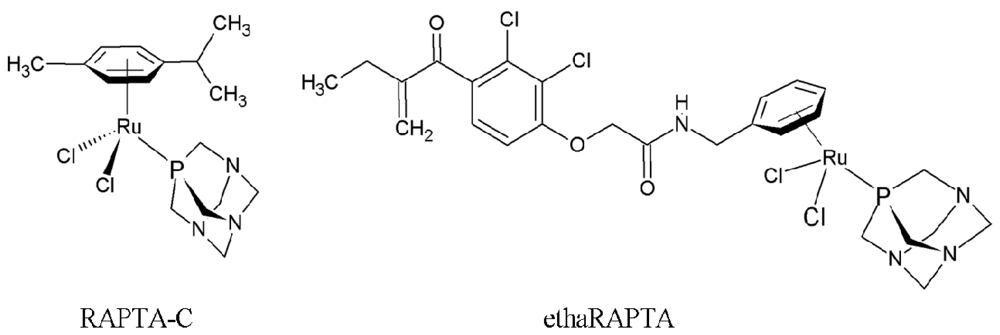
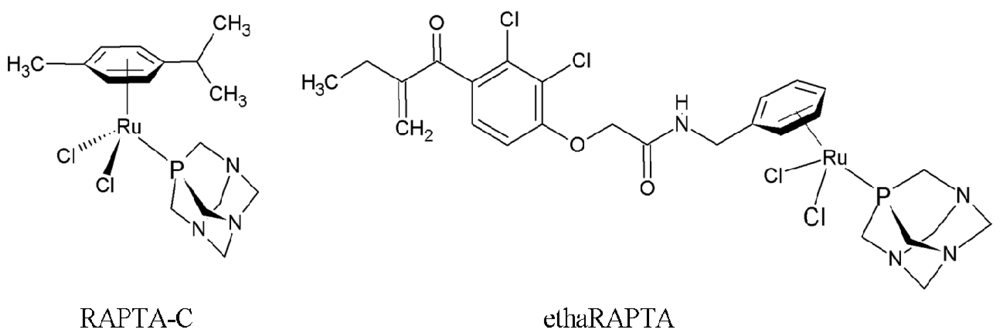
Altered DNA Binding and Amplification of Human Breast Cancer Suppressor Gene BRCA1 Induced by a Novel Antitumor Compound, [Ru(η6-p-phenylethacrynate)Cl2(pta)]

Abstract
:1. Introduction
2. Results and Discussion
2.1. EthaRAPTA-Mediated Conformational Changes of the Cell-Free BRCA1 Fragment
2.2. The Preference of ethaRAPTA Base Binding is in the Order A > G > T > C
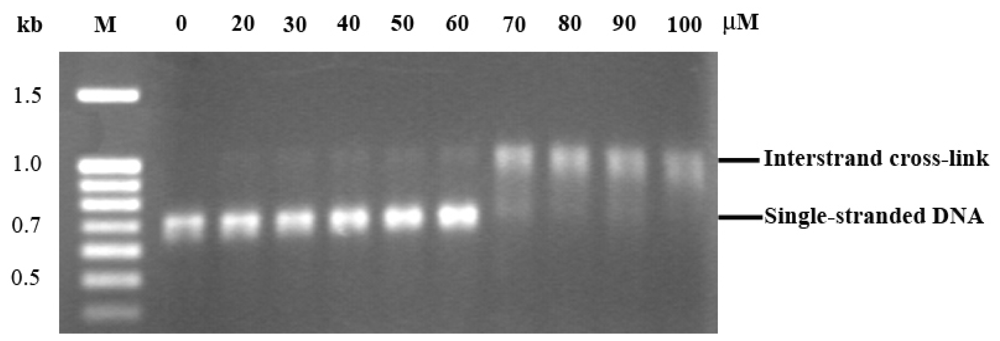
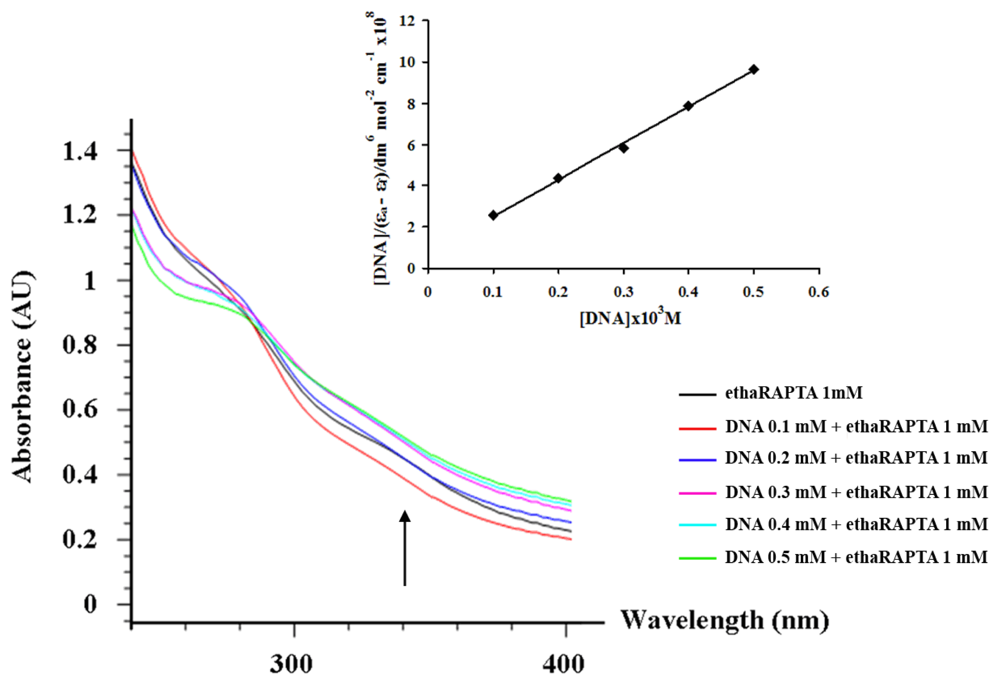
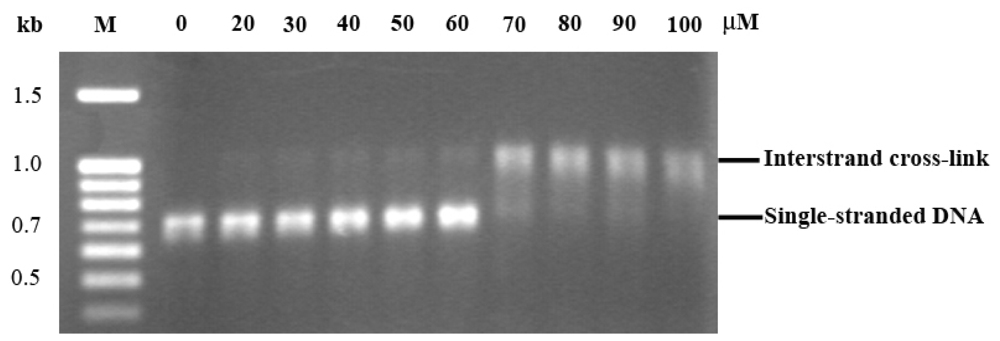
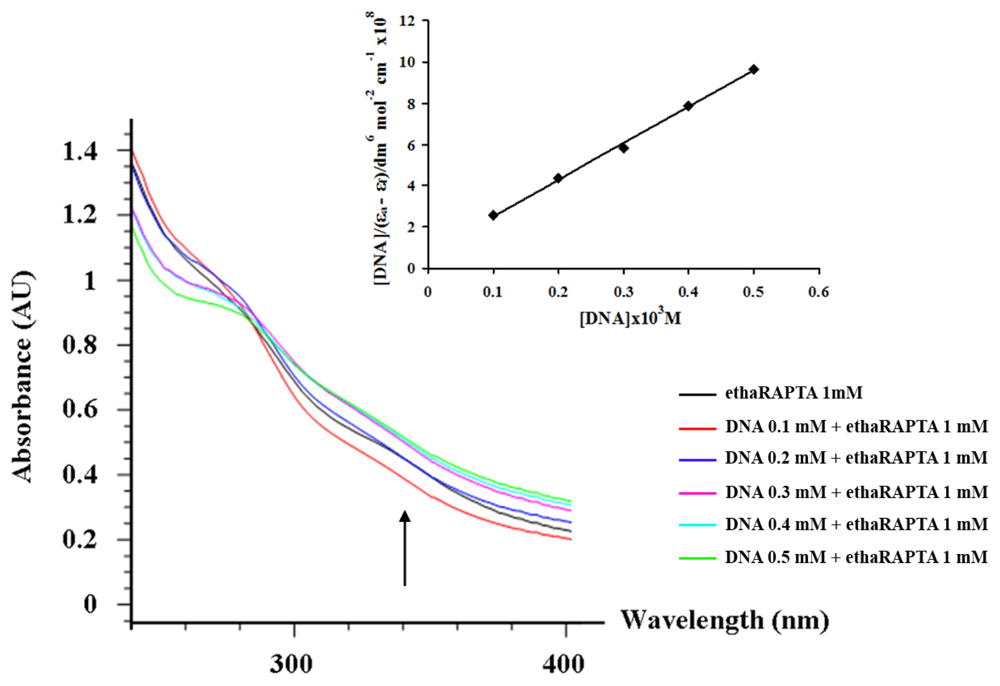
2.3. EthaRAPTA can also Bind to the BRCA1 Fragment through Intercalation
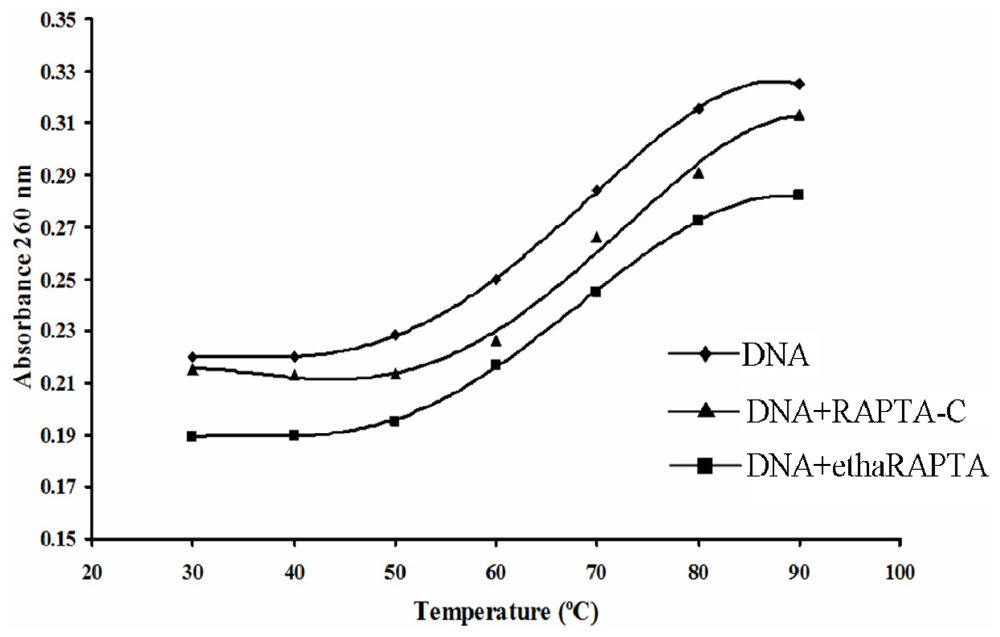
2.4. Altered Thermal Stability of EthaRAPTA Adducts
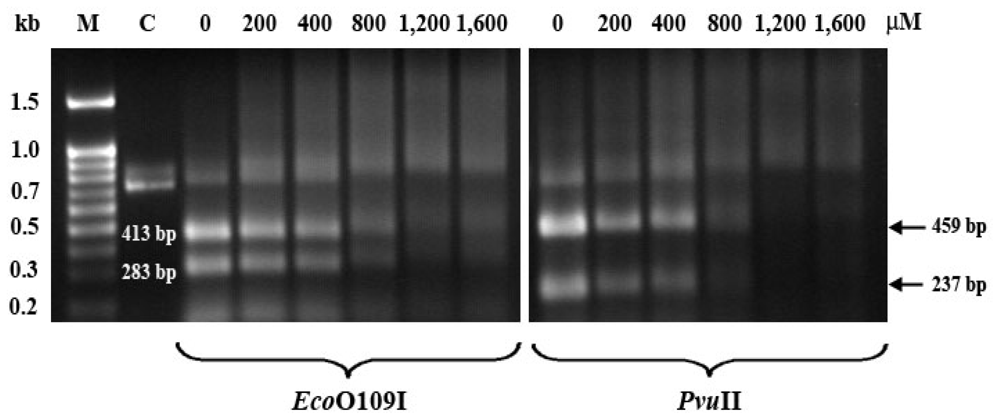
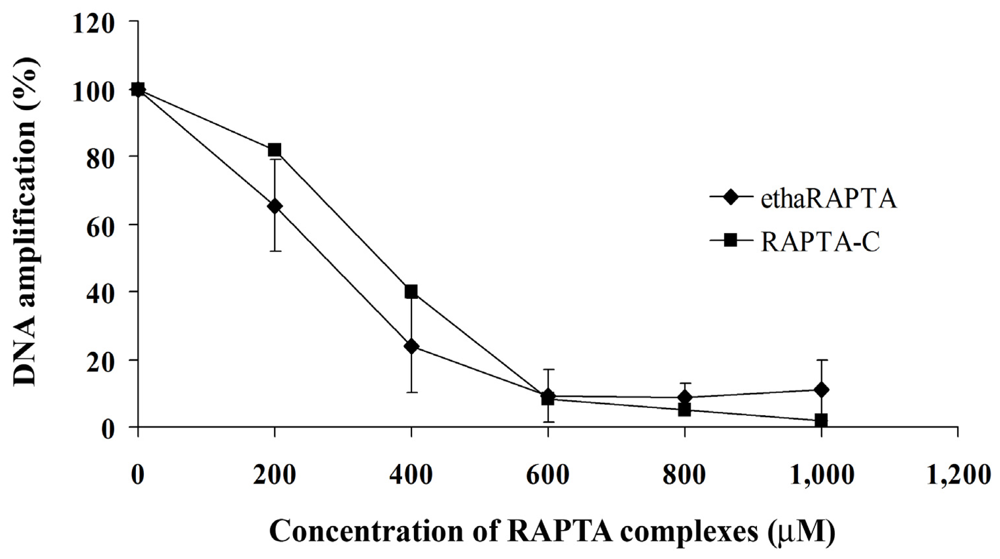
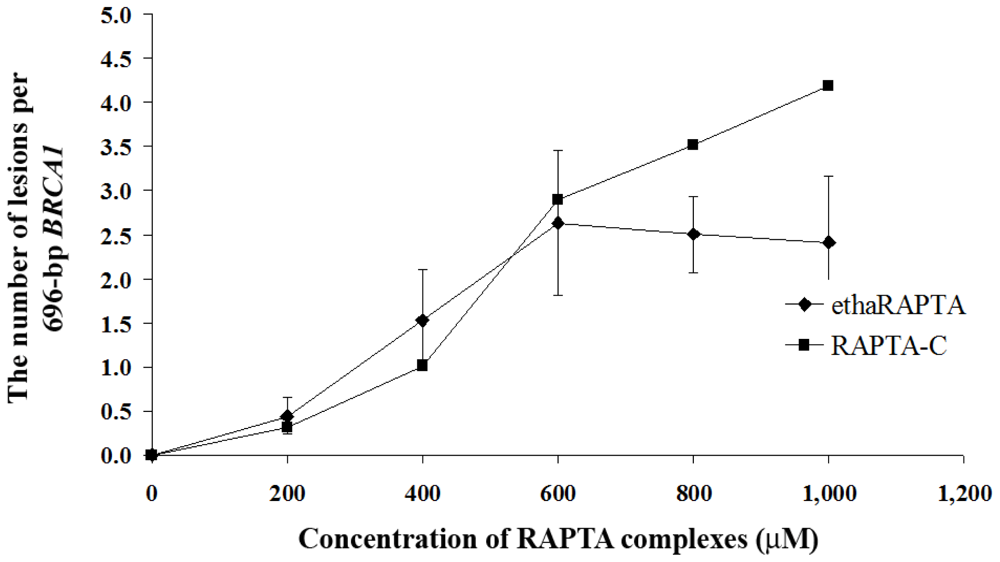
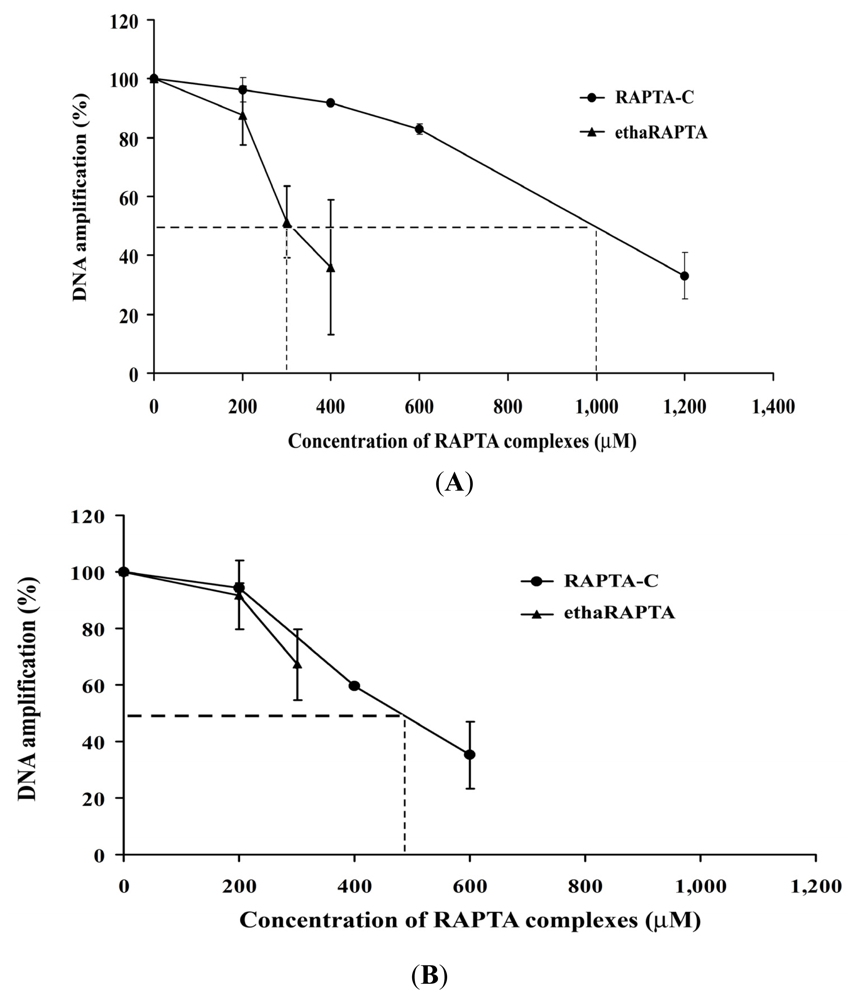
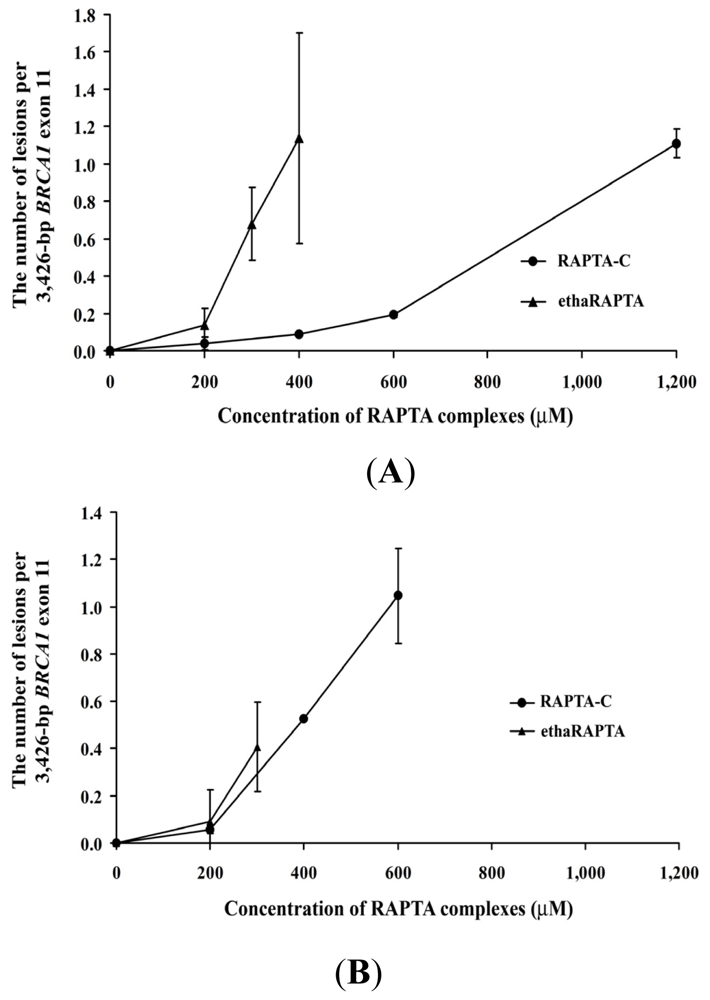
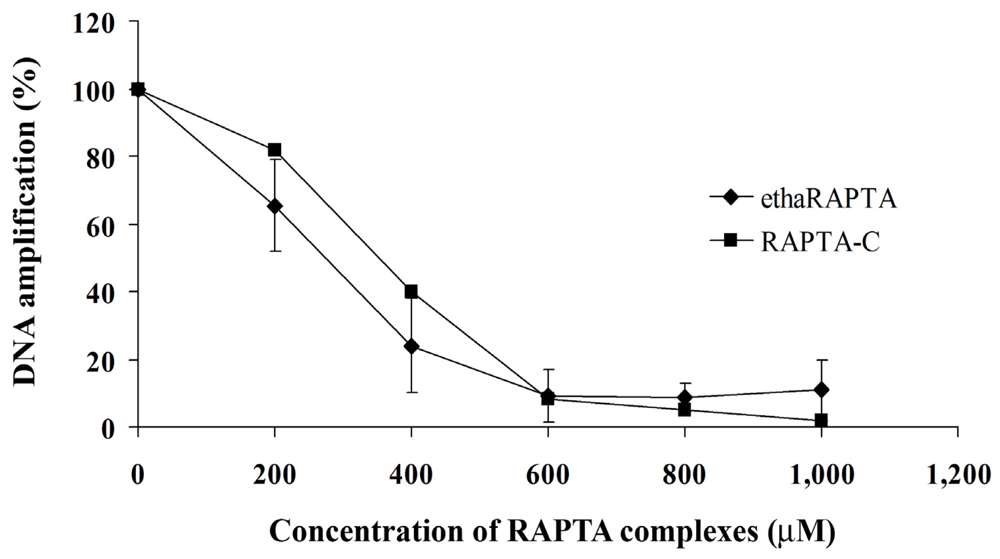
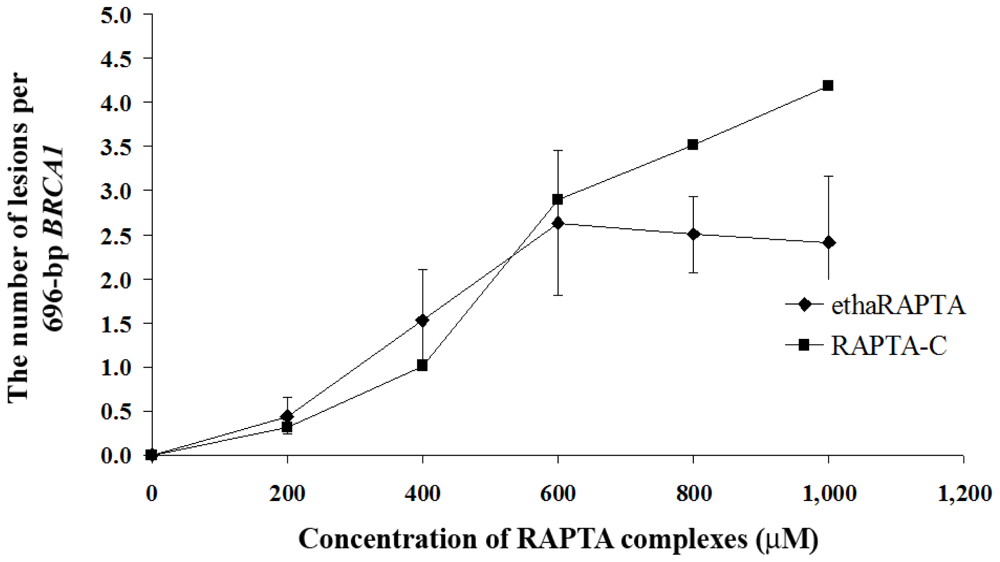
2.5. EthaRAPTA Exhibited a Higher Efficiency than RAPTA-C in Inhibiting BRCA1 Amplification
3. Experimental Section
3.1. Materials
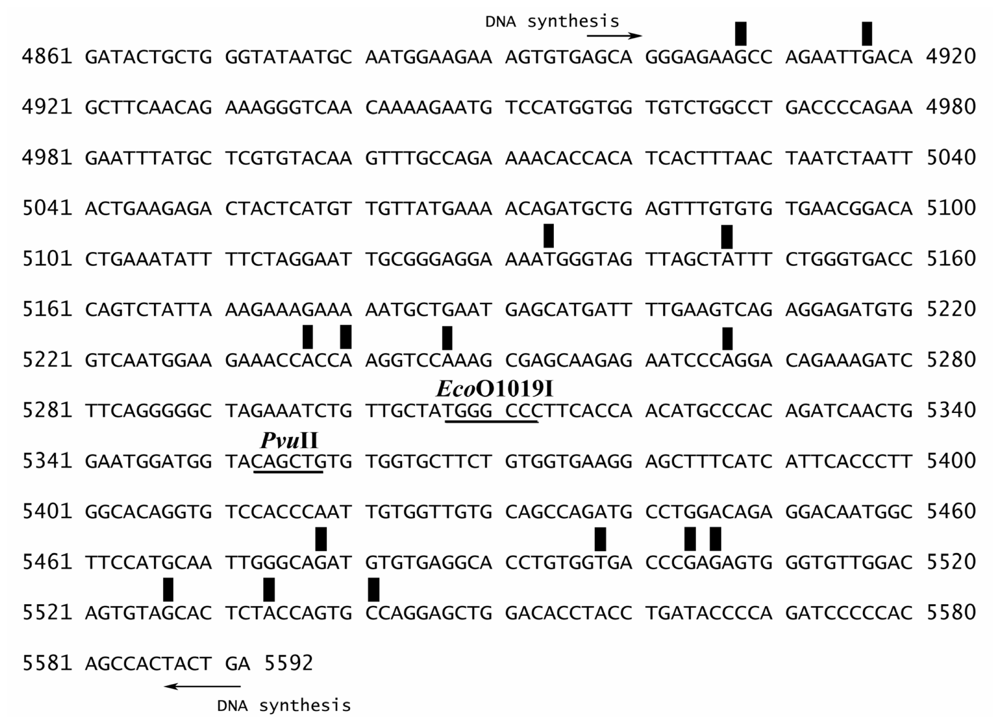
3.2. Preparation of the 696-bp BRCA1 Fragment (Exon 16–24, Nucleotide 4897–5592)
3.3. Sequence Preference for EthaRAPTA Binding to the BRCA1 Fragment
3.4. Absorption Titration
3.5. Thermal Denaturation of RAPTA-BRCA1 Adducts
3.6. Quantification of DNA Lesions Using QPCR
3.7. Cell Culture
3.8. Cell Treatment and Genomic DNA Preparation
3.9. Quantification of DNA Lesions in Cancer Cells
3.10. Cytotoxicity Assays
4. Conclusions
Acknowledgments
References
- Rosenberg, B.; van Camp, L.; Krigas, T. Inhibition of division in Escherichia coli by electrolysis products from a platinum electrode. Nature 1965, 205, 698–699. [Google Scholar]
- Rosenberg, B.; van Camp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumor agents. Nature 1969, 222, 385–386. [Google Scholar]
