The Properties of Sintered Calcium Phosphate with [Ca]/[P] = 1.50

Abstract
:1. Introduction
2. Results and Discussion
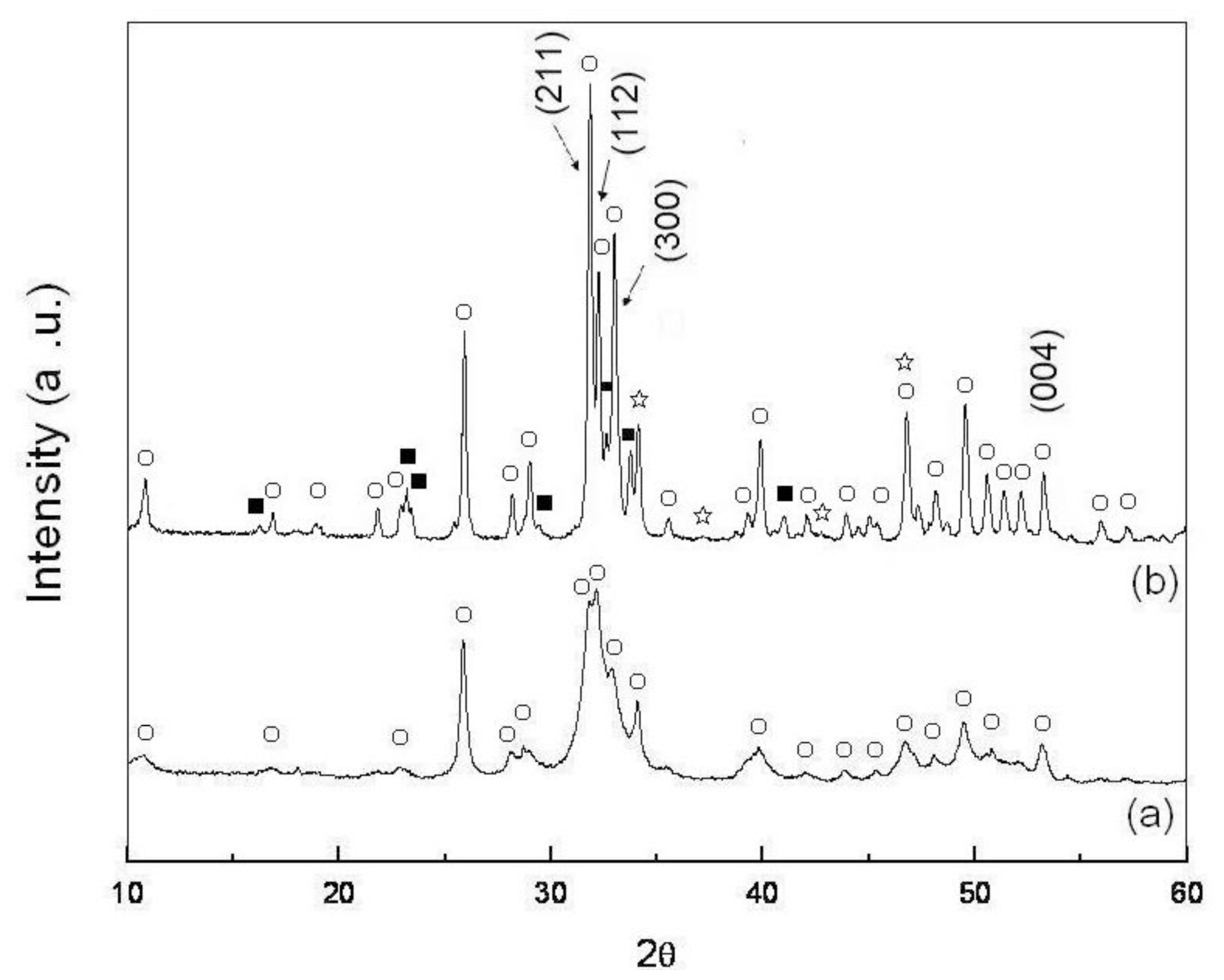
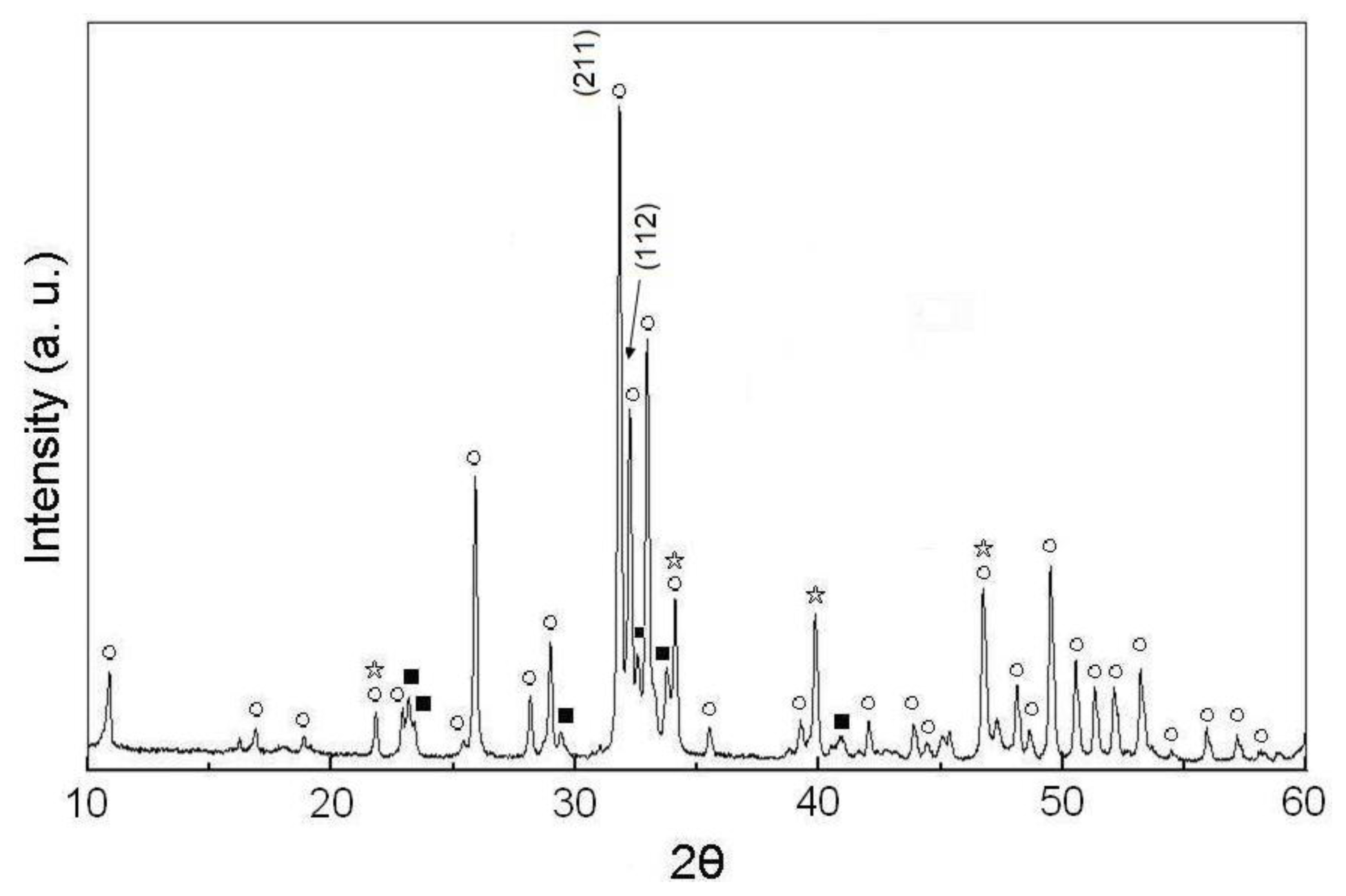
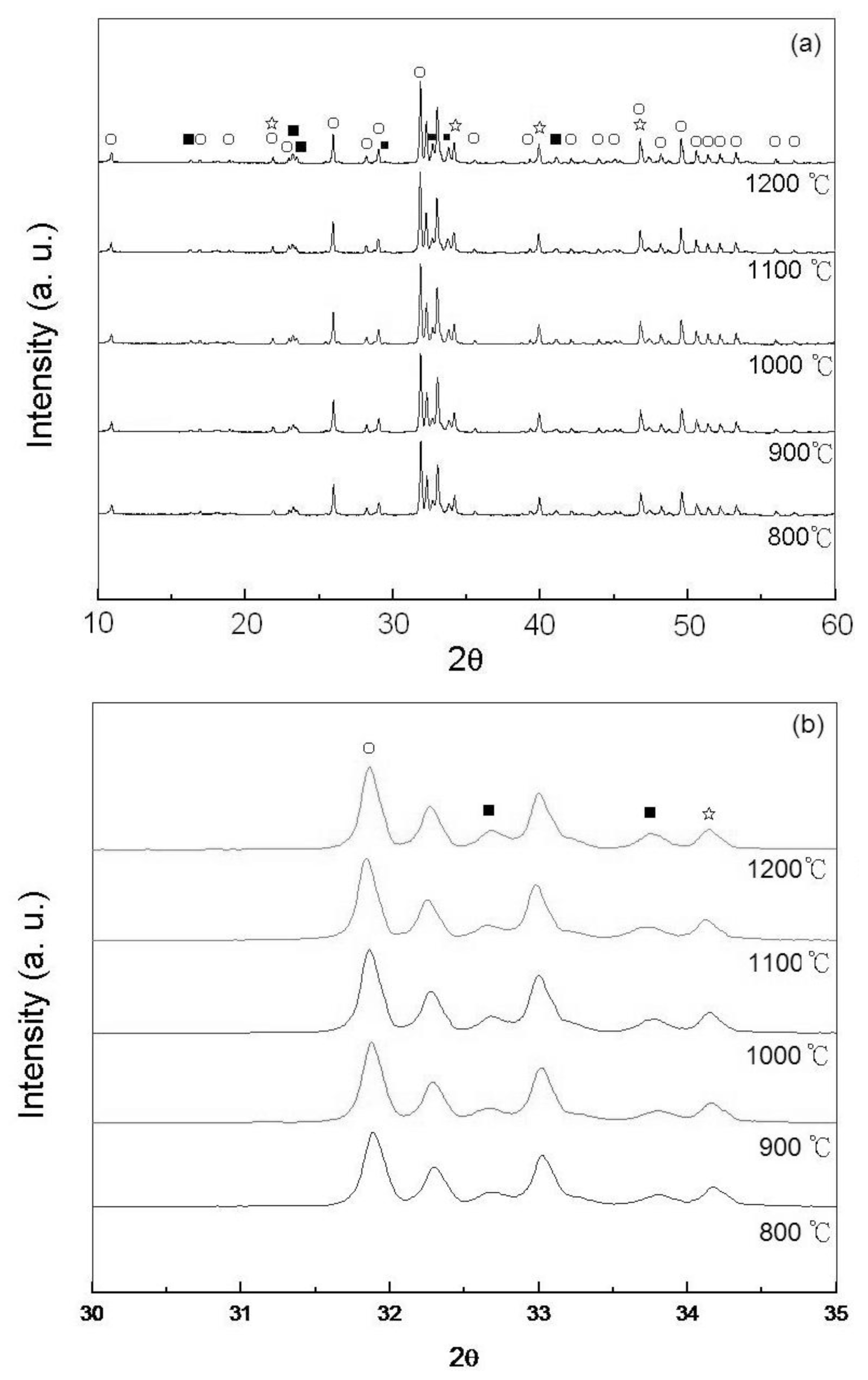
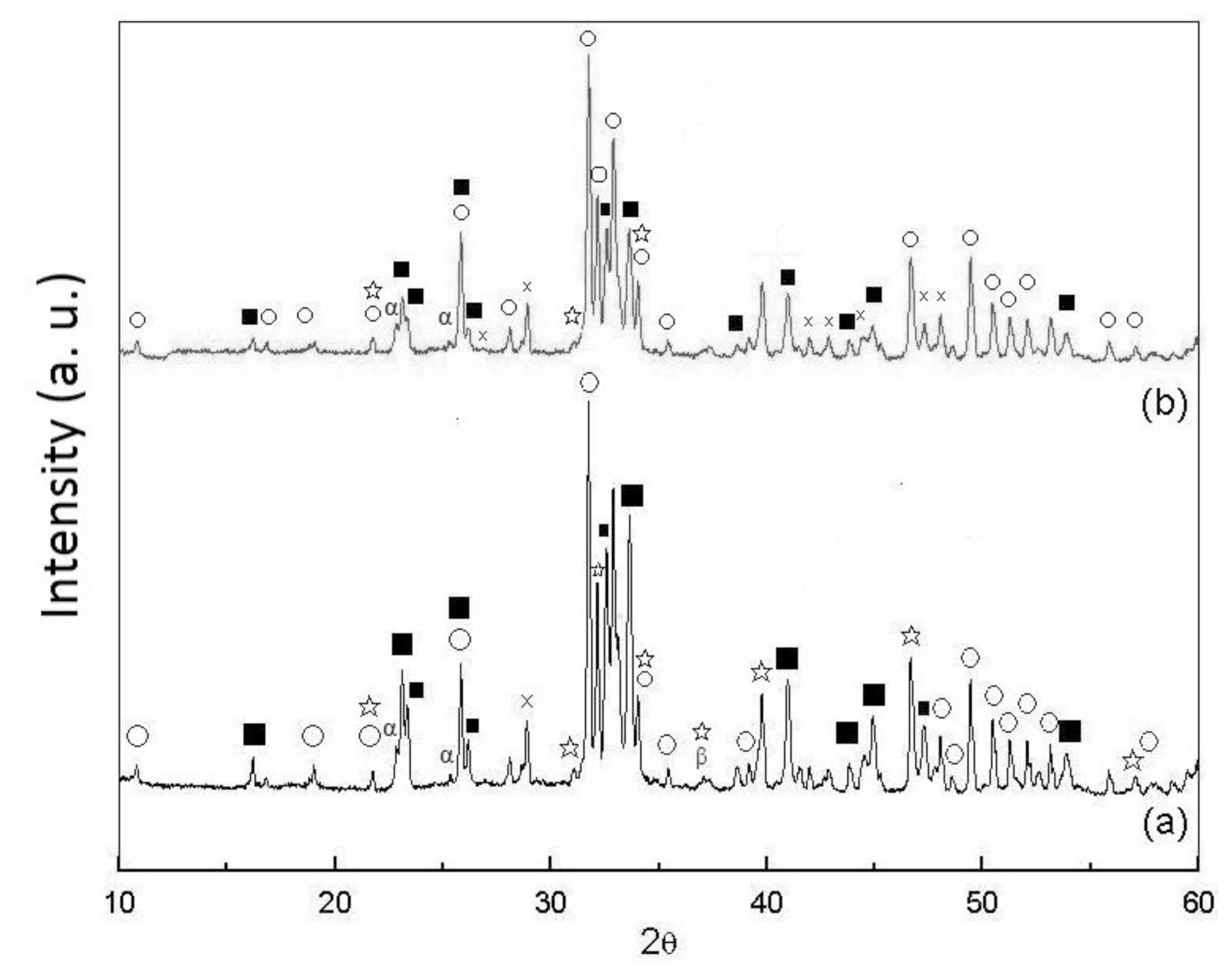
2.1. Phases of As-Dried Calcium Phosphate Powders with [Ca]/[P] = 1.50 after Sintering
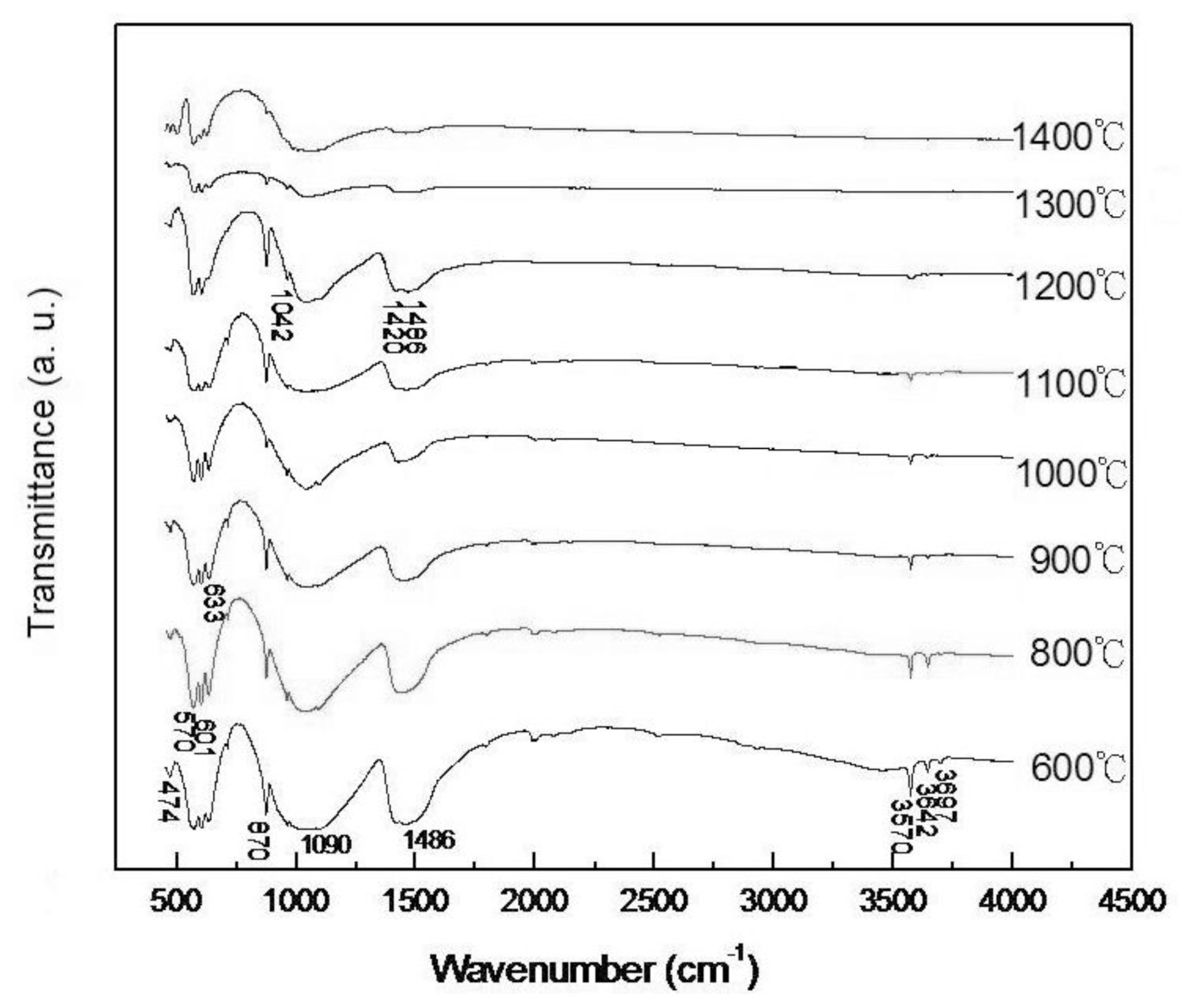
2.2. FT-IR Spectra of Pellet Samples of As-Dried Calcium Phosphate Powders with [Ca]/[P] = 1.50 as Sintered at Various Temperatures for 4h
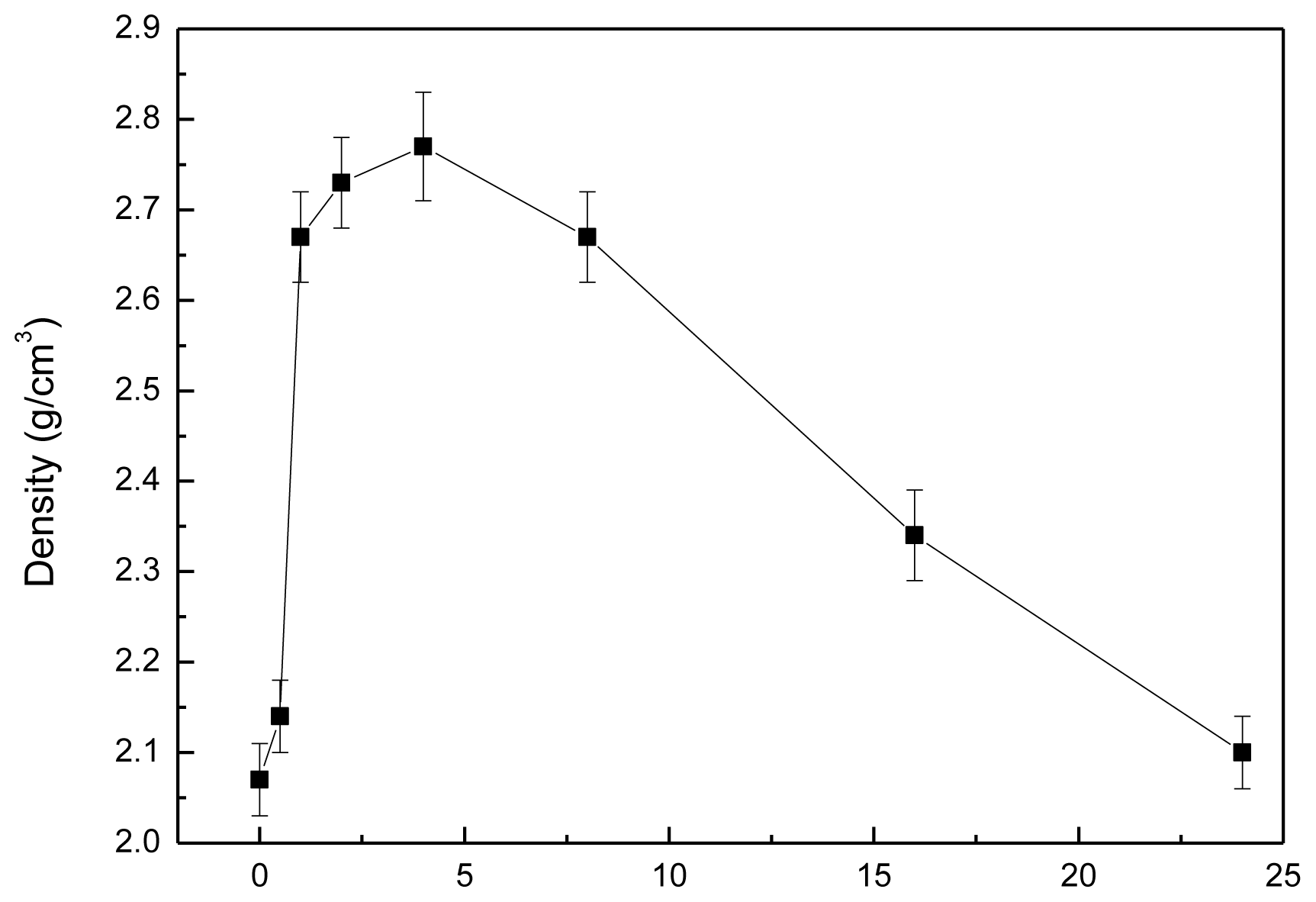
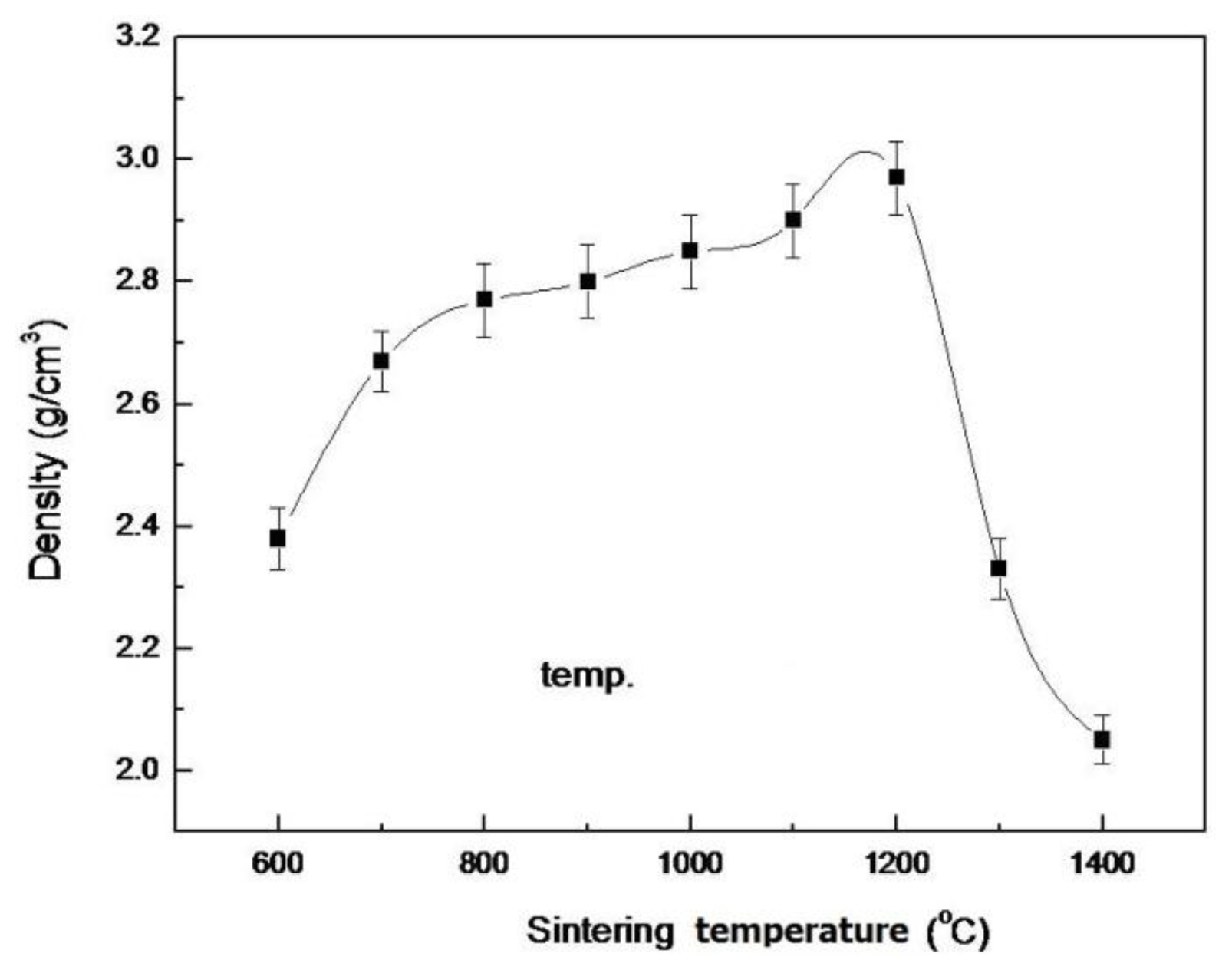
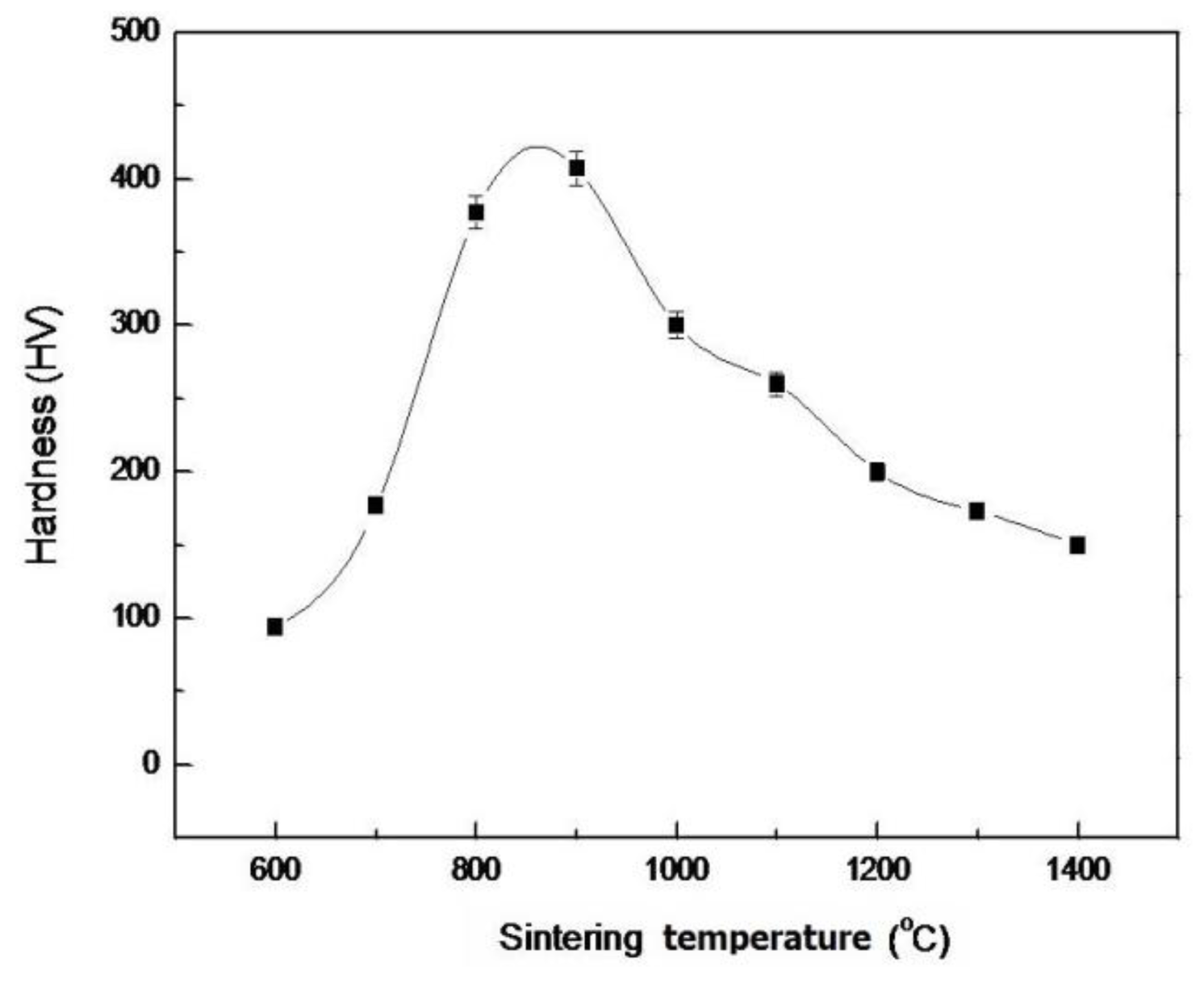
2.3. Density and Hardness of Pellet Samples of As-Dried Calcium Phosphate Powders with [Ca]/[P]=1.50 Sintered at Various Temperatures
2.4. SEM Microstructure of Pellet Samples of As-Dried Calcium Phosphate Powders after Sintering
3. Experimental Section
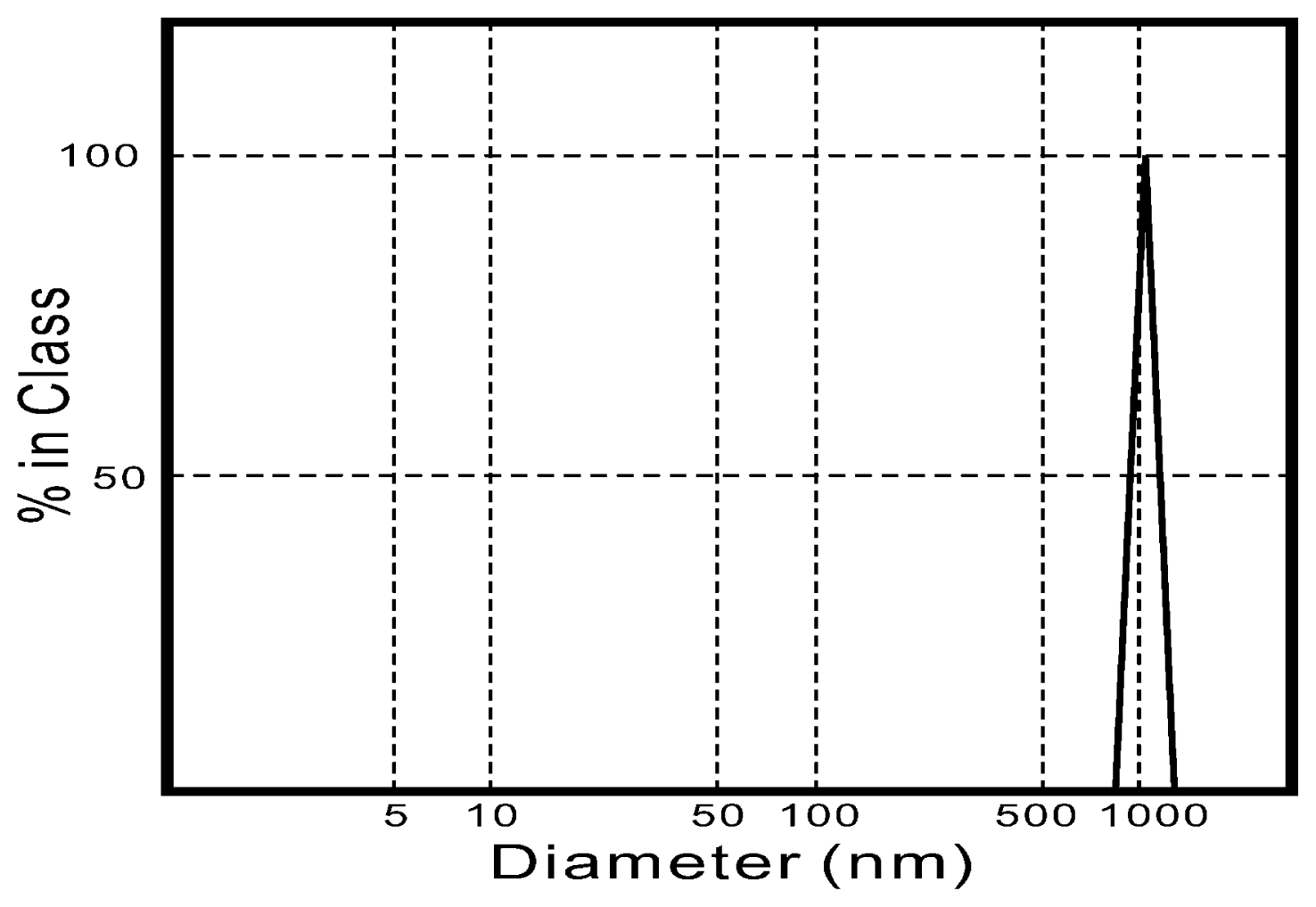
3.1. Sample Preparation
3.2. Sample Characterization
4. Conclusion
Acknowledgments
References
- Currey, J. Biomaterials: Sacrificial bonds heal bone. Nature 2001, 414, 699–708. [Google Scholar]
