Natural Biomolecules and Protein Aggregation: Emerging Strategies against Amyloidogenesis

Abstract
:1. Introduction
2. Protein Folding and Aggregation
2.1. Protein Folding
2.2. Protein Aggregation
3. Amyloid Fibrils and Disease
3.1. General Features
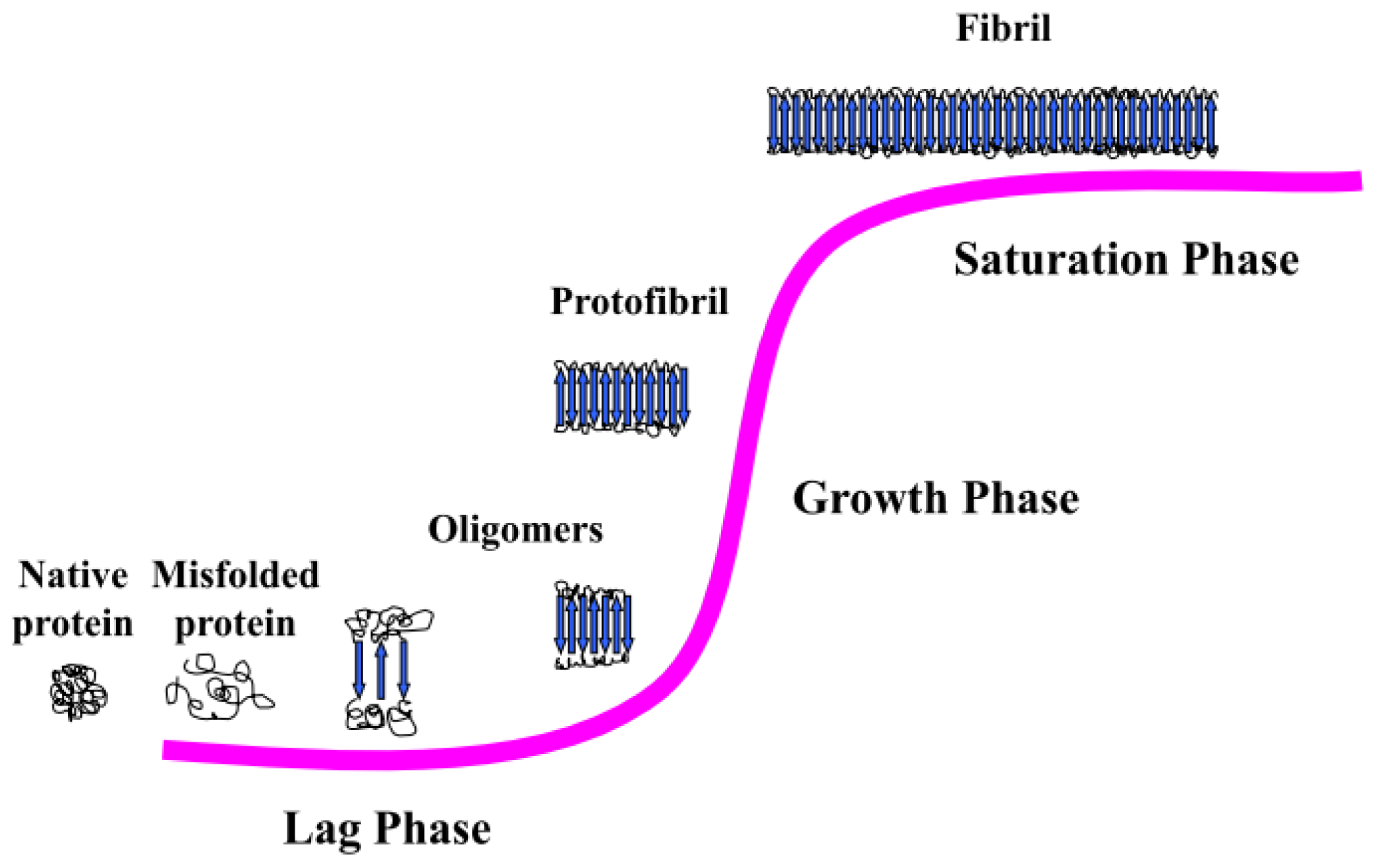
3.2. Amyloid Fibrillogenesis
3.3. Amyloid Cytotoxicity
3.4. Therapeutic Strategies
- Inhibition of the amyloidogenic form of proteins by stabilization of the native conformation or by reduction in the production of misfolded/unfoldded structures.
