A DNA Repair BRCA1 Estrogen Receptor and Targeted Therapy in Breast Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
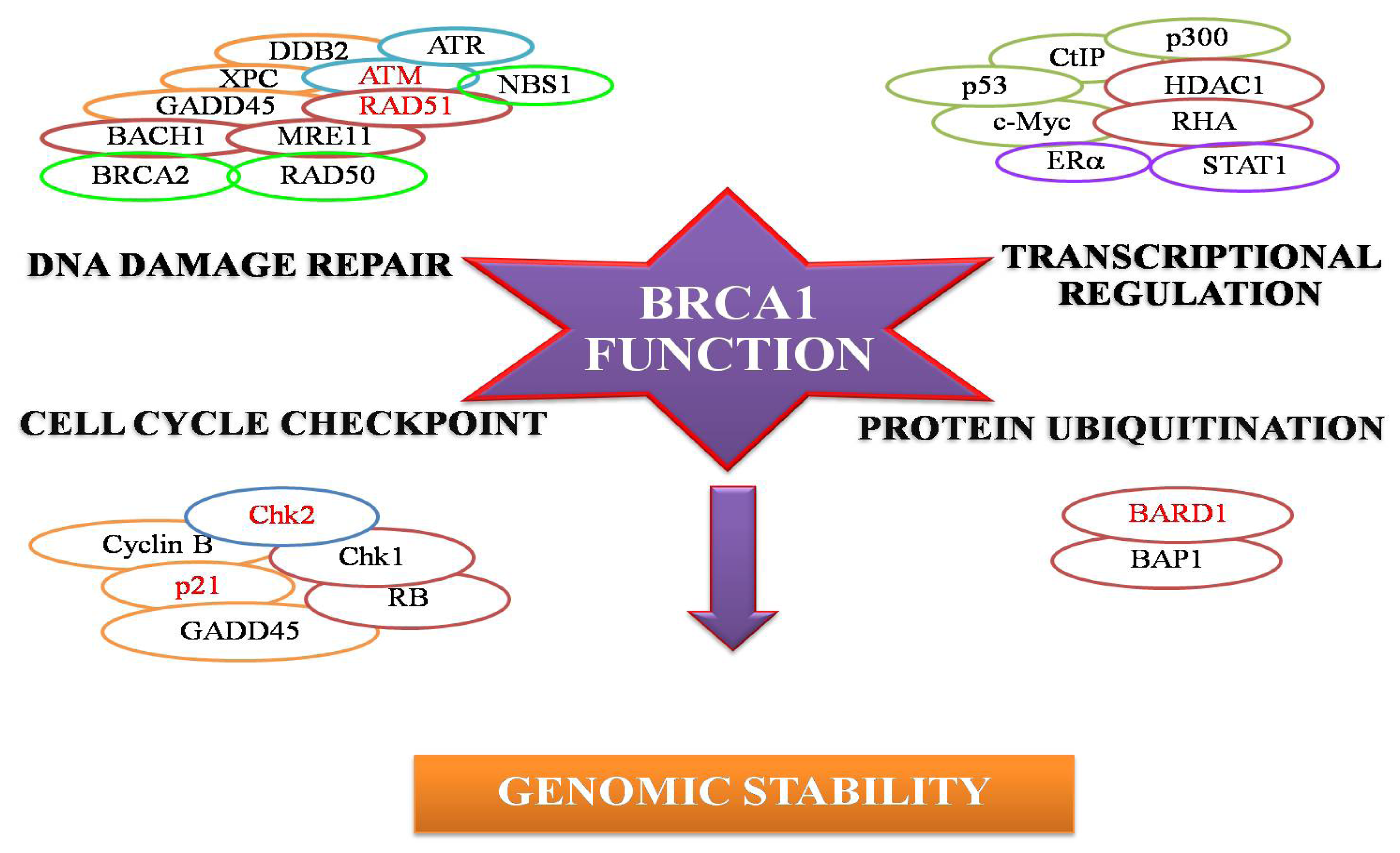
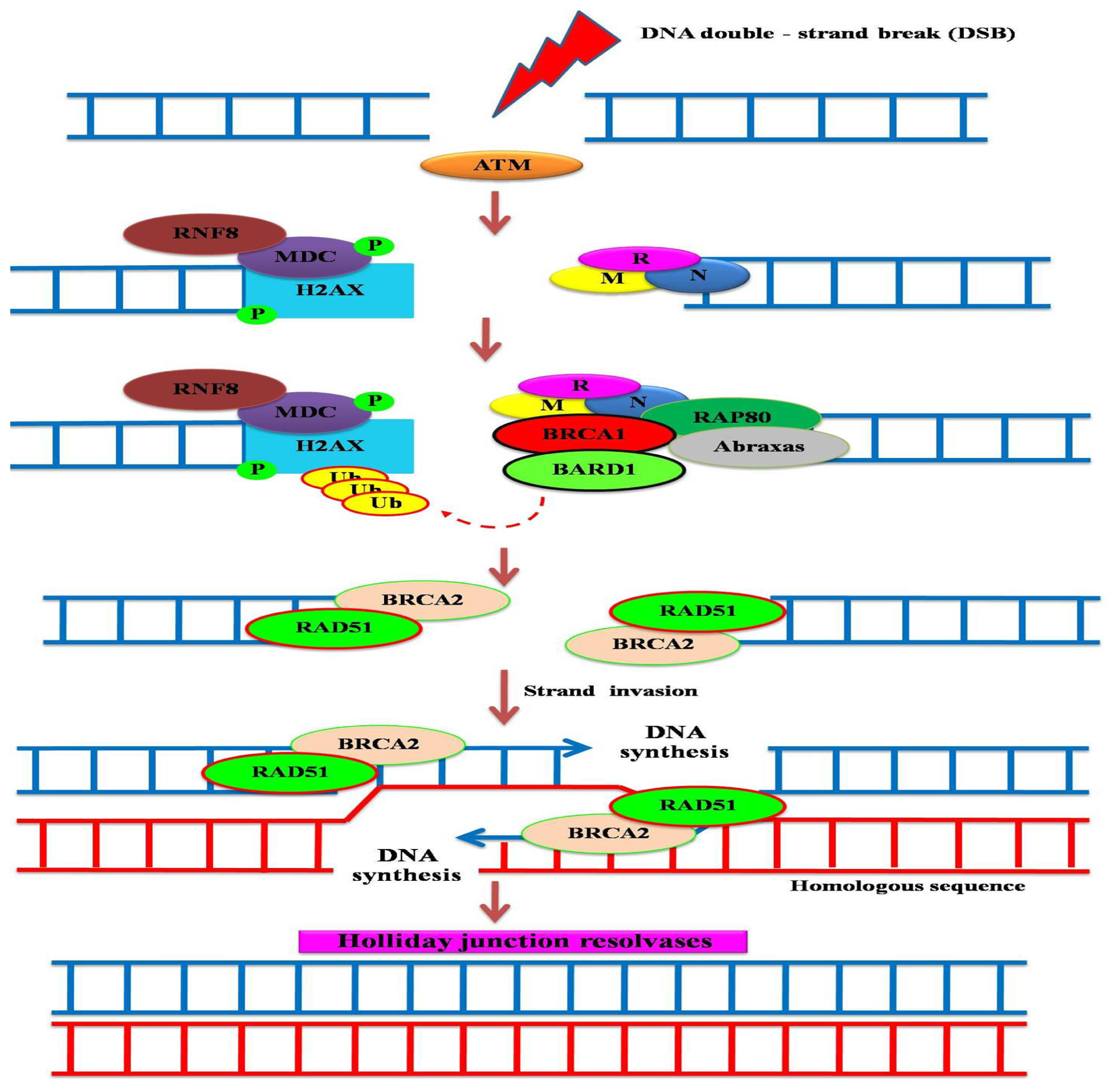
:1. Breast Cancer Suppressor Gene 1 (BRCA1) and its Encoded Protein
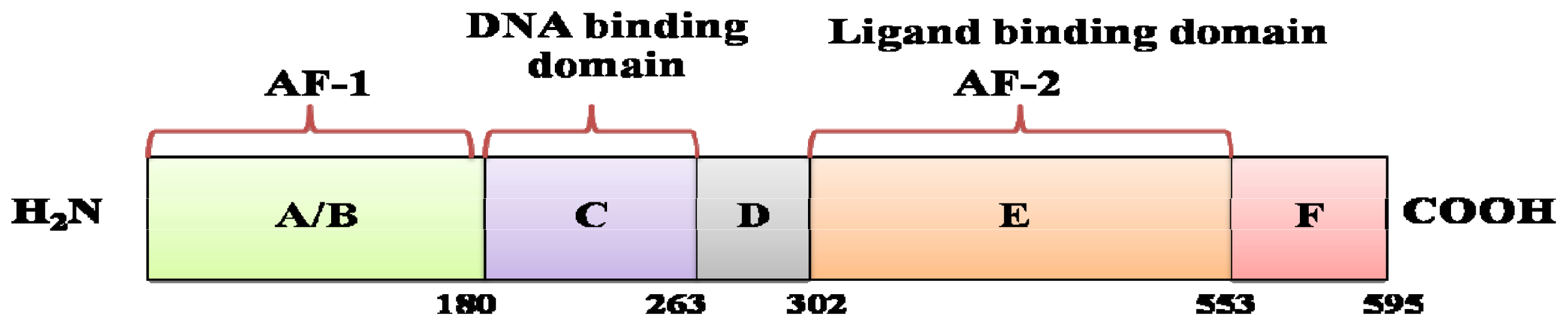
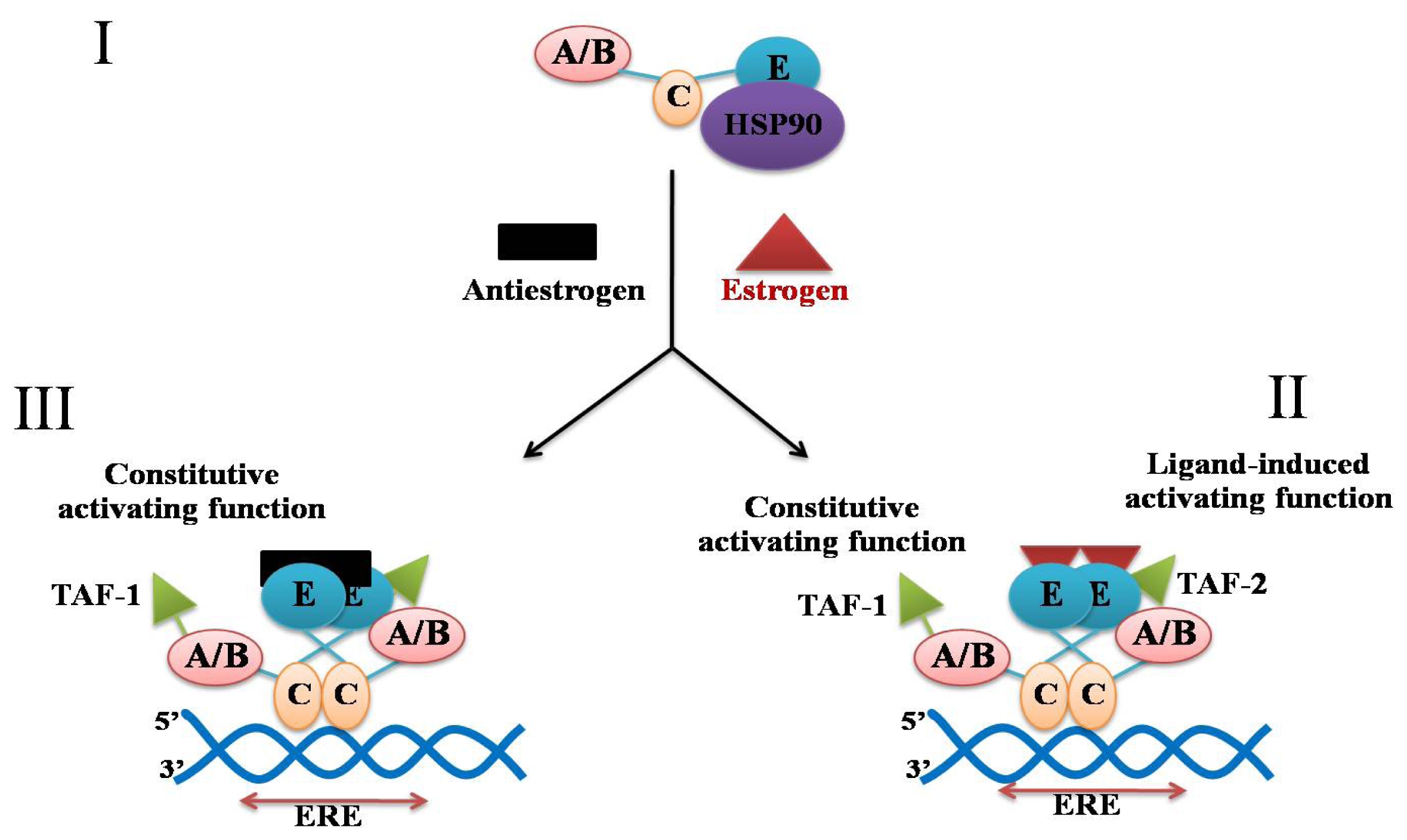
2. Estrogen, Estrogen Receptor and Breast Cancer
3. Association between BRCA1 Expression and Response to Antiestrogen Treatment
4. “Triple-Negative” Breast Cancer and Treatment
5. Conclusions
Acknowledgements
References
- Huen, M.S.; Sy, M.H.S.; Chen, J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol 2010, 11, 138–148. [Google Scholar]
- Rosen, E.M.; Fan, S.; Pestell, R.G.; Goldberg, I.D. BRCA1 gene in breast cancer. J. Cell. Physiol 2003, 196, 19–41. [Google Scholar]
- Hashizume, R.; Fukuda, M.; Maeda, I.; Nishikawa, H.; Oyake, D.; Yabuki, Y.; Ogata, H.; Ohta, T. The RING heterodomer BRCA1-BARD1 is ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem 2001, 276, 14537–14540. [Google Scholar]
- Jin, Y.; Xu, X.L.; Yang, M.C.; Wei, F.; Ayi, T.C.; Bowcock, A.M.; Baer, R. Cell cycle-dependent colocalization of BARD1 and BRCA1 proteins in discrete nuclear domains. Proc. Natl. Acad. Sci. USA 1997, 94, 12075–12080. [Google Scholar]
- Xia, Y.; Pao, G.M.; Chen, H.W.; Verma, I.M.; Hunter, T. Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J. Biol. Chem 2003, 278, 5255–5263. [Google Scholar]
- Mark, W.Y.; Liao, J.C.; Lu, Y.; Ayed, A.; Laister, R.; Szymczyna, B.; Chakrabartty, A.; Arrowsmith, C.H. Characterization of segments from the central region of BRCA1: An intrinsically disordered scaffold for multiple protein-protein and protein-DNA interactions? J. Mol. Biol 2005, 345, 275–285. [Google Scholar]
- Rosen, E.M.; Fan, S.; Pestell, R.G.; Goldberg, I.D. BRCA1 in hormone-responsive cancer. Trends Endocrinol. Metab 2003, 14, 378–385. [Google Scholar]
- Watts, F.Z.; Brissett, N.C. Linking up and interacting with BRCT domains. DNA Repair (Amst.) 2010, 9, 103–108. [Google Scholar]
- Liu, W.; Zong, W.; Wu, G.; Fujita, T.; Li, W.; Wu, J.; Wan, Y. Turnover of BRCA1 involves in radiation-induced apoptosis. PLoS One 2010, 12, e14484. [Google Scholar]
- Starita, L.M.; Parvin, J. The multiple nuclear functions of BRCA1: Transcription ubiquitination and DNA repair. Curr. Opin. Cell Biol 2003, 13, 345–350. [Google Scholar]
- Scully, R.; Chen, J.; Plug, A.; Xiao, Y.; Weaver, D.; Feunteun, J.; Ashley, T.; Livingston, D.M. Association of BRCA1 with RAD51 in mitotic and meiotic cell. Cell 1997, 88, 265–275. [Google Scholar]
- Ashworth, A. A synthetic lethal therapeutic approach poly (ADP) ribose polymeraseinhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol 2008, 26, 3785–3790. [Google Scholar]
- Ohta, T.; Wu, W.; Koike, A.; Asakawa, H.; Koizumi, H.; Fukuda, M. Contemplating chemosensitivity of basal-like breast cancer based on BRCA1 dysfunction. Breast Cancer 2009, 16, 268–274. [Google Scholar]
- Bassing, C.H.; Suh, H.; Ferguson, D.O.; Chua, K.F.; Manis, J.; Eckersdorff, M.; Gleason, M.; Bronson, R.; Lee, C.; Alt, F.W. Histone H2AX: A dosage-dependent suppressor of oncogenic translocations and tumors. Cell 2003, 114, 359–370. [Google Scholar]
