google Scholar]
- Christy, B.; Nathans, D. DNA binding site of the growth factor-inducible protein zif268. Proc. Natl. Acad. Sci. USA 1989, 86, 8737–8741. [Google Scholar]
- Pavletich, N.P.; Pabo, C.O. Zinc finger-DNA recognition: Crystal structure of a zif268-DNA complex at 2.1 a. Science 1991, 252, 809–817. [Google Scholar]
- Sakamoto, K.M.; Bardeleben, C.; Yates, K.E.; Raines, M.A.; Golde, D.W.; Gasson, J.C. 5′ upstream sequence and genomic structure of the human primary response gene, Egr-1/tis8. Oncogene 1991, 6, 867–871. [Google Scholar]
- Christy, B.; Nathans, D. Functional serum response elements upstream of the growth factor-inducible gene zif268. Mol. Cell. Biol 1989, 9, 4889–4895. [Google Scholar]
- Tsai-Morris, C.H.; Cao, X.M.; Sukhatme, V.P. 5′ flanking sequence and genomic structure of Egr-1, a murine mitogen inducible zinc finger encoding gene. Nucleic Acids Res 1988, 16, 8835–8846. [Google Scholar]
- Schwachtgen, J.L.; Campbell, C.J.; Braddock, M. Full promoter sequence of human early growth response factor-1 (Egr-1): Demonstration of a fifth functional serum response element. DNA Seq 2000, 10, 429–432. [Google Scholar]
- Meira, L.B.; Cheo, D.L.; Hammer, R.E.; Burns, D.K.; Reis, A.; Friedberg, E.C. Genetic interaction between hap1/ref-1 and p53. Nat. Genet 1997, 17, 145. [Google Scholar]
- Tell, G.; Quadrifoglio, F.; Tiribelli, C.; Kelley, M.R. The many functions of ape1/ref-1: Not only a DNA repair enzyme. Antioxid. Redox. Signal 2009, 11, 601–620. [Google Scholar]
- Tell, G.; Scaloni, A.; Pellizzari, L.; Formisano, S.; Pucillo, C.; Damante, G. Redox potential controls the structure and DNA binding activity of the paired domain. J. Biol. Chem 1998, 273, 25062–25072. [Google Scholar]
- Tell, G.; Zecca, A.; Pellizzari, L.; Spessotto, P.; Colombatti, A.; Kelley, M.R.; Damante, G.; Pucillo, C. An “environment to nucleus” signaling system operates in b lymphocytes: Redox status modulates bsap/pax-5 activation through ref-1 nuclear translocation. Nucleic Acids Res 2000, 28, 1099–1105. [Google Scholar]
- Tell, G.; Pines, A.; Pandolfi, M.; D’Elia, A.V.; Donnini, D.; Lonigro, R.; Manzini, G.; Russo, D.; di Loreto, C.; Damante, G. Ape/ref-1 is controlled by both redox and camp-dependent mechanisms in rat thyroid cells. Horm. Metab. Res 2002, 34, 303–310. [Google Scholar]
- Izumi, T.; Henner, W.D.; Mitra, S. Negative regulation of the major human ap-endonuclease, a multifunctional protein. Biochemistry 1996, 35, 14679–14683. [Google Scholar]
- Harrison, L.; Ascione, A.G.; Wilson, D.M., III; Demple, B. Characterization of the promoter region of the human apurinic endonuclease gene (ape). J. Biol. Chem 1995, 270, 5556–5564. [Google Scholar]
- Cao, X.; Mahendran, R.; Guy, G.R.; Tan, Y.H. Detection and characterization of cellular Egr-1 binding to its recognition site. J. Biol. Chem 1993, 268, 16949–16957. [Google Scholar]
- Aggeli, I.K.; Beis, I.; Gaitanaki, C. Erks and jnks mediate hydrogen peroxide-induced Egr-1 expression and nuclear accumulation in h9c2 cells. Physiol. Res 2009, 59, 443–454. [Google Scholar]
- Hartney, T.; Birari, R.; Venkataraman, S.; Villegas, L.; Martinez, M.; Black, S.M.; Stenmark, K.R.; Nozik-Grayck, E. Xanthine oxidase-derived ros upregulate Egr-1 via erk1/2 in pa smooth muscle cells; model to test impact of extracellular ros in chronic hypoxia. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Iyoda, T.; Zhang, F.; Sun, L.; Hao, F.; Schmitz-Peiffer, C.; Xu, X.; Cui, M.Z. Lysophosphatidic acid induces early growth response-1 (Egr-1) protein expression via protein kinase cdelta-regulated extracellular signal-regulated kinase (erk) and c-jun n-terminal kinase (jnk) activation in vascular smooth muscle cells. J. Biol. Chem 2012, 287, 22635–22642. [Google Scholar]