- Barralet, J.E.; Fleming, G.J.P.; Campion, C.; Harris, J.J. Formation of translucent hydroxyapatite ceramics by sintering in carbon dioxide atmospheres. J. Mater. Sci. Mater. Med 2003, 38, 3979–3993. [Google Scholar]
- Fathi, M.H.; Hanifi, A.; Mortazavi, V. Preparation and bioactivity evaluation of bone-like hydroxyapatite nanopowder. J. Mater. Process. Technol 2008, 202, 536–542. [Google Scholar]
- Kim, H.M. Ceramic bioactivity and related biomimetic strategy. J. Curr. Opin. Solid State Mater. Sci 2003, 7, 289–299. [Google Scholar]
- Vail, T.P.; Urbaniak, J.R. Donor-site morbidity with use of vascularized autogenous fibular grafts. J. Bone Joint Surg. Am 1996, 78, 204–211. [Google Scholar]
- Arrington, E.D.; Smith, W.J.; Chambers, H.G.; Bucknell, A.L.; Davino, N.A. Complications of iliac crest bone graft harvesting. Clin. Orthop. Relat. Res 1996, 329, 300–309. [Google Scholar]
- Bloemers, F.W.; Blokhuis, T.J.; Patka, P.; Bakker, F.C.; Wippermann, B.W.; Haarman, H.J.T.M. Autologous bone versus calcium-phosphate ceramics in treatment of experimental bone defects. J. Biomed. Mater. Res. B 2003, 66B, 526–531. [Google Scholar]
- Bergmann, C.; Lindner, M.; Zhang, W.; Koczur, K.; Kirsten, A.; Telle, R.; Fischer, H. 3D printing of bone substitute implants using calcium phosphate and bioactive glasses. J. Eur. Ceram. Soc 2010, 30, 2563–2567. [Google Scholar]