- Celeste, A.; Fernandez-Capetillo, O.; Kruhlak, M.J.; Pilch, D.R.; Staudt, D.W.; Lee, A.; Bonner, R.F.; Bonner, W.M.; Nussenzweig, A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol 2003, 5, 675–679. [Google Scholar]
- Lou, Z.; Minter-Dykhoise, K.; Franco, S.; Gostissa, M.; Rivera, M.A.; Celeste, A.; Manis, J.P.; van Deursen, J.; Nussenzweig, A.; Paull, T.T.; et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell 2006, 21, 187–200. [Google Scholar]
- Stucki, M.; Clapperton, J.A.; Moharmmad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular response to DNA double-strand breaks. Cell 2005, 123, 1213–1226. [Google Scholar]
- Stucki, M.; Jackson, S.P. gammaH2AX and MDC1: Anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst.) 2006, 5, 534–543. [Google Scholar]
- Huen, M.S.; Grant, R.; Manke, I.; Minn, K.; Yu, X.; Yaffe, M.B.; Chen, J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007, 131, 901–914. [Google Scholar]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007, 318, 1637–1640. [Google Scholar]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 ubiquitylate histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar]
- Plans, V.; Scheper, J.; Soler, M.; Loukili, N.; Okano, Y.; Thomson, T.M. The RING finger protein RNF8 recruits UBC13 for lysine 63-based self polyubiquitylation. J. Cell. Biochem 2006, 97, 572–582. [Google Scholar]
- Kim, H.; Chen, J.; Yu, X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 2007, 316, 1202–1205. [Google Scholar]
- Sobhian, B.; Shao, G.; Lilli, D.R.; Culhance, A.C.; Moreau, L.A.; Xia, B.; Livingston, D.M.; Greenberg, R.A. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 2007, 316, 1198–1202. [Google Scholar]
- Wang, B.; Matsuoka, S.; Ballif, B.A.; Zhang, D.; Smogirzewska, A.; Gygi, S.P.; Elledge, S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007, 316, 1194–1198. [Google Scholar]
- Greenberg, R.A.; Sobhian, B.; Pathania, S.; Cantor, S.B.; Nakatani, Y.; Livingston, D.M. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev 2006, 20, 34–46. [Google Scholar]
- Ransburgh, D.J.; Chiba, N.; Ishioka, C.; Toland, A.E.; Parvin, J.D. Identification of breast tumor mutations in BRCA1 that abolish its function in homologous DNA recombination. Cancer Res 2010, 70, 988–995. [Google Scholar]
- Yu, D.S.; Sonoda, E.; Takeda, S.; Huang, C.L.; Pellegrini, L.; Blundell, T.L.; Venkitaraman, A.R. Dynamic control of RAd51 recombinase by self-association and interaction with BRCA2. Mol. Cell 2003, 12, 1029–1041. [Google Scholar]
- Morris, J.R.; Boutell, C.; Keppler, M.; Densham, R.; Weekes, D.; Alamshah, A.; Butler, L.; Pangon, L.; Kiuchi, T.; Ng, T.; et al. The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature 2009, 462, 886–890. [Google Scholar]
- Morris, J.R.; Pangon, L.; Boutell, C.; Katagiri, T.; Keep, N.H.; Solomon, E. Genetic analysis of BRCA1 ubiquitin ligase activity and its relationship to breast cancer susceptibility. Hum. Mol. Genet 2006, 15, 599–606. [Google Scholar]
- Snouwaert, J.N.; Gowen, L.C.; Latour, A.M.; Mohn, A.R.; Xiao, A.; DiBiase, L.; Koller, B.H. BRCA1 deficient embryonic stem cell displays a decreased homologous recombination frequency and an increased frequency of non-homologous recombination that is corrected by expression of Brac1 transgene. Oncogene 1999, 18, 7900–7907. [Google Scholar]