- Keppler, B.K. Metal Complexes in Cancer Chemotherapy, 1st ed; VCH Publishers: New York, NY, USA, 1993; pp. 1–8. [Google Scholar]
- Orvig, C.; Abrams, M.J. Medicinal inorganic chemistry: Introduction. J. Chem. Rev 1999, 99, 2202–2203. [Google Scholar]
- Eastman, A. Reevaluation of interaction of cis-dichloro(ethylenediamine)-platinum(II) with DNA. Biochemistry 1986, 25, 3912–3915. [Google Scholar]
- Fichtinger-Schepman, A.M.J.; van Oosterom, A.T.; Lohman, P.H.M.; Berends, F. cis-Diamminedichloroplatinum(II)-induced DNA adducts in peripheral leukocytes from seven cancer patients. Cancer Res 1987, 47, 3000–3004. [Google Scholar]
- Wong, E.; Giandomenico, C.M. Current status of platinum-based antitumor drugs. Chem. Rev 1999, 99, 2451–2466. [Google Scholar]
- Holler, E. Mechanism of Action of Tumor-Inhibiting Metal Complexes. In Metal Complexes in Cancer Chemotherapy, 1st ed; Keppler, B.K., Ed.; VCH Publishers: New York, NY, USA, 1993; pp. 39–71. [Google Scholar]
- Wernyj, R.P.; Morin, P.J. Molecular mechanisms of platinum resistance: Still searching for the Achilles’ heel. Drug Resist. Updat 2004, 7, 227–232. [Google Scholar]
- Scolaro, C.; Bergamo, A.; Brescacin, L.; Delfino, R.; Cocchietto, M.; Laurenczy, G.; Geldbach, T.J.; Sava, G.; Dyson, P.J. In vitro and in vivo evaluation of ruthenium(II)-arene pta complexes. J. Med. Chem 2005, 48, 4161–4171. [Google Scholar]
- Allardyce, C.S.; Dyson, P.J. Ruthenium in medicine: Current clinical uses and future prospects. Platinum Metals Rev 2001, 45, 62–69. [Google Scholar]
- Ang, W.H.; Dyson, P.J. Classical and non-classical ruthenium-based anticancer drugs: Towards targeted chemotherapy. Eur. J. Inorg. Chem 2006, 2006, 4003–4018. [Google Scholar]
- Groessl, M.; Tsybin, Y.; Hartinger, C.; Keppler, B.K.; Dyson, P.J. Ruthenium versus platinum: Interactions of anticancer metallodrugs with duplex oligonucleotides characterized by electrospray ionisation mass spectrometry. J. Biol. Inorg. Chem 2010, 15, 677–688. [Google Scholar]