- Best, S.; Bonfield, W. Processing behavior of hydroxyapatite powders with contrasting morphology. J. Mater. Sci. Mater. Med 1994, 5, 516–521. [Google Scholar]
- Shih, W.J.; Chen, Y.F.; Hon, M.H.; Wang, M.C. Morphology and crystallinity of the nano-sized hydroxyapatite synthesized by hydrolysis using cetyltrimethylammonium bromide (CTAB) as a surfactant. J. Crystal Growth 2005, 275, e2339–e2344. [Google Scholar]
- LeGeros, R.Z. Biodegradation and bioresorption of calcium phosphate ceramics. Clin Mater 1993, 14, 65–68. [Google Scholar]
- Li, Y.; Wijn, J.R.D.; Klein, C.P.A.T.; Meer, S.V.D.; de Groot, K. Proparation and characterization of nanograde osteoapatite-like rod crystals. J. Mater. Sci. Mater. Med 1994, 5, 252–255. [Google Scholar]
- Ramesh, S.; Tan, C.Y.; Bhaduri, S.B.; Teng, W.D.; Sopyan, I. Densification behaviour of nanocrystalline hydroxyapatite bioceramics. J. Mater. Process. Technol 2008, 206, 221–230. [Google Scholar]
- Tang, C.Y.; Uskokovic, P.S.; Tsui, C.P.; Veljovic, Dj.; Petrovic, R.; Janackovic, Dj. Influence of microstructure and phase composition on the nanoindentation characterization of bioceramic materials based on hydroxyapatite. Ceram. Int 2009, 35, 2171–2178. [Google Scholar]