- Husain, A.; He, G.; Venkatraman, E.S.; Spriggs, D.R. BRCA1 up-regulation is associated with repair-mediated resistance to cis-diaminedichloroplatinum (II). Cancer Res 1998, 58, 1120–1123. [Google Scholar]
- Lafarge, S.; Sylvain, V.; Ferrara, M.; Bignon, Y.J. Inhibition of BRCA1 leads to increased chemoresistance to microtubule-interfering agents, and effects that involves the JNK pathway. Oncogene 2001, 20, 6597–6606. [Google Scholar]
- Quinn, J.E.; Kennedy, R.E.; Mullan, P.B.; Gilmore, P.M.; Carty, M.; Johnston, P.G.; Harkin, D.P. BRCA1 functions as a differential modulator of chemotherapy-induced apoptosis. Cancer Res 2003, 63, 6221–6228. [Google Scholar]
- James, C.R.; Quinn, J.E.; Mullan, P.B.; Johnston, P.G.; Harkin, D.P. BRCA1, a potential predictive biomarker in the treatment of breast cancer. Oncologist 2007, 12, 142–150. [Google Scholar]
- Quinn, J.E.; Carser, J.E.; James, C.R.; Kennedy, R.D.; Harkin, D.P. BRCA1 and implications for response to chemotherapy in ovarian cancer. Gynecol. Oncol 2009, 113, 134–142. [Google Scholar]
- Turner, N.C.; Reis-Filho, J.S.; Russell, A.M.; Springall, R.J.; Ryder, K.; Steele, D.; Savage, K.; Gillett, C.E.; Schmitt, F.C.; Ashworth, A.; et al. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene 2007, 26, 2126–2132. [Google Scholar]
- Moskwa, P.; Buffa, F.M.; Pan, Y.; Panchakshari, R.; Gottipati, P.; Muschel, R.J.; Beech, J.; Kulshrestha, R.; Abdelmohsen, K.; Weinstock, D.M.; et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol. Cell 2011, 41, 210–220. [Google Scholar]
- Tassone, P.; Tagliaferri, P.; Pericelli, P.; Blotta, S.; Quaresima, B.; Martelli, M.L.; Goel, A.; Barbieri, V.; Costanzo, F.; Boland, C.R.; et al. BRCA1 expression modulates chemosensitivity of BRCA1-defective HCC1937 human breast cancer cells. Br. J. Cancer 2003, 88, 1285–1291. [Google Scholar]
- Bhattacharyya, A.; Ear, U.S.; Koller, B.H.; Weicheselbaum, R.R.; Bishop, D.K. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J. Biol. Chem 2000, 275, 23899–23903. [Google Scholar]
- Moynahan, M.E.; Cui, T.Y.; Jasin, M. Homology-induced DNA repair, mitomycin C resistance, and chromosome stability is restored with correction of a BRCA1 mutation. Cancer Res 2001, 61, 4842–4850. [Google Scholar]
- Fedier, A.; Steiner, R.A.; Schwarz, V.A.; Lenherr, L.; Haller, U.; Fink, D. The effect of loss of BRCA1 on the sensitivity to anticancer agents in p53 deficient cells. Int. J. Oncol 2003, 22, 1169–1173. [Google Scholar]
- Chapman, M.S.; Verma, I.M. Transcription activation by BRCA1. Nature 1996, 382, 678–679. [Google Scholar]
- Monteiro, A.N.; August, A.; Hanafusa, H. Evidence for a transcriptional activation function of BRCA1 C-terminal region. Proc. Natl. Acad. Sci. USA 1996, 93, 13595–13599. [Google Scholar]
- Ratanaphan, A. A DNA Repair Protein BRCA1 as a Potentially Molecular Target for the Anticancer Platinum Drug Cisplatin, DNA Repair; Kruman, I., Ed.; InTech Open Access Publisher: Rijeka, Croatia, 2011. Available online: http://www.intechopen.com/books/dna-repair/a-dna-repair-protein-brca1-as-a-potentially-molecular-target-for-the-anticancer-platinum-drug-cispla accessed on 12 November 2012.
