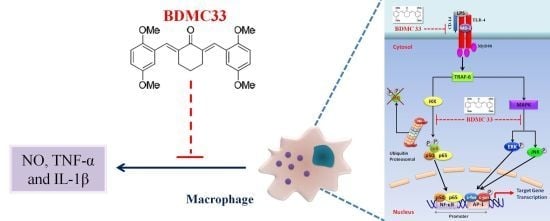
BDMC33, A Curcumin Derivative Suppresses Inflammatory Responses in Macrophage-Like Cellular System: Role of Inhibition in NF-κB and MAPK Signaling Pathways

Abstract
:
1. Introduction
2. Results
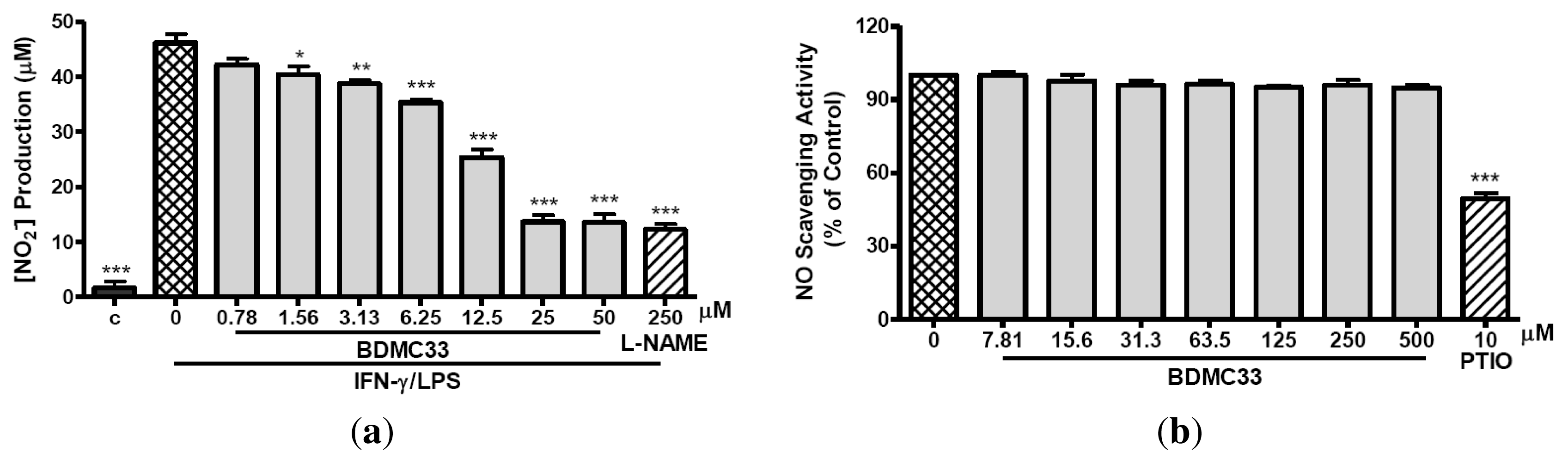
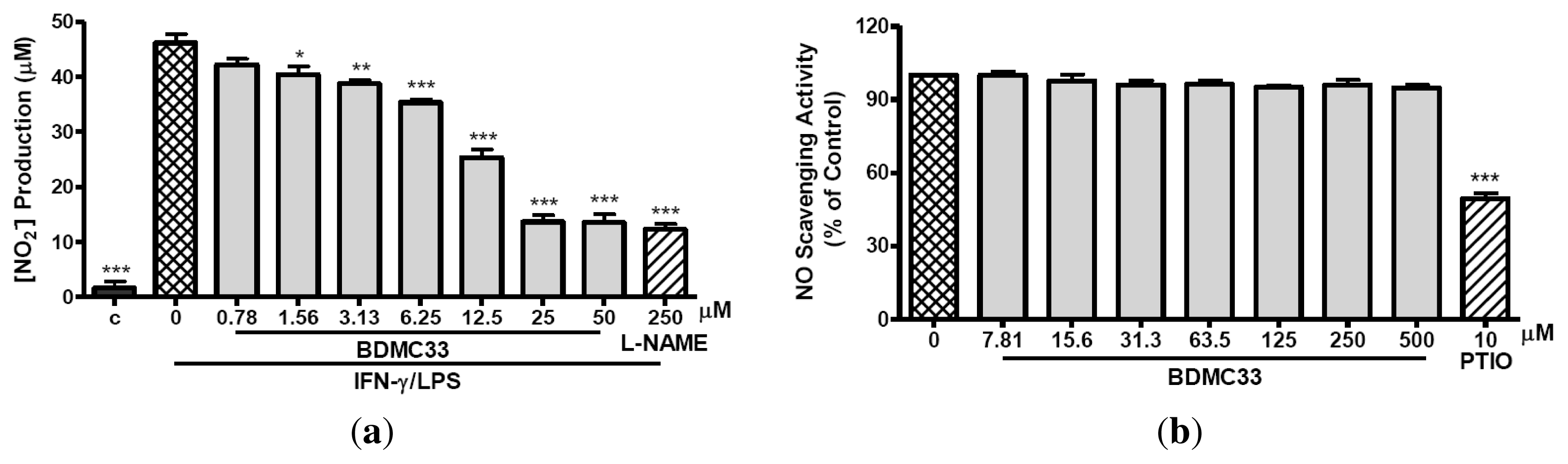
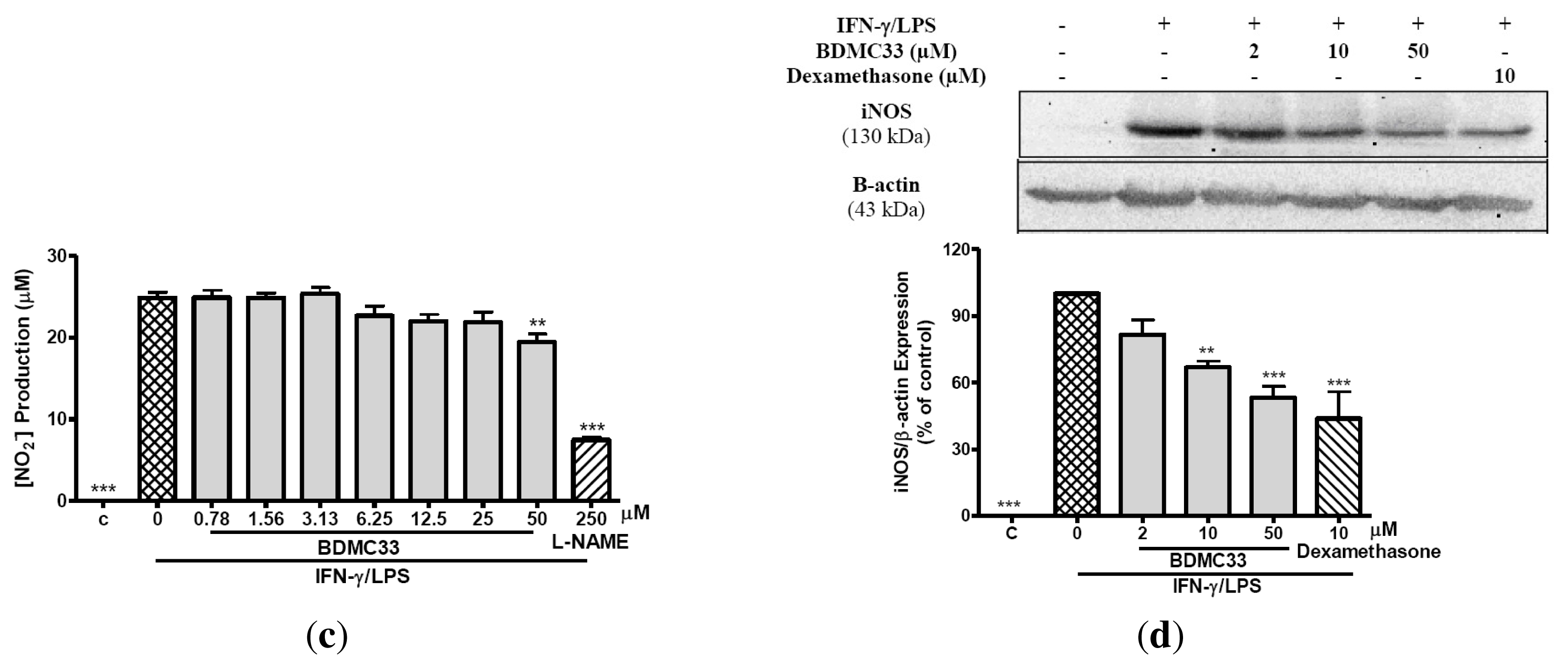