- Chen, C.A.; Chen, T.S.; Chen, H.C. Extracellular signal-regulated kinase plays a proapoptotic role in podocytes after reactive oxygen species treatment and inhibition of integrin-extracellular matrix interaction. Exp. Biol. Med. (Maywood) 2012, 237, 777–783. [Google Scholar]
- Treisman, R. Identification and purification of a polypeptide that binds to the c-fos serum response element. EMBO J 1987, 6, 2711–2717. [Google Scholar]
- Whitmarsh, A.J.; Shore, P.; Sharrocks, A.D.; Davis, R.J. Integration of map kinase signal transduction pathways at the serum response element. Science 1995, 269, 403–407. [Google Scholar]
- Arsenian, S.; Weinhold, B.; Oelgeschlager, M.; Ruther, U.; Nordheim, A. Serum response factor is essential for mesoderm formation during mouse embryogenesis. EMBO J 1998, 17, 6289–6299. [Google Scholar]
- Datta, R.; Taneja, N.; Sukhatme, V.P.; Qureshi, S.A.; Weichselbaum, R.; Kufe, D.W. Reactive oxygen intermediates target cc(a/t)6gg sequences to mediate activation of the early growth response 1 transcription factor gene by ionizing radiation. Proc. Natl. Acad. Sci. USA 1993, 90, 2419–2422. [Google Scholar]
- Marais, R.; Wynne, J.; Treisman, R. The srf accessory protein elk-1 contains a growth factor-regulated transcriptional activation domain. Cell 1993, 73, 381–393. [Google Scholar]
- Hill, C.S.; Marais, R.; John, S.; Wynne, J.; Dalton, S.; Treisman, R. Functional analysis of a growth factor-responsive transcription factor complex. Cell 1993, 73, 395–406. [Google Scholar]
- Vickers, E.R.; Kasza, A.; Kurnaz, I.A.; Seifert, A.; Zeef, L.A.; O’Donnell, A.; Hayes, A.; Sharrocks, A.D. Ternary complex factor-serum response factor complex-regulated gene activity is required for cellular proliferation and inhibition of apoptotic cell death. Mol. Cell. Biol 2004, 24, 10340–10351. [Google Scholar]
- Lim, C.P.; Jain, N.; Cao, X. Stress-induced immediate-early gene, Egr-1, involves activation of p38/jnk1. Oncogene 1998, 16, 2915–2926. [Google Scholar]
- Hebert, M.A.; Serova, L.I.; Sabban, E.L. Single and repeated immobilization stress differentially trigger induction and phosphorylation of several transcription factors and mitogen-activated protein kinases in the rat locus coeruleus. J. Neurochem 2005, 95, 484–498. [Google Scholar]
- Pagel, J.I.; Ziegelhoeffer, T.; Heil, M.; Fischer, S.; Fernandez, B.; Schaper, W.; Preissner, K.T.; Deindl, E. Role of early growth response 1 in arteriogenesis: Impact on vascular cell proliferation and leukocyte recruitment in vivo. Thromb. Haemost 2012, 107, 562–574. [Google Scholar]
- Hallahan, D.E.; Dunphy, E.; Virudachalam, S.; Sukhatme, V.P.; Kufe, D.W.; Weichselbaum, R.R. C-jun and Egr-1 participate in DNA synthesis and cell survival in response to ionizing radiation exposure. J. Biol. Chem 1995, 270, 30303–30309. [Google Scholar]
- Lee, C.G.; Cho, S.J.; Kang, M.J.; Chapoval, S.P.; Lee, P.J.; Noble, P.W.; Yehualaeshet, T.; Lu, B.; Flavell, R.A.; Milbrandt, J.; et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor β1-induced pulmonary fibrosis. J. Exp. Med 2004, 200, 377–389. [Google Scholar]