- Inhibition of protein self-assembly in oligomers and fibrils.
- Enhancement in the clearance of toxic aggregates.
4. Natural Molecules
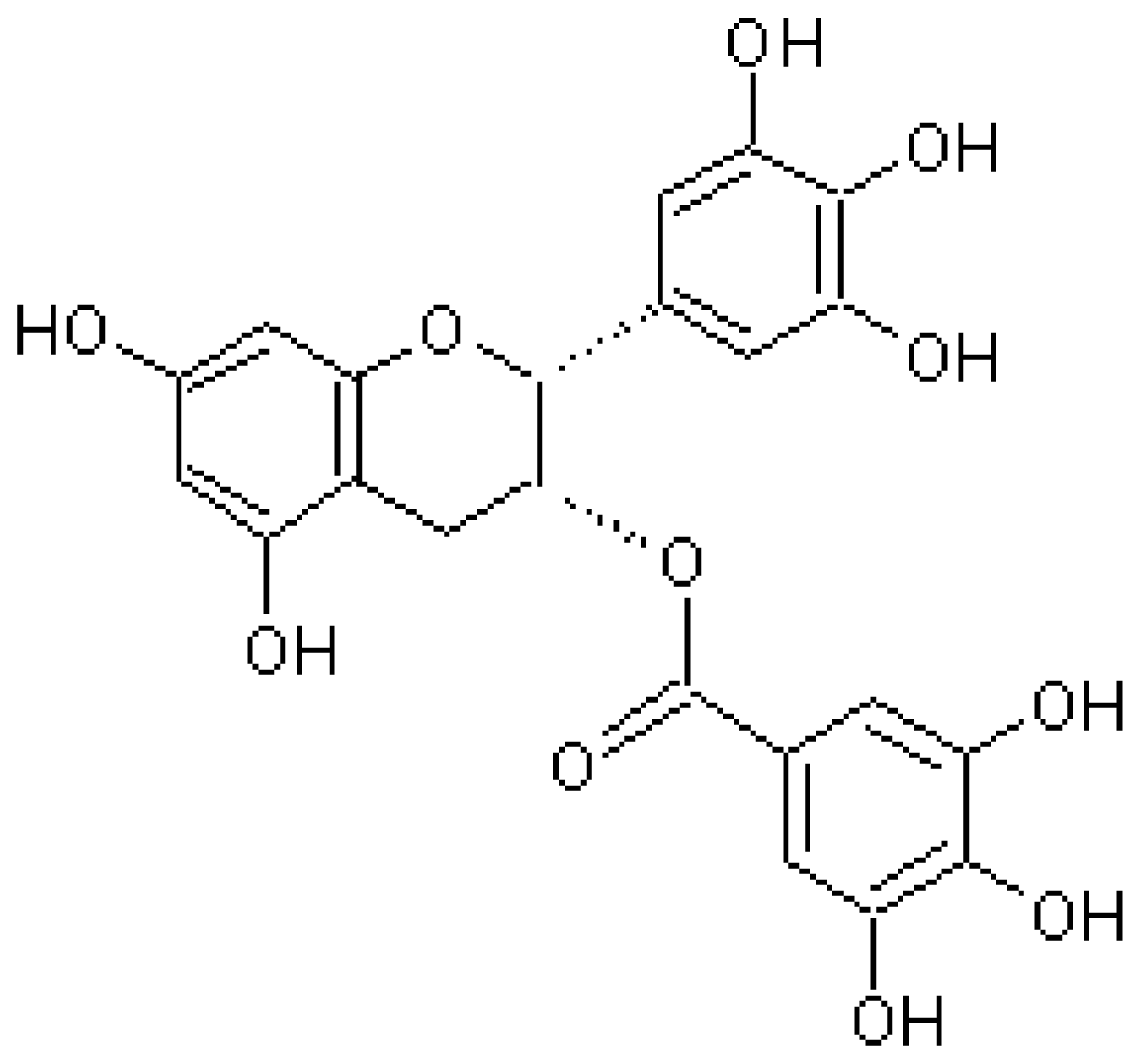
4.1. Epigallocatechin-3-Gallate (EGCG)