- Liu, D.M.; Yang, Q.; Troczynski, T.; Tseng, W.J. Structural evolution of sol-gel-derived hydroxyapatite. Biomaterials 2002, 23, 1679–1687. [Google Scholar]
- Liu, J.; Ye, X.; Wang, H.; Zhu, M.; Wang, B.; Yan, H. The influence of pH and temperature on the morphology of hydroxyapatite synthesized by hydrothermal method. Ceram. Int 2003, 29, 629–633. [Google Scholar]
- Ma, T.Y.; Xia, Z.G.; Liao, L.B. Effect of reaction systems and surfactant additives on the morphology evolution of hydroxyapatite nanorods obtained via a hydrothermal route. Appl. Surf. Sci 2011, 257, 4384–4388. [Google Scholar]
- Liu, D.M.; Troczynski, T.; Tseng, W.J. Water-based sol–gel synthesis of hydroxyapatite: Process development. Biomaterials 2001, 22, 1721–1730. [Google Scholar]
- Koumoulidis, G.C.; Katsoulidis, A.P.; Ladavos, A.K.; Pomonis, P.J.; Trapalis, C.C.; Sdoukos, A.T.; Vaimukis, T.C. Preparation of hydroxylapatite via microemulsion route. J. Colloid. Interface Sci 2003, 259, 253–260. [Google Scholar]
- Kothapallia, C.; Wei, M.; Vasilieva, A.; Shaw, M.T. Influence of temperature and concentration on the sintering behavior and mechanical properties of hydroxyapatite. Acta Mater 2004, 52, 5655–5663. [Google Scholar]
- Zhou, J.; Zhang, X.; Chen, J.; Zeng, S.; de Groot, K. High temperature characteristics of synthetic hydroxyapatite. J. Mater. Sci. Mater. Med 1993, 4, 83–85. [Google Scholar]