- Baron, V.; Adamson, E.D.; Calogero, A.; Ragona, G.; Mercola, D. The transcription factor Egr1 is a direct regulator of multiple tumor suppressors including tgfbeta1, pten, p53, and fibronectin. Cancer Gene Ther 2006, 13, 115–124. [Google Scholar]
- Ohba, M.; Shibanuma, M.; Kuroki, T.; Nose, K. Production of hydrogen peroxide by transforming growth factor-β1 and its involvement in induction of Egr-1 in mouse osteoblastic cells. J. Cell Biol 1994, 126, 1079–1088. [Google Scholar]
- Nozik-Grayck, E.; Suliman, H.B.; Majka, S.; Albietz, J.; Van Rheen, Z.; Roush, K.; Stenmark, K.R. Lung ec-sod overexpression attenuates hypoxic induction of Egr-1 and chronic hypoxic pulmonary vascular remodeling. Am. J. Physiol. Lung Cell. Mol. Physiol 2008, 295, L422–L430. [Google Scholar]
- Banks, M.F.; Gerasimovskaya, E.V.; Tucker, D.A.; Frid, M.G.; Carpenter, T.C.; Stenmark, K.R. Egr-1 antisense oligonucleotides inhibit hypoxia-induced proliferation of pulmonary artery adventitial fibroblasts. J. Appl. Physiol 2005, 98, 732–738. [Google Scholar]
- Stuart, J.R.; Kawai, H.; Tsai, K.K.; Chuang, E.Y.; Yuan, Z.M. C-abl regulates early growth response protein (Egr1) in response to oxidative stress. Oncogene 2005, 24, 8085–8092. [Google Scholar]
- Fahmy, R.G.; Khachigian, L.M. Antisense Egr-1 RNA driven by the cmv promoter is an inhibitor of vascular smooth muscle cell proliferation and regrowth after injury. J. Cell. Biochem 2002, 84, 575–582. [Google Scholar]
- Liao, Y.; Shikapwashya, O.N.; Shteyer, E.; Dieckgraefe, B.K.; Hruz, P.W.; Rudnick, D.A. Delayed hepatocellular mitotic progression and impaired liver regeneration in early growth response-1-deficient mice. J. Biol. Chem 2004, 279, 43107–43116. [Google Scholar]
- Fahmy, R.G.; Dass, C.R.; Sun, L.Q.; Chesterman, C.N.; Khachigian, L.M. Transcription factor Egr-1 supports fgf-dependent angiogenesis during neovascularization and tumor growth. Nat. Med 2003, 9, 1026–1032. [Google Scholar]
- Sattler, M.; Verma, S.; Shrikhande, G.; Byrne, C.H.; Pride, Y.B.; Winkler, T.; Greenfield, E.A.; Salgia, R.; Griffin, J.D. The bcr/abl tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J. Biol. Chem 2000, 275, 24273–24278. [Google Scholar]
- Gao, Y.; Howard, A.; Ban, K.; Chandra, J. Oxidative stress promotes transcriptional up-regulation of fyn in bcr-abl1-expressing cells. J. Biol. Chem 2009, 284, 7114–7125. [Google Scholar]
- Abdulkadir, S.A.; Carbone, J.M.; Naughton, C.K.; Humphrey, P.A.; Catalona, W.J.; Milbrandt, J. Frequent and early loss of the Egr1 corepressor nab2 in human prostate carcinoma. Hum. Pathol 2001, 32, 935–939. [Google Scholar]
- Abdulkadir, S.A.; Qu, Z.; Garabedian, E.; Song, S.K.; Peters, T.J.; Svaren, J.; Carbone, J.M.; Naughton, C.K.; Catalona, W.J.; Ackerman, J.J.; et al. Impaired prostate tumorigenesis in Egr1-deficient mice. Nat. Med 2001, 7, 101–107. [Google Scholar]
- Yang, S.Z.; Abdulkadir, S.A. Early growth response gene 1 modulates androgen receptor signaling in prostate carcinoma cells. J. Biol. Chem 2003, 278, 39906–39911. [Google Scholar]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar]
- Yang, S.Z.; Eltoum, I.A.; Abdulkadir, S.A. Enhanced Egr1 activity promotes the growth of prostate cancer cells in an androgen-depleted environment. J. Cell. Biochem 2006, 97, 1292–1299. [Google Scholar]