4.2. Curcumin
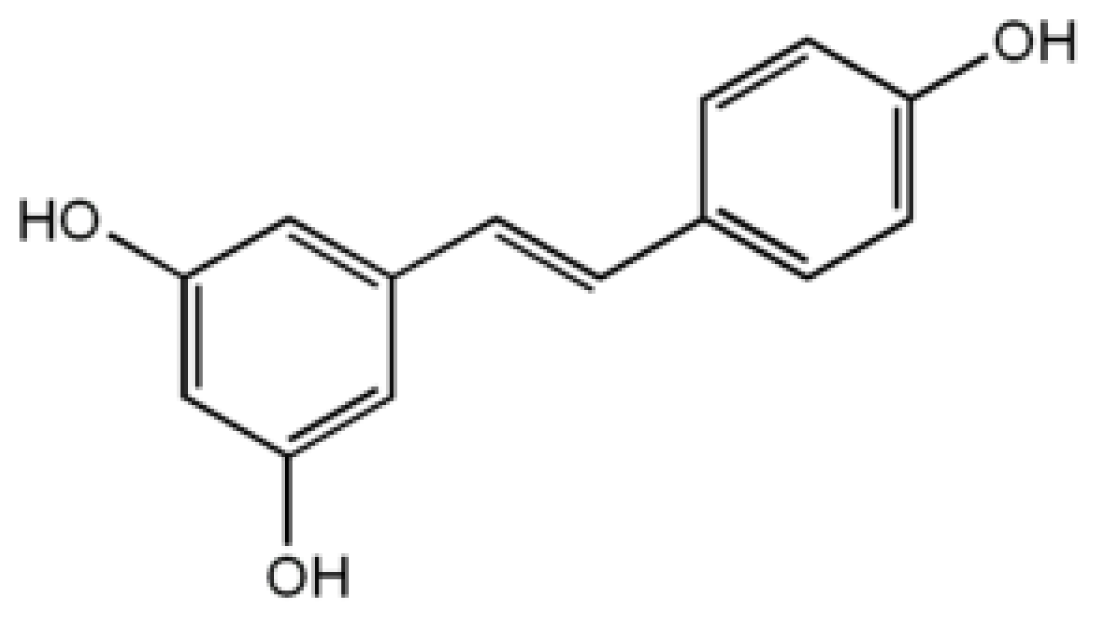
4.3. Resveratrol
4.4. Hypericin
4.5. Ferulic Acid
5. Conclusions
Acknowledgments
- Conflict of InterestThe author declares no conflict of interest.