- Dorcier, A.; Hartinger, C.G.; Scopelliti, R.; Fish, R.H.; Keppler, B.K.; Dyson, P.J. Studies on the reactivity of organometallic Ru-, Rh- and Os-pta complexes with DNA model compounds. J. Inorg. Biochem 2008, 102, 1066–1076. [Google Scholar]
- Allardyce, C.S.; Dyson, P.J.; Ellis, D.J.; Heath, S.L. [Ru(eta(6)-p-cymene)Cl2(pta)] (pta=1,3,5-triaza-7-phosphatricyclo[3.3.1.1]decane): A water soluble compound that exhibits pH dependent DNA binding providing selectivity for diseased cells. Chem. Commun 2001, 1396–1397. [Google Scholar]
- Groessl, M.; Hartinger, C.G.; Dyson, P.J.; Keppler, B.K. CZE-ICP-MS as a tool for studying the hydrolysis of ruthenium anticancer drug candidates and their reactivity towards the DNA model compound dGMP. J. Inorg. Biochem 2008, 102, 1060–1065. [Google Scholar]
- Bergamo, A.; Masi, A.; Dyson, P.J.; Sava, G. Modulation of the metastatic progression of breast cancer with an organometallic ruthenium compound. Int. J. Oncol 2008, 33, 1281–1289. [Google Scholar]
- Chatterjee, S.; Kundu, S.; Bhattacharyya, A.; Hartinger, C.G.; Dyson, P.J. The ruthenium(II)-arene compound RAPTA-C induces apoptosis in EAC cells through mitochondrial and p53-JNK pathways. J. Biol. Inorg. Chem 2008, 13, 1149–1155. [Google Scholar]
- Ang, W.H.; de Luca, A.; Chapuis-Bernasconi, C.; Juillerat-Jeanneret, L.; Lo Bello, M.; Dyson, P.J. Organometallic ruthenium inhibitors of glutathione-S-transferase P1-1 as anticancer drugs. Chem Med Chem 2007, 2, 1799–1806. [Google Scholar]
- Huen, M.S.Y.; Sy, S.M.H.; Chen, J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell. Biol 2010, 11, 138–148. [Google Scholar]
- O’Donovan, P.J.; Livingston, D.M. BRCA1 and BRCA2: Breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis 2010, 31, 961–967. [Google Scholar]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar]