- Kawamura, S.; Sato, I.; Wada, T.; Yamaguchi, K.; Li, Y.; Li, D.; Zhao, X.; Ueno, S.; Aoki, H.; Tochigi, T.; et al. Plasma membrane-associated sialidase (neu3) regulates progression of prostate cancer to androgen-independent growth through modulation of androgen receptor signaling. Cell Death Differ 2012, 19, 170–179. [Google Scholar]
- Katsarou, K.; Lavdas, A.A.; Tsitoura, P.; Serti, E.; Markoulatos, P.; Mavromara, P.; Georgopoulou, U. Endocytosis of hepatitis c virus non-enveloped capsid-like particles induces mapk-erk1/2 signaling events. Cell. Mol. Life Sci 2010, 67, 2491–2506. [Google Scholar]
- Bedadala, G.R.; Palem, J.R.; Graham, L.; Hill, J.M.; McFerrin, H.E.; Hsia, S.C. Lytic hsv-1 infection induces the multifunctional transcription factor early growth response-1 (Egr-1) in rabbit corneal cells. Virol. J 2011, 8, 262. [Google Scholar]
- Calogero, A.; Cuomo, L.; D’Onofrio, M.; de Grazia, U.; Spinsanti, P.; Mercola, D.; Faggioni, A.; Frati, L.; Adamson, E.D.; Ragona, G. Expression of Egr-1 correlates with the transformed phenotype and the type of viral latency in ebv genome positive lymphoid cell lines. Oncogene 1996, 13, 2105–2112. [Google Scholar]
- Sudenga, S.L.; Kempf, M.C.; McGwin, G., Jr; Wilson, C.M.; Hook, E.W., III; Shrestha, S. Incidence, prevalence, and epidemiology of herpes simplex virus-2 in HIV-1-positive and hiv-1-negative adolescents. Sex. Transm. Dis 2012, 39, 300–305. [Google Scholar]
- Ye, F.; Zhou, F.; Bedolla, R.G.; Jones, T.; Lei, X.; Kang, T.; Guadalupe, M.; Gao, S.J. Reactive oxygen species hydrogen peroxide mediates kaposi’s sarcoma-associated herpesvirus reactivation from latency. PLoS Pathog 2011, 7, e1002054. [Google Scholar]
- Preston, C.M.; Nicholl, M.J. Induction of cellular stress overcomes the requirement of herpes simplex virus type 1 for immediate-early protein icp0 and reactivates expression from quiescent viral genomes. J. Virol 2008, 82, 11775–11783. [Google Scholar]
- Dyson, O.F.; Traylen, C.M.; Akula, S.M. Cell membrane-bound kaposi’s sarcoma-associated herpesvirus-encoded glycoprotein b promotes virus latency by regulating expression of cellular Egr-1. J. Biol. Chem 2010, 285, 37491–37502. [Google Scholar]
- Lukac, D.M.; Kirshner, J.R.; Ganem, D. Transcriptional activation by the product of open reading frame 50 of kaposi’s sarcoma-associated herpesvirus is required for lytic viral reactivation in b cells. J. Virol 1999, 73, 9348–9361. [Google Scholar]
- Dyson, O.F.; Walker, L.R.; Whitehouse, A.; Cook, P.P.; Akula, S.M. Resveratrol inhibits kshv reactivation by lowering the levels of cellular Egr-1. PLoS One 2012, 7, e33364. [Google Scholar]
- Chang, Y.; Lee, H.H.; Chen, Y.T.; Lu, J.; Wu, S.Y.; Chen, C.W.; Takada, K.; Tsai, C.H. Induction of the early growth response 1 gene by epstein-barr virus lytic transactivator zta. J. Virol 2006, 80, 7748–7755. [Google Scholar]
- Bedard, K.; Krause, K.H. The nox family of ros-generating nadph oxidases: Physiology and pathophysiology. Physiol. Rev 2007, 87, 245–313. [Google Scholar]
- Terashima, M.; Inoue, N.; Ohashi, Y.; Yokoyama, M. Relationship between coronary plaque formation and nad(p)h oxidase-derived reactive oxygen species—Comparison of intravascular ultrasound finding of atherosclerotic lesions with histochemical characteristics. Kobe J. Med. Sci 2007, 53, 107–117. [Google Scholar]
- Hasan, R.N.; Schafer, A.I. Hemin upregulates Egr-1 expression in vascular smooth muscle cells via reactive oxygen species erk-1/2-elk-1 and nf-kappab. Circ. Res 2008, 102, 42–50. [Google Scholar]