2.1. Inhibitory Action on NO Production via Down-Regulation of iNOS Expression
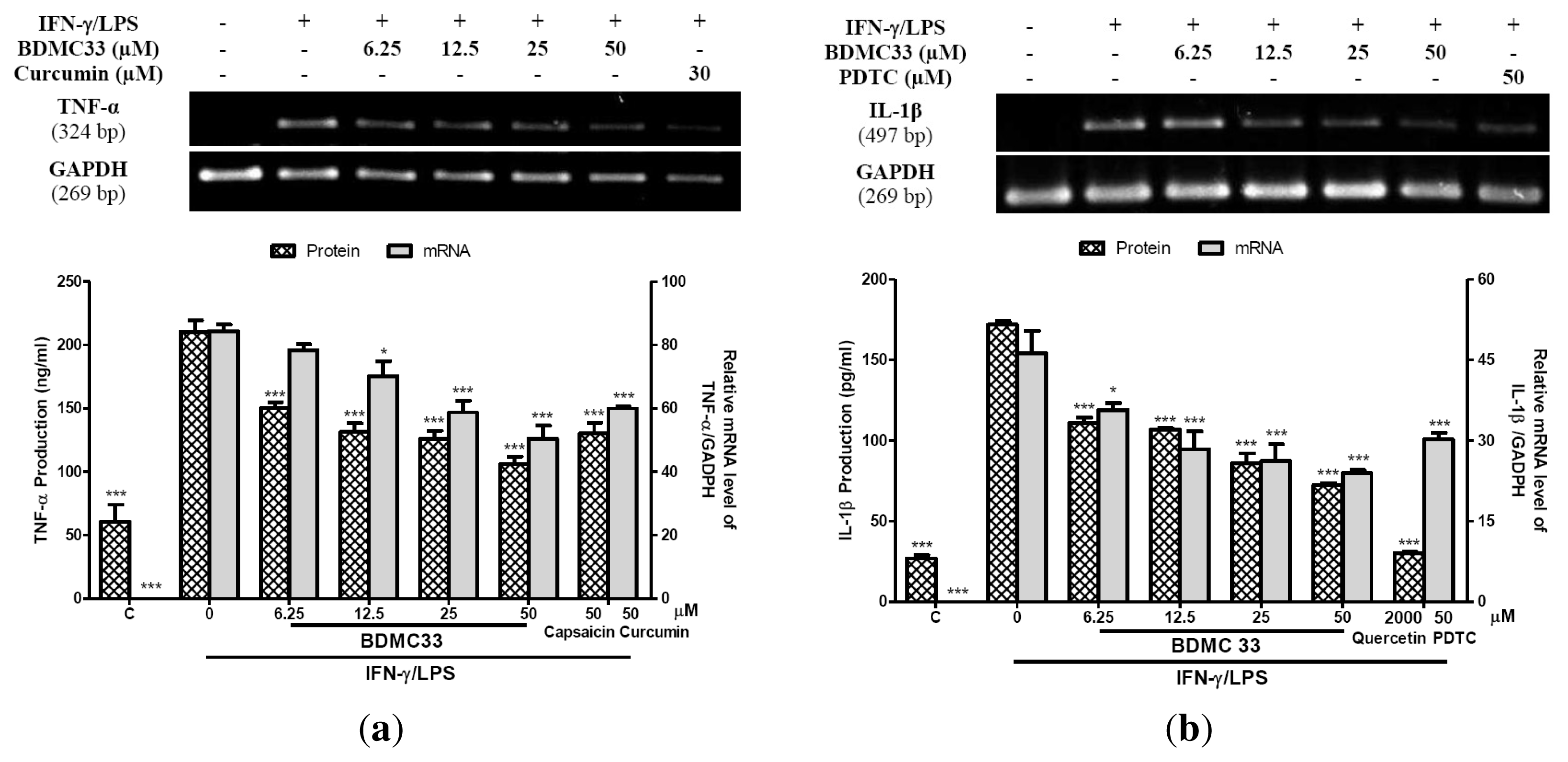