- Ratanaphan, A.; Temboot, P.; Dyson, P.J. In vitro ruthenation of human breast cancer suppressor gene1 (BRCA1) by the antimetastasis compound RAPTA-C and its analogue carboRAPTA-C. Chem. Biodivers 2010, 7, 1290–1302. [Google Scholar]
- Ratanaphan, A.; Wasiksiri, S.; Canyuk, B.; Prasertsan, P. Cisplatin-damaged BRCA1 exhibits altered thermostability and transcriptional transactivation. Cancer Biol. Ther 2009, 8, 890–898. [Google Scholar]
- Hartinger, C.G.; Timerbaev, A.R.; Keppler, B.K. Capillary electrophoresis in anti-cancer metallodrug research: Advances and future challenges. Electrophoresis 2003, 24, 2023–2037. [Google Scholar]
- Mei, W.J.; Liu, J.; Chao, H.; Ji, L.N.; Li, A.X.; Liu, J.Z. DNA-binding and cleavage studies of a novel porphyrin ruthenium mixed complex [MPyTPP-Ru(pip)2Cl]+. Transit. Met. Chem 2003, 28, 852–857. [Google Scholar]
- Arjmand, F.; Mohani, B.; Ahmad, S. Synthesis, antibacterial, antifungal activity and interaction of CT-DNA with a new benzimidazole derived Cu(II) complex. Eur. J. Med. Chem 2005, 40, 1103–1110. [Google Scholar]
- Mei, W.J.; Liu, Y.X.; Liu, J.; Li, J.; Zheng, K.C.; Ji, L.N. Synthesis, characterization and DNA-binding properties of mixed porphyrin-polypyridyl ruthenium(II) complexes. Transit. Met. Chem 2005, 30, 82–88. [Google Scholar]
- Lu, X.L.; Zhang, L.; Lou, J.D.; Yan, J.; Nong, P.-S.; Chen, X.-H.; Yang, J.-J.; Gao, M. Synthesis, characterization and DNA binding studies of two cyclopentadienyl ruthenium(II) complexes with amino acid ligands. Transit. Met. Chem 2010, 35, 513–519. [Google Scholar]
- Ang, W.H.; Casini, A.; Sava, G.; Dyson, P.J. Organometallic ruthenium-based antitumor compounds with novel modes of action. J. Organomet. Chem 2011, 696, 989–998. [Google Scholar]
- Messori, L.; Casini, A.; Vullo, D.; Haroutiunian, S.G.; Dalian, E.B.; Orioli, P. Effects of two representative antitumor ruthenium(III) complexes on thermal denaturation profiles of DNA. Chim. Acta 2000, 303, 283–286. [Google Scholar]