References
- Clardy, J.; Walsh, C. Lessons from natural molecules. Nature 2004, 432, 829–837. [Google Scholar]
- Carocho, M.; Ferreira, I.C.F.R. A review on antioxidants, prooxidants and related controversy: Natural and synthetic compounds, screening and analysis methodologies and future perspectives. Food Chem. Toxicol 2013, 51, 15–25. [Google Scholar]
- Mecocci, P.; Polidori, M.C. Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 631–638. [Google Scholar]
- Hawkes, C.A.; Ng, V.; McLaurin, J.A. Small molecule inhibitors of Aβ-aggregation and neurotoxicity. Drug Dev. Res 2009, 70, 111–124. [Google Scholar]
- Joynera, P.M.; Cichewicz, R.H. Bringing natural products into the fold—Exploring the therapeutic lead potential of secondary metabolites for the treatment of protein-misfolding related neurodegenerative diseases. Nat. Prod. Rep 2011, 28, 26–47. [Google Scholar]
- Stefani, M. Protein misfolding and aggregation: New examples in medicine and biology of the dark side of the protein world. Biochim. Biophys. Acta 2004, 1739, 5–25. [Google Scholar]
- Berg, T. Modulation of protein-protein interactions with small organic molecules. Angew. Chem. Int. Ed 2003, 42, 2462–2481. [Google Scholar]
- Lindsey, J.S. Self-assembly in synthetic routes to molecular devices. Biological principles and chemical perspectives: A review. New J. Chem 1991, 15, 153–180. [Google Scholar]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar]
- Dill, K.A. The meaning of hydrophobicity. Science 1990, 250, 297–298. [Google Scholar]
- Herzfeld, J. Understanding hydrophobic behavior. Science 1991, 253, 88. [Google Scholar]
- Dobson, C.M.; Karplus, M. The fundamentals of protein folding: Bringing together theory and experiment. Curr. Opin. Struct. Biol 1999, 9, 92–101. [Google Scholar]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 18–25. [Google Scholar]
- Dobson, CM. Principles of protein folding, misfolding and aggregation. Semin. Cell Dev. Biol. 2004, 15, 3–16. [Google Scholar]
- Dobson, C.M. Experimental investigation of protein folding and misfolding. Methods 2004, 34, 4–14. [Google Scholar]
- Hartl, F.U.; Hayer-Hartl, M. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 2002, 295, 1852–1858. [Google Scholar]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med 2003, 81, 678–699. [Google Scholar]
- Invernizzi, G.; Papaleo, E.; Sabate, R.; Ventura, S. Protein aggregation: Mechanisms and functional consequences. Int. J. Biochem. Cell Biol 2012, 44, 1541–1554. [Google Scholar]
- Pastore, A.; Temussi, P. Protein aggregation and misfolding: Good or evil? J. Phys. Condens. Matter 2012, 24, 1–9. [Google Scholar]
- Maury, C.P.J. The emerging concept of functional amyloid. J. Intern. Med. 2009, 265, 329–334. [Google Scholar]
- Zerovnik, E. Amyloid fibril formation. Proposed mechanisms and relevance to conformational disease. Eur. J. Biochem 2002, 269, 3362–3371. [Google Scholar]
- Greenwald, J.; Riek, R. Biology of amyloid: Structure, function, and regulation. Structure 2010, 18, 1244–1260. [Google Scholar]
- Astbury, W.T.; Dickinson, S.; Bailey, K. The X-ray interpretation of denaturation and the structure of the seed globulins. Biochem. J 1935, 29, 2351–2360. [Google Scholar]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem 2006, 75, 333–366. [Google Scholar]
- Chiti, F.; Dobson, C.M. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol 2009, 5, 15–22. [Google Scholar]