- Wang, P.E.; Chaki, T.K. Sintering behavior and mechanical properties of hydroxyapatite and dicalcium phosphate. J. Mater. Sci. Mater. Med 1993, 4, 150–158. [Google Scholar]
- Liu, H.S.; Chin, T.S.; Lai, L.S.; Chiu, S.Y.; Chung, K.H.; Chang, C.S.; Lui, M.T. Hydroxyapatite synthesized by a simplified hydrothermal method. Ceram. Int 1997, 23, 19–25. [Google Scholar]
- Lin, F.H.; Liao, C.J.; Chen, K.S.; Sun, J.S. Thermal reconstruction behavior of the quenched hydroxyapatite during reheating in air. Mater. Sci. Eng. C 2000, 13, 97–104. [Google Scholar]
- Ishikawa, K.; Ducheyne, P.; Radin, S. Determination of the C/P ration in calcium-deficient hydroxyapatite using X-ray diffraction analysis. J. Mater. Sci. Mater. Med 1993, 4, 165–168. [Google Scholar]
- Shih, W.J.; Wang, J.W.; Wang, M.C.; Hon, M.H. A study on the phase transformation of the nanosized hydroxyapatite synthesized by hydrolysis using in situ high temperature X-ray diffraction. Mater. Sci. Eng. C 2006, 26, 1434–1438. [Google Scholar]
- Trombe, J.C.; Montel, G. Some features of the incorporation of oxygen in different oxidation states in the apatitic lattice: I. J. Inorg. Nucl. Chem 1978, 40, 15–21. [Google Scholar]