- Grover, N.; Welch, T.W.; Fairley, T.A.; Cory, M.; Thorp, H.H. Covalent binding of aquaruthenium complexes to DNA. Inorg. Chem 1994, 33, 3544–3548. [Google Scholar]
- Ratanaphan, A.; Canyuk, B.; Wasiksiri, S.; Mahasawat, P. In vitro platination of human breast cancer suppressor gene 1 (BRCA1) by the anticancer drug carboplatin. Biochim. Biophys. Acta 2005, 1725, 145–151. [Google Scholar]
- Grimaldi, K.A.; Bingham, J.P.; Souhami, R.L.; Hartley, J.A. DNA damage by anticancer agents and its repair: Mapping in cells at the subgene level with quantitative polymerase chain reaction. Anal. Biochem 1994, 222, 236–242. [Google Scholar]
- Jennerwein, M.M.; Eastman, A. A polymerase chain reaction-based method to detect cisplatin adducts in specific genes. Nucleic Acids Res 1991, 19, 6209–6214. [Google Scholar]
- Govan, H.L.; Valles-Ayoub, Y.; Braun, J. Fine-mapping of DNA damage and repair in specific genomic segments. Nucleic Acids Res 1990, 18, 3823–3830. [Google Scholar]
- Honma, M.; Hayashi, M.; Hackman, P.; Sofuni, T. Chlorambucil-induced structural changes in the gpt gene of AS 52 cells. Mutat. Res. Toxicol. Environ. Mutagen 1997, 389, 199–205. [Google Scholar]
- Hickson, I.; Fairbairn, L.J.; Chinnasamy, N.; Lashford, L.S.; Thatcher, N.; Margison, G.P.; Dexter, T.M.; Rafferty, J.A. Chemoprotective gene transfer I: Transduction of human haemopoietic progenitors with O6-benzylguanine-resistant O6 alkylating-DNA alkyltransferase attenuates the toxic effects of O6-alkylating agents in vitro. Gene Ther 1998, 5, 835–841. [Google Scholar]
- Yuh, S.H.; Tibudan, M.; Hentosh, P. Analysis of 2-chloro-2′-deoxy-adenosine incorporation into cellular DNA by quantitative polymerase chain reaction. Anal. Biochem 1998, 262, 1–8. [Google Scholar]
- Ang, W.H. Development of organometallic ruthenium(II) anticancer (RAPTA) drugs. Chimia 2007, 61, 140–142. [Google Scholar]