- Ratanaphan, A.; Wasiksiri, S.; Canyuk, B.; Prasertsan, P. Cisplatin-damaged BRCA1 exhibits altered thermostability and transcriptional transactivation. Cancer Biol. Ther 2009, 8, 890–898. [Google Scholar]
- Mullan, P.B.; Quinn, J.E.; Harkin, D.P. The role of BRCA1 in transcriptional regulation and cell cycle control. Oncogene 2006, 25, 5854–5863. [Google Scholar]
- Somasundaram, K.; Zhang, H.; Zeng, Y.X.; Houvras, Y.; Peng, Y.; Zhang, H.; Wu, G.S.; Licht, J.D.; Weber, B.L.; El-Deiry, W.S. Arrest of the cell cycle by the tumor-suppressor BRCA1 requires the CDK-inhibitor p21WAF1/CiP1. Nature 1997, 389, 187–190. [Google Scholar]
- Zhang, H.; Somasundaram, K.; Peng, Y.; Tian, H.; Bi, D.; Weber, B.L.; El-Deiry, W.S. BRCA1 physically associates with p53 and stimulates its transcriptional activity. Oncogene 1998, 16, 1713–1721. [Google Scholar]
- Dubrovska, A.; Kanamoto, T.; Lomnyyska, M.; Heldin, C.H.; Volodko, N.; Souchelnytskyi, S. TGF beta1/Smad3 counteracts BRCA1-dependent repair of DNA damage. Oncogene 2005, 14, 2289–2297. [Google Scholar]
- Fan, S.; Wang, J.; Yuan, R.; Ma, Y.; Meng, Q.; Erdos, M.R.; Pestell, R.G.; Yuan, F.; Auborn, K.S.; Goldberg, I.D.; et al. BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science 1999, 284, 1354–1356. [Google Scholar]
- Atipairin, A.; Canyuk, B.; Ratanaphan, A. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by the platinum-based anticancer drugs. Breast Cancer Res. Treat 2011, 126, 203–209. [Google Scholar]
- Eakin, C.M.; Maccoss, M.J.; Finney, G.L.; Klevit, R.E. Estrogen receptor α is putative substrate for the BRCA1 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2007, 104, 5794–5799. [Google Scholar]
- Osborne, C.K.; Zhao, H.; Fuqua, S.A.W. Selective estrogen receptor modulators: Structure, function, and clinical use. J. Clin. Oncol 2000, 18, 3172–3186. [Google Scholar]
- Schwabe, J.W.; Chapman, L.; Finch, J.T.; Rhodes, D.; Neuhaus, D. DNA recognition by the oestrogen receptor: from solution to the crystal. Structure 1993, 1, 187–204. [Google Scholar]
- Chen, X.; Danes, C.; Lowe, M.; Herliczek, T.W.; Keyomarsi, K. Activation of the estrogen-signalling pathway by p21WAF1/CIP1 in estrogen receptor-negative breast cancer cells. J. Natl. Cancer Inst 2000, 92, 1403–1431. [Google Scholar]
- Osborne, C.K. Steroid hormone receptors in breast cancer management. Breast Cancer Res. Treat 1998, 51, 227–238. [Google Scholar]
- Hosey, A.M.; Gorski, J.J.; Murray, M.M.; Quinn, J.E.; Chung, W.Y.; Gail, E.C.; Colin, R.S.; Susan, M.J.; Jude, M.F.; Alistair, N.M.; et al. Molecular basis for estrogen receptor α deficiency in BRCA1-linked breast cancer. J. Natl. Cancer Inst 2007, 99, 1683–1694. [Google Scholar]
- Macgregor, J.; Jordan, C. Basic guide to the mechanisms of antiestrogen action. Pharm. Rev 1998, 50, 152–187. [Google Scholar]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acid Res 2001, 29, 2905–2919. [Google Scholar]
- Schwabe, J.W.; Chapman, L.; Finch, J.T.; Rhodes, D. The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell 1993b, 75, 567–578. [Google Scholar]
- Schwabe, J.W.; Chapman, L.; Rhodes, D. The oestrogen receptor recognizes an imperfectly palindromic response element through an alternative side-chain conformation. Structure 1995, 3, 201–213. [Google Scholar]