- Baldwin, A.J.; Knowles, T.P.J.; Tartaglia, G.G.; Fizpatrick, A.W.; Devlin, G.L.; Shammas, S.L.; Waudby, C.A.; Mossuto, M.F.; Meehan, S.; Gras, S.; et al. Metastability of native proteins and the phenomenon of amyloid formation. J. Am. Chem. Soc 2011, 133, 14160–14163. [Google Scholar]
- Lee, C.C.; Nayak, A.; Sethuraman, A.; Belfort, G.; McRae, G.J. A three-stage kinetic model of amyloid fibrillation. Biophys. J 2007, 92, 3448–3458. [Google Scholar]
- Nguyen, H.D.; Hall, H.K. Molecular dynamics simulations of spontaneous fibril formation by random-coil peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 16180–16185. [Google Scholar]
- Bhak, G.; Choe, Y.J.; Paik, S.R. Mechanism of amyloidogenesis: Nucleation-dependent fibrillation versus double-concerted fibrillation. BMB Rep 2009, 42, 541–551. [Google Scholar]
- Chiti, F.; Stefani, M.; Taddei, N.; Ramponi, G.; Dobson, C.M. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature 2003, 424, 805–808. [Google Scholar]
- Gazit, E. A possible role for π-stacking in self-assembly of amyloid fibrils. FASEB J 2002, 16, 77–83. [Google Scholar]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol 2007, 8, 101–112. [Google Scholar]
- Klein, W.L.; Lacor, P.N.; De Felice, F.G.; Ferreira, S.T. Molecules that Disrupt Memory Circuits in Alzheimer’s Disease: The Attack on Synapses by Aβ Oligomers (ADDLs). In Memories: Molecules and Circuits; Springer: Berlin, Germany, 2007; pp. 155–179. [Google Scholar]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci 2005, 8, 79–84. [Google Scholar]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T., Jr. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar]
- Stine, B.W., Jr; Dahlgren, K.N.; Krafft, G.A; LaDu, M.J. In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J. Biol. Chem. 2003, 282, 11612–11622. [Google Scholar]
- Conway, K.A.; Lee, S.-J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T. Acceleration of oligomerization not fibrillization is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease. Implication for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar]
- Sousa, M.M.; Cardoso, I.; Fernandes, R.; Guimaraes, A.; Saraiva, M.J. Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy. Am. J. Pathol 2001, 159, 1993–2000. [Google Scholar]
- Poirier, M.A.; Li, H.; Macosko, J.; Cail, S.; Amzel, M.; Ross, C.A. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrillization. J. Biol. Chem 2002, 277, 41032–41037. [Google Scholar]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar]
- Bucciantini, M.; Calloni, G.; Chiti, F.; Formigli, L.; Nosi, D.; Dobson, C.M.; Stefani, M. Pre-fibrillar amyloid protein aggregates share common features of cytotoxicity. J. Biol. Chem 2004, 279, 31374–31382. [Google Scholar]
- Novitskaya, V.; Bocharova, O.V.; Bronstein, I.; Baskakov, I.V. Amyloid fibrils of mammalian prion protein are highly toxic to cultured cells and primary neurons. J. Biol. Chem 2006, 281, 13828–13836. [Google Scholar]
- Bucciantini, M.; Nosi, D.; Forzan, M.; Russo, E.; Calamai, M.; Pieri, L.; Formigli, L.; Quercioli, F.; Soria, S.; Pavone, F.; et al. Toxic effects of amyloid fibrils on cell membranes: The importance of ganglioside GM1. FASEB J 2012, 26, 818–831. [Google Scholar]