- Garve, R.C.; Nicholson, P.S. Phase analysis in zirconia system. J. Am. Ceram. Soc 1972, 55, 303–305. [Google Scholar]
- Ando, J.; Matsuno, S. Ca3(PO4)2-NaCaPO4 system. Bull. Chem. Soc. Jpn 1968, 4, 342–350. [Google Scholar]
- Moseke, C.; Gbureck, U. Tetracalcium phosphate: Synthesis, properties and biomedical applications. Acta Biomater 2010, 6, 3815–3823. [Google Scholar]
- Brown, W.E.; Epstein, E.F. Crystallography of tetracalcium phosphate. J. Res. National Bureau Standards A Phys. Chem 1965, 69A, 547–551. [Google Scholar]
- Fowler, B.O. Infrared studies of apatite: I. Inorg. Chem 1974, 13, 194–297. [Google Scholar]
- Varma, H.K.; Babu, S.S. Synthesis of calcium phosphate bioceramics by citrate gel pyrolysis method. Ceram. Int 2005, 31, 109–114. [Google Scholar]
- Mansc, M.; Manglet, M.; Jimenez, C.; Martinez-duarte, J.M. Hydroxyapatite coating obtained by the thermal activation of polymeric sols. Int. J. Ionrg. Mater 2001, 3, 1153–1155. [Google Scholar]
- Chang, M.C.; Tanaka, J. FTIR study for hydroxyapatite. Collagen nanocomposite cross-linked by glutaralde hyde. Biomaterials 2002, 23, 4811–4818. [Google Scholar]