- Khachigian, L.M. Early growth response-1 in cardiovascular pathobiology. Circ. Res 2006, 98, 186–191. [Google Scholar]
- Yan, S.F.; Lu, J.; Zou, Y.S.; Kisiel, W.; Mackman, N.; Leitges, M.; Steinberg, S.; Pinsky, D.; Stern, D. Protein kinase c-beta and oxygen deprivation. A novel Egr-1-dependent pathway for fibrin deposition in hypoxemic vasculature. J. Biol. Chem 2000, 275, 11921–11928. [Google Scholar]
- Heo, K.S.; Kim, D.U.; Kim, L.; Nam, M.; Baek, S.T.; Park, S.K.; Park, Y.; Myung, C.S.; Hwang, S.O.; Hoe, K.L. Activation of pkcbeta(ii) and pkctheta is essential for ldl-induced cell proliferation of human aortic smooth muscle cells via gi-mediated erk1/2 activation and Egr-1 upregulation. Biochem. Biophys. Res. Commun 2008, 368, 126–131. [Google Scholar]
- Konishi, H.; Tanaka, M.; Takemura, Y.; Matsuzaki, H.; Ono, Y.; Kikkawa, U.; Nishizuka, Y. Activation of protein kinase c by tyrosine phosphorylation in response to H2O2. Proc. Natl. Acad. Sci. USA 1997, 94, 11233–11237. [Google Scholar]
- Harja, E.; Chang, J.S.; Lu, Y.; Leitges, M.; Zou, Y.S.; Schmidt, A.M.; Yan, S.F. Mice deficient in pkcbeta and apolipoprotein e display decreased atherosclerosis. FASEB J 2009, 23, 1081–1091. [Google Scholar]
- Cramer, T.; Juttner, S.; Plath, T.; Mergler, S.; Seufferlein, T.; Wang, T.C.; Merchant, J.; Hocker, M. Gastrin transactivates the chromogranin a gene through mek-1/erk- and pkc-dependent phosphorylation of sp1 and creb. Cell. Signal 2008, 20, 60–72. [Google Scholar]
- Hasan, R.N.; Phukan, S.; Harada, S. Differential regulation of early growth response gene-1 expression by insulin and glucose in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol 2003, 23, 988–993. [Google Scholar]
- Wang, C.C.; Sharma, G.; Draznin, B. Early growth response gene-1 expression in vascular smooth muscle cells effects of insulin and oxidant stress. Am. J. Hypertens 2006, 19, 366–372. [Google Scholar]
- Yu, X.; Shen, N.; Zhang, M.L.; Pan, F.Y.; Wang, C.; Jia, W.P.; Liu, C.; Gao, Q.; Gao, X.; Xue, B.; et al. Egr-1 decreases adipocyte insulin sensitivity by tilting pi3k/akt and mapk signal balance in mice. EMBO J 2011, 30, 3754–3765. [Google Scholar]
- Eto, K.; Kaur, V.; Thomas, M.K. Regulation of pancreas duodenum homeobox-1 expression by early growth response-1. J. Biol. Chem 2007, 282, 5973–5983. [Google Scholar]
- Eto, K.; Kaur, V.; Thomas, M.K. Regulation of insulin gene transcription by the immediate-early growth response gene Egr-1. Endocrinology 2006, 147, 2923–2935. [Google Scholar]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol 1956, 11, 298–300. [Google Scholar]
- Bjelakovic, G.; Nikolova, D.; Simonetti, R.G.; Gluud, C. Systematic review: Primary and secondary prevention of gastrointestinal cancers with antioxidant supplements. Aliment. Pharmacol. Ther 2008, 28, 689–703. [Google Scholar]
- Vivekananthan, D.P.; Penn, M.S.; Sapp, S.K.; Hsu, A.; Topol, E.J. Use of antioxidant vitamins for the prevention of cardiovascular disease: Meta-analysis of randomised trials. Lancet 2003, 361, 2017–2023. [Google Scholar]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. JAMA 2007, 297, 842–857. [Google Scholar]
- Eidelman, R.S.; Hollar, D.; Hebert, P.R.; Lamas, G.A.; Hennekens, C.H. Randomized trials of vitamin e in the treatment and prevention of cardiovascular disease. Arch. Intern. Med 2004, 164, 1552–1556. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).