- Gharibyan, A.L.; Zamotin, V.; Yanamandra, K.; Moskaleva, O.S.; Margulis, B.A.; Kostanyan, I.A.; Morozova-Roche, L.A. Lysozyme amyloid oligomers and fibrils induce cellular death via different apoptotic/necrotic pathways. J. Mol. Biol 2007, 365, 1337–13349. [Google Scholar]
- Giorgetti, S.; Raimondi, S.; Pagano, K.; Relini, A.; Bucciantini, M.; Corazza, A.; Fogolari, F.; Codutti, L.; Salmona, M.; Mangione, P.; et al. Effect of tetracyclines on the dynamics of formation and destructuration of β2-microglobulin amyloid fibrils. J. Biol. Chem 2011, 286, 2121–2131. [Google Scholar]
- Bemporad, F.; Taddei, N.; Stefani, M.; Chiti, F. Assessing the role of aromatic residues in the amyloid aggregation of human muscle acylphosphatase. Protein Sci 2006, 15, 862–870. [Google Scholar]
- Porat, Y.; Abramowitz, A.; Gazit, E. Inhibition of amyloid fibril formation by polyphenols: Structural similarity and aromatic interactions as a common inhibition mechanism. Chem. Biol. Drug. Des 2006, 67, 27–37. [Google Scholar]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol 2008, 15, 558–566. [Google Scholar]
- Ehrnhoefer, D.E.; Duennwald, M.; Markovic, P.; Wacker, J.L.; Engemann, S.; Roark, M.; Legleiter, J.; Marsh, J.L.; Thompson, L.M.; Lindquist, S.; et al. Green tea (−)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models. Hum. Mol. Genet 2006, 15, 2743–2751. [Google Scholar]
- Meng, F.; Abedini, A.; Plesner, A.; Verchere, C.B.; Raleigh, D.P. The flavanol (−)-epigallocatechin 3-gallate inhibits amyloid formation by islet amyloid polypeptide, disaggregates amyloid fibrils, and protects cultured cells against IAPP-induced toxicity. Biochemistry 2010, 49, 8127–8133. [Google Scholar]
- Miyata, M.; Sato, T.; Kugimiya, M.; Sho, M.; Nakamura, T.; Ikemizu, S.; Chirifu, M.; Mizuguchi, M.; Nabeshima, Y.; Suwa, Y.; et al. The crystal structure of the green tea polyphenol (−)-epigallocatechin gallate-transthyretin complex reveals a novel binding site distinct from the thyroxine binding site. Biochemistry 2010, 49, 6104–6114. [Google Scholar]
- Rezai-Zadeh, K.; Arendash, G.W.; Fernandez, F.; Jensen, M.; Runfeldt, M.; Shytle, D.; Tan, J. Green tea epigallocatechin-3-gallate (EGCG) reduces β-amyloid mediated cognitive impairment and modulates tau pathology in Alzheimer transgenic mice. Brain Res 2008, 1214, 177–187. [Google Scholar]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar]
- Cao, P.; Raleigh, D.P. Analysis of the inhibition and remodeling of islet amyloid polypeptide amyloid fibers by flavanols. Biochemistry 2012, 51, 2670–2683. [Google Scholar]
- Hamaguchi, T.; Ono, K.; Yamada, M. Curcumin and Alzheimer’s disease. CNS Neurosci. Ther 2010, 16, 285–297. [Google Scholar]
- Ahmad, B.; Lapidus, L.J. Curcumin prevents aggregation in α-synuclein by increasing reconfiguration rate. J. Biol. Chem 2012, 287, 9193–9199. [Google Scholar]
- Baskakov, I.V.; Legname, G.; Baldwin, M.A.; Prusiner, S.B.; Cohen, F.E. Pathway complexity of prion protein assembly into amyloid. J. Biol. Chem 2002, 277, 21140–21148. [Google Scholar]
- Hafner-Bratkovič, I.; Gašperšič, J.; Šmid, L.M.; Bresjanac, M.; Jerala, R. Curcumin binds to the α-helical intermediate and to the amyloid form of prion protein—A new mechanism for the inhibition of PrPSc accumulation. J. Neurochem 2008, 104, 1553–1564. [Google Scholar]
- Sahebkar, A. Neuroprotective effects of resveratrol: Potential mechanisms. Neurochem. Int 2010, 57, 621–622. [Google Scholar]