- Schwabe, J.W.; Neuhaus, D.; Rhodes, D. Solution structure of the DNA-binding domain of the oestrogen receptor. Nature 1990, 348, 458–461. [Google Scholar]
- Ruff, M.; Gangloff, M.; Wurtz, J.M.; Moras, D. Estrogen receptor transcription and transactivation structure function relationship in DNA and ligand binding domains of estrogen receptor. Breast Cancer Res 2000, 2, 353–359. [Google Scholar]
- Cano, A.; Hermenegildo, C. Modulation of the oestrogen receptor: A process with distinct susceptible steps. Hum. Reprod. Update 2000, 6, 207–211. [Google Scholar]
- Cherlet, T.; Murphy, L.C. Estrogen receptors inhibit Smad3 transcriptional activity throughAp-1 transcription factors. Mol. Cell Biochem 2007, 306, 33–42. [Google Scholar]
- Matsuda, T.; Yamamoto, T.; Muraguchi, A.; Saatcioglu, F. Cross-talk between transforming growth factor-beta and estrogen receptor signaling through Smad3. J. Biol. Chem 2001, 276, 42908–42914. [Google Scholar]
- Herman, M.E.; Katzenellenbogen, B.S. Response-specific antiestrogen resistance in a newly characterized MCF-7 human breast cancer cell line resulting from long-term exposure to trans-hydroxytamoxifen. J. Steroid Biochem. Mol. Biol 1996, 59, 121–134. [Google Scholar]
- Berry, D.A.; Cirrincione, C.; Citron, M.L.; Budman, D.R.; Goldstein, L.J.; Martino, S.; Perez, E.A.; Muss, H.B.; Norton, L.; Hudis, C.; et al. Estrogen-receptor status and outcomes of modern chemotherapy for patients with node-positive breast cancer. JAMA 2006, 295, 1658–1667. [Google Scholar]
- Gross, P.E.; Ingle, J.N.; Martino, S.; Robert, N.J.; Muss, H.B.; Piccart, M.J.; Castigliome, M.; Tu, D.; Shepherd, L.E.; Pritchard, K.I.; et al. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N. Engl. J. Med. 2003, 3491, 1793–1802. [Google Scholar]
- McDonnell, D.P.; Clemm, D.L.; Hermann, T.; Goldman, M.E.; Pike, J.W. Analysis of estrogen receptor function in vitro reveals three distinct classes of antiestrogens. Mol. Endocrinol 1995, 9, 659–669. [Google Scholar]
- Tzukerman, M.T.; Esty, A.; Santiso, M.D.; Danielian, P.; Parker, M.G.; Stein, R.B.; Pike, J.W.; McDonnell, D.P. Human estrogen receptor transactivational capacity is determined by both cellular and promoter context and mediated by two functionally distinct intramolecular regions. Mol. Endocrinol 1994, 8, 21–30. [Google Scholar]
- Foulkes, W.D.; Metcalfe, K.; Sun, P.; Hanna, W.M.; Lynch, H.T.; Ghadirian, P.; Tung, N.; Olopade, O.I.; Weber, B.L.; McLennan, J.; et al. Estrogen receptor status in BRCA1- and BRCA2-related breast cancer: the influence of age, grade, and histological type. Clin. Cancer Res 2004, 10, 2029–2034. [Google Scholar]
- Rosen, E.M.; Fan, S.; Isaacs, C. BRCA1 in hormonal carcinogenesis: basis and clinical research. Endocr. Relat. Cancer 2005, 12, 533–548. [Google Scholar]
- Hu, Y.; Ghosh, S.; Amleh, A.; Yue, W.; Lu, Y.; Katz, A.; Li, R. Modulation of aromatase expression by BRCA1: A possible link to tissue-specific tumor suppression. Oncogene 2005, 24, 8343–8348. [Google Scholar]
- Howell, A.; Osborne, C.K.; Morris, C.; Wakeling, A.E. ICI 182,780 (Faslodex): Development of a novel, “pure” antiestrogen. Cancer 2000, 89, 817–825. [Google Scholar]