- Feki, H.E.; Savariault, J.M.; Salah, A.B. ChemInform Abstract: Structure refinements by the rietveld method of partially substituted hydroxyapatite: Ca9Na0.5(PO4)4.5 (CO3)1.5(OH)2. J. Alloys Compd 1999, 287, 114–120. [Google Scholar]
- Palmer, D.A.; Eldik, R.V. The chemistry of metal carbonato and carbon dioxide complexes. Chem. Rev 1983, 83, 651–731. [Google Scholar]
- Kalita, S.J.; Bose, S.; Hosick, H.L.; Bandyopadhyay, A. CaO–P2O5–Na2O-based sintering additives for hydroxyapatite (HAp) ceramics. Biomaterials 2004, 25, 2331–2339. [Google Scholar]
- Olsen, E.; Erlichman, J.; Bunch, T.E.; Moore, P.B. Buchwaldite, a new meteoritic phosphate mineral. Am. Mineral 1977, 62, 362–364. [Google Scholar]
- Tsuyoshi, M.; Masayuki, T. Preparation and thermal properties of dense polycrystalline oxyhydroxyapatite. J. Am. Ceram. Soc 1979, 62, 455–460. [Google Scholar]
- Jarcho, M.; Bolen, C.H.; Thomas, M.B.; Bobick, J.; Kay, J.F.; Doremus, R.H. Hydroxylapatite synthesis and characterization in dense polycrystalline form. J. Mater. Sci 1976, 11, 2027–2035. [Google Scholar]
- Kingery, W.D.; Bowen, H.K.; Uhlmann, D.R. Introduction to Ceramics, 2nd ed; John Wiley Sons, Inc: Singapore, 1976; p. 448. [Google Scholar]
- Wang, C.K.; Ju, C.P.; Chern Lin, J.H. Effect of doped bioactive glass on structure and properties of sintered hydroxyapatite. Mater. Chem. Phys 1998, 53, 138–149. [Google Scholar]