- Huang, T.C.; Lu, K.T.; Wo, Y.Y.; Wu, Y.J.; Yang, Y.L. Resveratrol protects rats from Abeta-induced neurotoxicity by the reduction of iNOS expression and lipid peroxidation. PLoS One 2011, 6, e29102. [Google Scholar]
- Marambaud, P.; Zhao, H.; Davies, P. Resveratrol promotes clearance of Alzheimer’s disease amyloid-beta peptides. J. Biol. Chem 2005, 280, 37377–37382. [Google Scholar]
- Karuppagounder, S.S.; Pinto, J.T.; Xu, H.; Chen, H.L.; Beal, M.F.; Gibson, G.E. Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer’s disease. Neurochem. Int 2009, 54, 111–118. [Google Scholar]
- Robb, E.L.; Winkelmolen, L.; Visanji, N.; Brotchie, J.; Stuart, J.A. Dietary resveratrol administration increases MnSOD expression and activity in mouse brain. Biochem. Biophys. Res. Commun 2008, 372, 254–259. [Google Scholar]
- Ge, J.-F.; Qiao, J.-P.; Qi, C.-C.; Wang, C.-W.; Zhou, J.-N. The binding of resveratrol to monomer and fibril amyloid beta. Neurochem. Int 2012, 61, 1192–1201. [Google Scholar]
- Ladiwala, A.R.; Lin, J.C.; Bale, S.S.; Marcelino-Cruz, A.M.; Bhattacharya, M.; Dordick, J.S.; Tessier, P.M. Resveratrol selectively remodels soluble oligomers and fibrils of amyloid Abeta into off-pathway conformers. J. Biol. Chem 2010, 285, 24228–24237. [Google Scholar]
- Bourgault, S.; Choi, S.; Buxbaum, J.N.; Kelly, J.W.; Price, J.L.; Reixach, N. Mechanisms of transthyretin cardiomyocyte toxicity inhibition by resveratrol analogs. Biochem. Biophys. Res. Commun 2011, 410, 707–713. [Google Scholar]
- Mishra, R.; Sellin, D.; Radovan, D.; Gohlke, A.; Winter, R. Inhibiting islet amyloid polypeptide fibril formation by the red wine compound resveratrol. ChemBioChem 2009, 10, 445–449. [Google Scholar]
- Jiang, P.; Li, W.; Shea, J.-E.; Mu, Y. Resveratrol inhibits the formation of multiple-layered β-sheet oligomers of the human islet amyloid polypeptide segment 22–27. Biophys. J 2011, 100, 1550–1558. [Google Scholar]
- Silva, B.A.; Dias, A.C.P.; Ferreres, F.; Malva, J.O.; Oliveira, C.R. Neuroprotective effect of H. perforatum extracts on β amyloid-induced neurotoxicty. Neurotoxic. Res 2004, 6, 119–130. [Google Scholar]
- Kraus, B.; Wolff, H.; Heilmann, J.; Elstner, E.F. Influence of Hypericum perforatum extract and its single compounds on amyloid-β mediated toxicity in microglial cells. Life Sci 2007, 81, 884–894. [Google Scholar]
- Karioti, A.; Bilia, A.R. Hypericins as potential leads for new therapeutics. Int. J. Mol. Sci 2010, 11, 562–594. [Google Scholar]
- Sgarbossa, A.; Lenci, F. Spectroscopic study of visible light effects on hypericin-lens proteins systems. Photochem. Photobiol 2001, 74, 196–200. [Google Scholar]
- Sgarbossa, A.; Buselli, D.; Lenci, F. In vitro perturbation of aggregation processes in beta-amyloid peptides: A spectroscopic study. FEBS Lett 2008, 582, 3288–3292. [Google Scholar]
- Bramanti, E.; Lenci, F.; Sgarbossa, A. Effects of hypericin on the structure and aggregation properties of β-amyloid peptides. Eur. Biophys. J 2010, 39, 1493–1501. [Google Scholar]
- Di Carlo, M.; Giacomazza, D.; San Biagio, P.L. Alzheimer’s disease: Biological aspects, therapeutic perspectives and diagnostic tools. J. Phys. Condens. Matter 2012, 24, 244102. [Google Scholar]