- Vendrell, J.A.; Magnino, F.; Danis, E.; Duchesne, M.J.; Pinloche, S.; Pons, M.; Birnbaum, D.; Nguyen, C.; Theillet, C.; Cohen, P.A. Estrogen regulation in human breast cancer cells of new downstream gene targets involved in estrogen metabolism, cell proliferation and cell transformation. J. Mol. Endocrinol 2004, 32, 397–414. [Google Scholar]
- Wakeling, A.E. Similarities and distinctions in the mode of action of different classes of antiestrogens. Endocr. Relat. Cancer 2000, 7, 17–28. [Google Scholar]
- Walkeling, A.E.; Bowler, J. Steroidal pure antioestrogens. J. Endocrinol 1987, 112, R7–R10. [Google Scholar]
- Wijayaratne, A.L.; McDonnell, D.P. The human estrogen receptor-alpha is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J. Biol. Chem 2001, 276, 35684–35692. [Google Scholar]
- Alarid, E.T.; Bakopoulos, N.; Solodin, N. Proteasome-mediated proteolysis of estrogen receptor: A novel component in autologous down-regulation. Mol. Endocrinol 1999, 13, 1522–1534. [Google Scholar]
- Lonard, D.M.; Nawaz, Z.; Smith, C.L.; O’Malley, B.W. The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen-alpha transcription. Mol. Cell 2000, 5, 939–948. [Google Scholar]
- Nawaz, Z.; Lonard, D.M.; Dennis, A.P.; Smith, C.L.; O’Malley, B.W. Proteasome-dependent degradation of the human estrogen receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 1858–1862. [Google Scholar]
- McKenna, N.J.; Lanz, R.B.; O’Malley, B.W. Nuclear receptor coregulators: cellular and molecular biology. Endocr. Rev 1999, 20, 321–344. [Google Scholar]
- Scott, S.M.; Brown, M.; Come, S.E. Emerging data on the efficacy and safety of fulvestrant, a unique antiestrogen therapy for advanced breast cancer. Expert Opin. Drug Saf 2011, 10, 819–826. [Google Scholar]
- Litton, J.K.; Arun, B.K.; Brown, P.H.; Hortobagyi, G.N. Aromatase inhibitors and breast cancer prevention. Expert. Opin. Pharmacother 2012, 13, 325–331. [Google Scholar]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijin, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumors. Nature 2000, 406, 747–752. [Google Scholar]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijin, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar]
- Hu, Z.; Fan, C.; Oh, D.S.; Marron, J.S.; He, X.; Qaqish, B.F.; Livasy, C.; Carey, L.A.; Reynolds, E.; Dressler, L.; et al. The molecular portraits of breast tumors are conserved across microarray platform. BMC Genomics 2006, 7, 96. [Google Scholar]
- Sorlie, T.; Wang, Y.; Xiao, C.; Johnsen, H.; Naume, B.; Samaha, R.R.; Borresen-Dale, A.L. Distinct molecular mechanisms underlying clinically relevant subtypes of breast cancer: Gene expression analyses across three different platforms. BMC Genomics 2006, 7, 127. [Google Scholar]
- Fan, C.; Oh, D.S.; Wessels, L.; Weigelt, B.; Nuyten, D.S.; Nobel, A.B.; van’t Veer, L.J.; Perou, C.M. Concordance among gene-expression-based predictors for breast cancer. N. Eng. J. Med 2006, 355, 560–569. [Google Scholar]
- Bauer, K.R.; Brown, M.; Cress, R.D.; Parise, C.A.; Caggiano, V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: A population-based study from the California Cancer Registry. Cancer 2007, 109, 1721–1728. [Google Scholar]
- Cleator, S.; Heller, W.; Coombes, R.C. Triple-negative breast cancer: Therapeutic options. Lancet Oncol 2007, 8, 235–244. [Google Scholar]
- Cleere, D.W. Triple-negative breast cancer: A clinical update. Commun. Oncol 2010, 7, 203–211. [Google Scholar]