- Ang, W.H.; Parker, L.J.; de Luca, A.; Juillerat-Jeanneret, L.; Morton, C.J.; Lo Bello, M.; Parker, M.W.; Dyson, P.J. Rational design of an organometallic glutathione transferase inhibitor. Angew. Chem. Int. Ed 2009, 48, 3854–3857. [Google Scholar]
- Mahajan, S.; Atkins, W.M. The chemistry and biology of inhibitors and pro-drugs targeted to glutathione S-transferases. Cell. Mol. Life Sci 2005, 62, 1221–1233. [Google Scholar]
- Van Iersel, M.L.P.S.; Ploemen, J.P.H.T.M.; Struik, I.; van Amersfoort, C.; Keyzer, A.E.; Schefferlie, J.G.; van Bladeren, P.J. Inhibition of glutathione S-transferase activity in human melanoma cells by α,β-unsaturated carbonyl derivatives. Effects of acrolein, cinnamaldehyde, citral, crotonaldehyde, curcumin, ethacrynic acid, and trans-2-hexenal. Chem. Biol. Interact 1996, 102, 117–132. [Google Scholar]
- Lo, H.W.; Ali-Osman, F. Genetic polymorphism and function of glutathione S-transferases in tumor drug resistance. Curr. Opin. Pharmacol 2007, 7, 367–374. [Google Scholar]
- Casini, A.; Hartinger, C.; Nazarov, A.; Dyson, P.J. Organometallic antitumour agents with alternative modes of action. Top. Organomet. Chem 2010, 32, 57–80. [Google Scholar]
- Chatterjee, S.; Biondi, I.; Dyson, P.J.; Bhattacharyya, A. A bifunctional organometallic ruthenium drug with multiple modes of inducing apoptosis. J. Biol. Inorg. Chem 2011, 16, 715–724. [Google Scholar]
- Meggers, E.; Atilla-Gokcumen, G.E.; Grundler, K.; Frias, C.; Prokop, A. Inert ruthenium half-sandwich complexes with anticancer activity. Dalton Trans 2009. [Google Scholar] [CrossRef]
- Adler, V.; Yin, Z.; Fuchs, S.Y.; Benezra, M.; Rosario, L.; Tew, K.D.; Pincus, M.R.; Sardana, M.; Henderson, C.J.; Wolf, C.R.; et al. Regulation of JNK signaling by GSTp. EMBO J 1999, 18, 1321–1334. [Google Scholar]
- Wu, B.; Ong, M.S.; Groessl, M.; Adhireksan, Z.; Hartinger, C.G.; Dyson, P.J.; Davey, C.A. A ruthenium antimetastasis agent forms specific histone protein adducts in the nucleosome core. Chem. Eur. J 2011, 17, 3562–3566. [Google Scholar]