- Gibson, I.R.; Ke, S.; Best, S.M.; Bonfield, W. Effect of powder characteristics on the sinterability of hydroxyapatite powders. J. Mater. Sci. Mater. Med 2001, 12, 163–171. [Google Scholar]
- Yeong, K.C.B.; Wang, J.; Ng, S.C. Mechanochemical synthesis of nanocrystalline hydroxyapatite from CaO and CaHPO4. Biomaterials 2001, 22, 2705–2712. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Concentration (mg/L) | Concentration (mmol/L) |
|---|---|---|
| Na | 57 | 2.5 |
| Ca | 3971 | 99.27 |
| P | 2026 | 67.53 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hung, I.-M.; Shih, W.-J.; Hon, M.-H.; Wang, M.-C. The Properties of Sintered Calcium Phosphate with [Ca]/[P] = 1.50. Int. J. Mol. Sci. 2012, 13, 13569-13586. https://doi.org/10.3390/ijms131013569
Hung I-M, Shih W-J, Hon M-H, Wang M-C. The Properties of Sintered Calcium Phosphate with [Ca]/[P] = 1.50. International Journal of Molecular Sciences. 2012; 13(10):13569-13586. https://doi.org/10.3390/ijms131013569
Chicago/Turabian StyleHung, I-Ming, Wei-Jen Shih, Min-Hsiung Hon, and Moo-Chin Wang. 2012. "The Properties of Sintered Calcium Phosphate with [Ca]/[P] = 1.50" International Journal of Molecular Sciences 13, no. 10: 13569-13586. https://doi.org/10.3390/ijms131013569