- Irvin, W.J., Jr; Carey, L.A. What is triple-negative breast cancer? Eur. J. Cancer 2008, 44, 2799–2805. [Google Scholar]
- Isakoff, S.J. Triple-negative breast cancer: Role of specific chemotherapy agents. Cancer J 2010, 16, 53–61. [Google Scholar]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol 2008, 26, 1275–1281. [Google Scholar]
- Hugh, J.; Hanson, J.; Cheang, M.C.U.; Nielsen, T.O.; Perou, C.M.; Dumontet, C.; Reed, J.; Krajewska, M.; Treilleux, I.; Rupin, M.; et al. Breast cancer subtypes and response to docetaxel in node-positive breast cancer: Use of immunohistochemical definition in the BCIRG 001 trial. J. Clin. Oncol 2009, 27, 1168–1176. [Google Scholar]
- Ellis, P.; Barrett-Lee, P.; Johnson, L.; Cameron, D.; Wardley, A.; O’Reilly, S.; Verrli, M.; Smith, I.; Yarnold, J.; Coleman, R.; et al. The TACT trial management group and the TACT trialists. Sequential docetaxel as adjuvant chemotherapy for early breast cancer (TACT): An open-label, phase III, randomized controlled trial. Lancet 2009, 373, 1681–1692. [Google Scholar]
- Hayes, D.; Thor, A.D.; Dressler, L.G.; Weaver, D.; Ederton, S.; Cowan, D.; Broadwater, G.; Goldstein, L.J.; Martino, S.; Ingle, J.; et al. The cancer and leukemia group B (CALGB) investigator. HER2 and response to paclitaxel in node-positive breast cancer. N. Engl. J. Med 2007, 357, 1496–1506. [Google Scholar]
- Sikov, W.M.; Dizon, D.S.; Strenger, R.; Legare, R.D.; Theall, K.P.; Graves, T.A.; Gass, J.S. Frequent pathologic complete responses in aggressive stage II to III breast cancers with every-4-week-carboplatin and weekly paclitaxel with or without trastuzumab: A Brown University Oncology Group study. J. Clin. Oncol 2008, 927, 4693–4700. [Google Scholar]
- Foulkes, W.D. Traffic control for BRCA1. N. Engl. J. Med 2010, 326, 755–756. [Google Scholar]
- Silver, D.P.; Richardson, A.L.; Eklund, A.C.; Wang, Z.C.; Szallasi, Z.; Li, Q.; Juul, N.; Leong, C.O.; Calogrias, D.; Buraimoh, A.; et al. Efficacy of neoadjuvant cisplatin in triple-negative breast cancer. J. Clin. Oncol 2010, 28, 1145–1153. [Google Scholar]
- Koshy, N.; Quispe, D.; Shi, R.; Mansour, R.; Burton, G.V. Cisplatin-gemcitabine therapy in metastatic breast cancer: Improved outcome in triple-negative breast cancer patients compared to non-triple negative patients. Breast 2010, 19, 246–248. [Google Scholar]
- Leong, C.O.; Vidnovic, N.; DeYoung, M.P.; Sgroi, D.; Ellisen, L.W. The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J. Clin. Invest 2007, 117, 1370–1380. [Google Scholar]
- Wong, S.W.; Tiong, K.H.; Kong, W.Y.; Yue, Y.C.; Chua, C.H.; Lim, J.Y.; Lee, C.Y.; Quah, S.I.; Fow, C.; Chung, C.; et al. Rapamycin synergizes cisplatin sensitivity in basal-like breast cancer through up-regulation of p73. Breast Cancer Res. Treat 2011, 128, 301–313. [Google Scholar]
- Gong, J.G.; Costanzo, A.; Yang, H.-Q.; Melinos, G.; Kaelin, W.G.; Levrero, M.; Wang, J.Y.J. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature 1999, 399, 806–809. [Google Scholar]
- Hastak, K.; Elizabeth, A.; Ford, J.M. Synergistic chemosensitivity of triple-negative breast cancer cell line to poly (ADP-ribose) polymerase inhibition, gemcitabine, and cisplatin. Cancer Res 2010, 70, 7970–7980. [Google Scholar]