- Messori, L.; Orioli, P.; Vullo, D.; Alessio, E.; Iengo, E. A spectroscopic study of the reaction of NAMI, a novel ruthenium(III) anti-neoplastic complex, with bovine serum albumin. Eur. J. Biochem 2000, 267, 1206–1213. [Google Scholar]
- Bergamo, A.; Messori, L.; Piccioli, F.; Cocchietto, M.; Sava, G. Biological role of adduct formation of the ruthenium(III) complex NAMI-A with serum albumin and serum transferrin. Invest. New Drug 2003, 21, 401–411. [Google Scholar]
- Piccioli, F.; Sabatini, S.; Messori, L.; Orioli, P.; Hartinger, C.G.; Keppler, B.K. A comparative study of adduct formation between the anticancer ruthenium(III) compound HInd trans-[RuCl4(Ind)2] and serum proteins. J. Inorg. Biochem 2004, 98, 1135–1142. [Google Scholar]
- Smith, C.A.; Sutherland-Smith, A.J.; Keppler, B.K.; Kratz, F.; Baker, E.N. Binding of ruthenium(III) anti-tumor drugs to human lactoferrin probed by high resolution X-ray crystallographic structure analyses. J. Biol. Inorg. Chem 1996, 1, 424–431. [Google Scholar]
- Casini, A.; Mastrobuoni, G.; Ang, W.H.; Gabbiani, C.; Pieraccini, G.; Moneti, G.; Dyson, P.J.; Messori, L. ESI-MS characterisation of protein adducts of anticancer ruthenium(II)-arene PTA (RAPTA) complexes. Chem Med Chem 2007, 2, 631–635. [Google Scholar]
- Hartinger, C.G.; Casini, A.; Duhot, C.; Tsybin, Y.O.; Messori, L.; Dyson, P.J. Stability of an organometallic ruthenium-ubiquitin adduct in the presence of glutathione: Relevance to antitumour activity. J. Inorg. Biochem 2008, 102, 2136–2141. [Google Scholar]
- Scolaro, C.; Chaplin, A.B.; Hartinger, C.G.; Bergamo, A.; Cocchietto, M.; Keppler, B.K.; Sava, G.; Dyson, P.J. Tuning the hydrophobicity of ruthenium(II)-arene (RAPTA) drugs to modify uptake, biomolecular interactions and efficacy. Dalton Trans 2007, 43, 5065–5072. [Google Scholar]