- Kim, H.S.; Cho, J.Y.; Kim, D.H.; Yan, J.J.; Lee, H.K.; Suh, H.W. Inhibitory effects of long term administration of ferulic acid on microgilal activation induced by intercerebroventricular injection of beta amyloid peptide (1-42) in mice. Biol. Pharm. Bull 2004, 27, 120–121. [Google Scholar]
- Sultana, R.; Ravagna, A.; Mohmmad-Abdul, H.; Calabrese, V.; Butterfield, D. Ferulic acid ethyl ester protects neurons against amyloid beta peptide (1–42)-induced oxidative stress and neurotoxicity: Relationship to antioxidant activity. J. Neurochem 2005, 92, 749–758. [Google Scholar]
- Picone, P.; Bondi, M.L.; Montana, G.; Bruno, A.; Pitarresi, G.; Giammona, G.; Di Carlo, M. Ferulic acid inhibits oxidative stress and cell death induced by Aβ oligomers: Improved delivery by solid lipid nanoparticles. Free Radic. Res. 2009, 43, 1133–1145. [Google Scholar]
- Ono, K.; Yoshiike, Y.; Takashima, A.; Hasegawa, K.; Naiki, H.; Yamada, M. Potent antiamyloidogenic and fibril-destabilizing effects of polyphenols in vitro: Implications for the prevention and therapeutics of Alzheimer’s disease. J. Neurochem 2003, 87, 172–181. [Google Scholar]
- Ono, K.; Hasegawa, K.; Yoshiike, Y.; Takashima, A.; Yamada, M.; Naiki, H. Nordihydroguaiaretic acid potently breaks down pre-formed Alzheimer’s beta-amyloid fibrils in vitro. J. Neurochem 2002, 81, 434–440. [Google Scholar]
- Ono, K.; Hirohata, M.; Yamada, M. Ferulic acid destabilizes preformed beta-amyloid fibrils in vitro. Biochem. Biophys. Res. Commun 2005, 336, 444–449. [Google Scholar]
- Ono, K.; Li, L.; Takamura, Y.; Yoshiike, Y.; Zhu, L.; Han, F.; Mao, X.; Ikeda, T.; Takasaki, J.; Nishijo, H.; et al. Phenolic compounds prevent amyloid beta-protein oligomerization and synaptic dysfunction by site-specific binding. J. Biol. Chem 2012, 287, 14631–14643. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathology | Peptides/Proteins |
|---|---|
| Alzheimer’s Disease | Amyloid β peptide, Tau protein |
| Parkinson’s Disease | α-synuclein |
| Transmissible Spongiform Encephalopathies | Prion protein |
| Huntington’s Disease | Huntingtin (poliQ expansion) |
| Amyotrophic Lateral Sclerosis | Superoxide dismutase |
| Cerebellar Ataxias | Ataxins (poliQ expansion) |
| Type II Diabetes | Islet amyloid polypeptide/amylin |
| Insulin-Related Amyloidosis | Insulin |
| Familial Amyloid Polyneuropathies | Transthyretin |
| Dialysis-Related Amyloidosis | β2-Microglobulin |
| Primary Systemic Amyloidosis | Immunoglobulin light chain |
| Finnish Hereditary Systemic Amyloidosis | Gelsolin |
| Lysozyme Systemic Amyloidosis | Lysozyme |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sgarbossa, A. Natural Biomolecules and Protein Aggregation: Emerging Strategies against Amyloidogenesis. Int. J. Mol. Sci. 2012, 13, 17121-17137. https://doi.org/10.3390/ijms131217121
Sgarbossa A. Natural Biomolecules and Protein Aggregation: Emerging Strategies against Amyloidogenesis. International Journal of Molecular Sciences. 2012; 13(12):17121-17137. https://doi.org/10.3390/ijms131217121
Chicago/Turabian StyleSgarbossa, Antonella. 2012. "Natural Biomolecules and Protein Aggregation: Emerging Strategies against Amyloidogenesis" International Journal of Molecular Sciences 13, no. 12: 17121-17137. https://doi.org/10.3390/ijms131217121
APA StyleSgarbossa, A. (2012). Natural Biomolecules and Protein Aggregation: Emerging Strategies against Amyloidogenesis. International Journal of Molecular Sciences, 13(12), 17121-17137. https://doi.org/10.3390/ijms131217121