- Santana-Davila, R.; Perez, E.A. Treatment options for patients with triple-negative breast cancer. J. Hematol. Oncol 2010, 3, 42. [Google Scholar]
- Byrski, T.; Huzarski, T.; Dent, R.; Gronwald, J.; Zuziak, D.; Cybulski, C.; Kladny, J.; Gorski, B.; Lubinski, J.; Narod, S.A. Response to neoadjuvant therapy with cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res. Treat 2009, 115, 359–363. [Google Scholar]
- Caray, L.; Winer, E.; Viale, G.; Cameron, D.; Gianni, L. Triple-negative breast cancer: Disease entity or title of convenience? Nat. Rev. Clin. Oncol 2010, 7, 683–692. [Google Scholar]
- Frasci, G.; Comella, P.; Rinaldo, M.; Iodice, G.; Di Bonito, M.; D’Aiuto, M.; Lastoria, S.; Sini, C.; Comella, G.; D’Aiuto, G. Preoperative weekly cisplatin-epirubicin-paclitaxel with G-CSF support in triple-negative large operable breast cancer. Ann. Oncol 2009, 20, 1185–1192. [Google Scholar]
- Anders, C.K.; Carey, L.A. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin. Breast Cancer 2009, 9, S73–S81. [Google Scholar]
- Nofech-Mozes, S.; Trudeau, M.; Kahn, H.K.; Dent, R.; Rawlinson, E.; Sun, P.; Narod, S.A.; Hanna, W.M. Patterns of recurrence in the basal and non-basal subtypes of triple-negative breast cancers. Breast Cancer Res. Treat 2009, 118, 131–137. [Google Scholar]
- Dedes, K.J.; Wilkerson, P.M.; Wetterskog, D.; Weigelt, B.; Ashworth, A.; Reis-Filho, J.S. Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle 2011, 10, 1192–1199. [Google Scholar]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.; Zander, S.A.; Derksen, P.W.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17089. [Google Scholar]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med 2009, 361, 123–134. [Google Scholar]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar]
- Rajan, A.; Carter, C.A.; Kelly, R.J.; Gutierrez, M.; Kummar, S.; Szabo, E.; Yancey, M.A.; Ji, J.; Mannargudi, B.; Woo, S.; et al. A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumors. Clin. Cancer Res 2012, 18, 2344–2351. [Google Scholar]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 429–301. [Google Scholar]
- Goncalves, A. PARP inhibitors and breast cancer: Update and perspectives. Bull. Cancer 2012, 99, 441–451. [Google Scholar]
- Comen, E.A.; Robson, M. Poly(ADP-ribose) polymerase inhibitors in triple-negative breast cancer. Cancer J 2010, 16, 48–52. [Google Scholar]






© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ratanaphan, A. A DNA Repair BRCA1 Estrogen Receptor and Targeted Therapy in Breast Cancer. Int. J. Mol. Sci. 2012, 13, 14898-14916. https://doi.org/10.3390/ijms131114898
Ratanaphan A. A DNA Repair BRCA1 Estrogen Receptor and Targeted Therapy in Breast Cancer. International Journal of Molecular Sciences. 2012; 13(11):14898-14916. https://doi.org/10.3390/ijms131114898
Chicago/Turabian StyleRatanaphan, Adisorn. 2012. "A DNA Repair BRCA1 Estrogen Receptor and Targeted Therapy in Breast Cancer" International Journal of Molecular Sciences 13, no. 11: 14898-14916. https://doi.org/10.3390/ijms131114898
APA StyleRatanaphan, A. (2012). A DNA Repair BRCA1 Estrogen Receptor and Targeted Therapy in Breast Cancer. International Journal of Molecular Sciences, 13(11), 14898-14916. https://doi.org/10.3390/ijms131114898