- Selvi, P.T.; Stoeckli-Evans, H.; Palaniandavar, M. Synthesis, structure and DNA interaction of cobalt (III) bis-complexes of 1,3-bis(2-pyridylimino)isoindoline and 1,4,7-triazacyclo-nonane. J. Inorg. Biochem 2005, 99, 2110–2118. [Google Scholar]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from nucleated cells. Nucleic Acids Res 1988, 16, 1215. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal complexes | 50% Inhibition (μM) of DNA amplification | 50% Inhibition (μM) of cancer cell growth | ||
|---|---|---|---|---|
| MCF-7 | HT-29 | MCF-7 | HT-29 | |
| Cisplatin | 25 [33] | ND | 36 | 17 |
| ethaRAPTA | 300 | >300 * | 20 | 50 |
| RAPTA-C | 1000 | 500 * | >1600 | 500 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chakree, K.; Ovatlarnporn, C.; Dyson, P.J.; Ratanaphan, A. Altered DNA Binding and Amplification of Human Breast Cancer Suppressor Gene BRCA1 Induced by a Novel Antitumor Compound, [Ru(η6-p-phenylethacrynate)Cl2(pta)]. Int. J. Mol. Sci. 2012, 13, 13183-13202. https://doi.org/10.3390/ijms131013183
Chakree K, Ovatlarnporn C, Dyson PJ, Ratanaphan A. Altered DNA Binding and Amplification of Human Breast Cancer Suppressor Gene BRCA1 Induced by a Novel Antitumor Compound, [Ru(η6-p-phenylethacrynate)Cl2(pta)]. International Journal of Molecular Sciences. 2012; 13(10):13183-13202. https://doi.org/10.3390/ijms131013183
Chicago/Turabian StyleChakree, Korawan, Chitchamai Ovatlarnporn, Paul J. Dyson, and Adisorn Ratanaphan. 2012. "Altered DNA Binding and Amplification of Human Breast Cancer Suppressor Gene BRCA1 Induced by a Novel Antitumor Compound, [Ru(η6-p-phenylethacrynate)Cl2(pta)]" International Journal of Molecular Sciences 13, no. 10: 13183-13202. https://doi.org/10.3390/ijms131013183
APA StyleChakree, K., Ovatlarnporn, C., Dyson, P. J., & Ratanaphan, A. (2012). Altered DNA Binding and Amplification of Human Breast Cancer Suppressor Gene BRCA1 Induced by a Novel Antitumor Compound, [Ru(η6-p-phenylethacrynate)Cl2(pta)]. International Journal of Molecular Sciences, 13(10), 13183-13202. https://doi.org/10.3390/ijms131013183