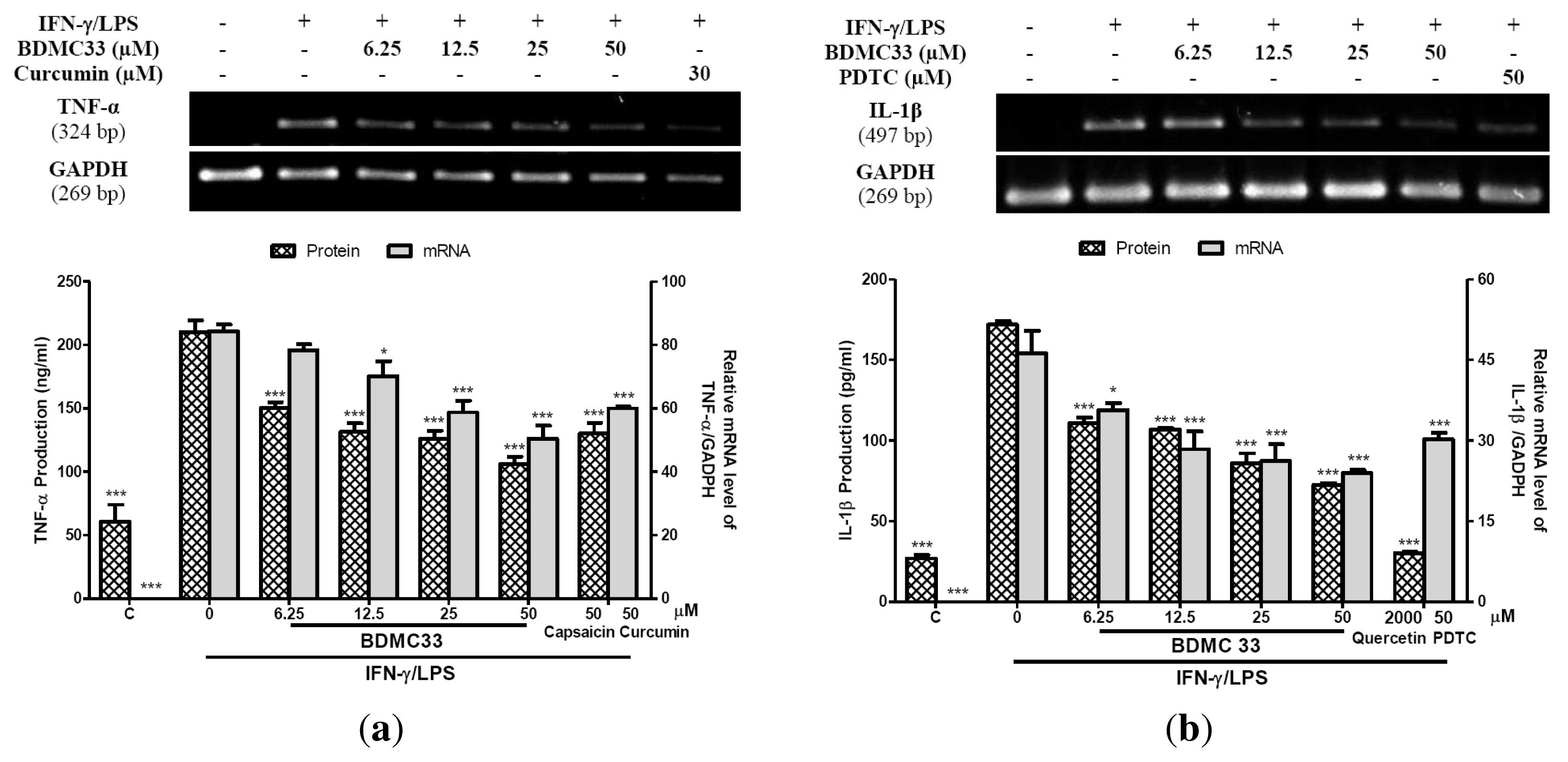
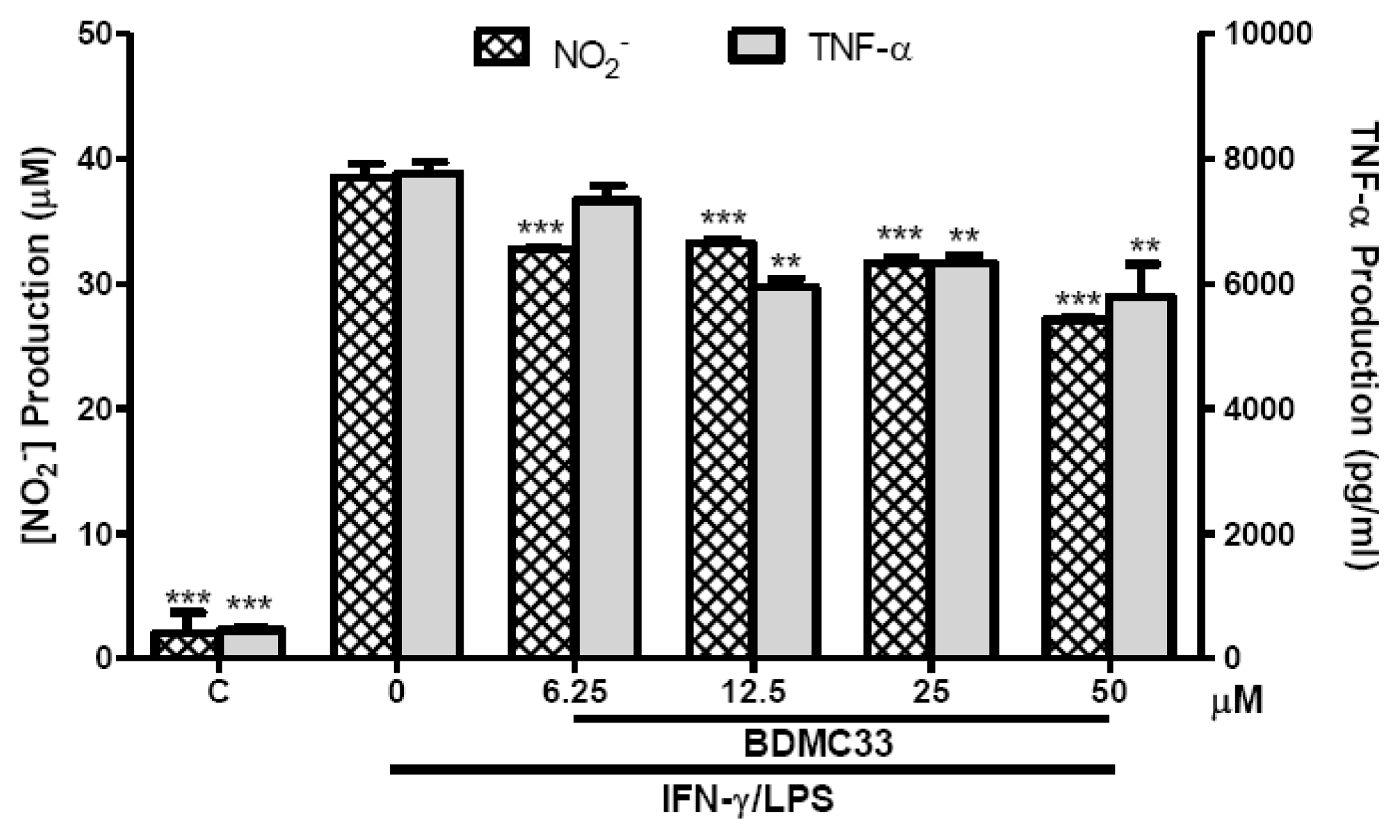
2.2. Interference in Pro-Inflammatory Cytokine Production
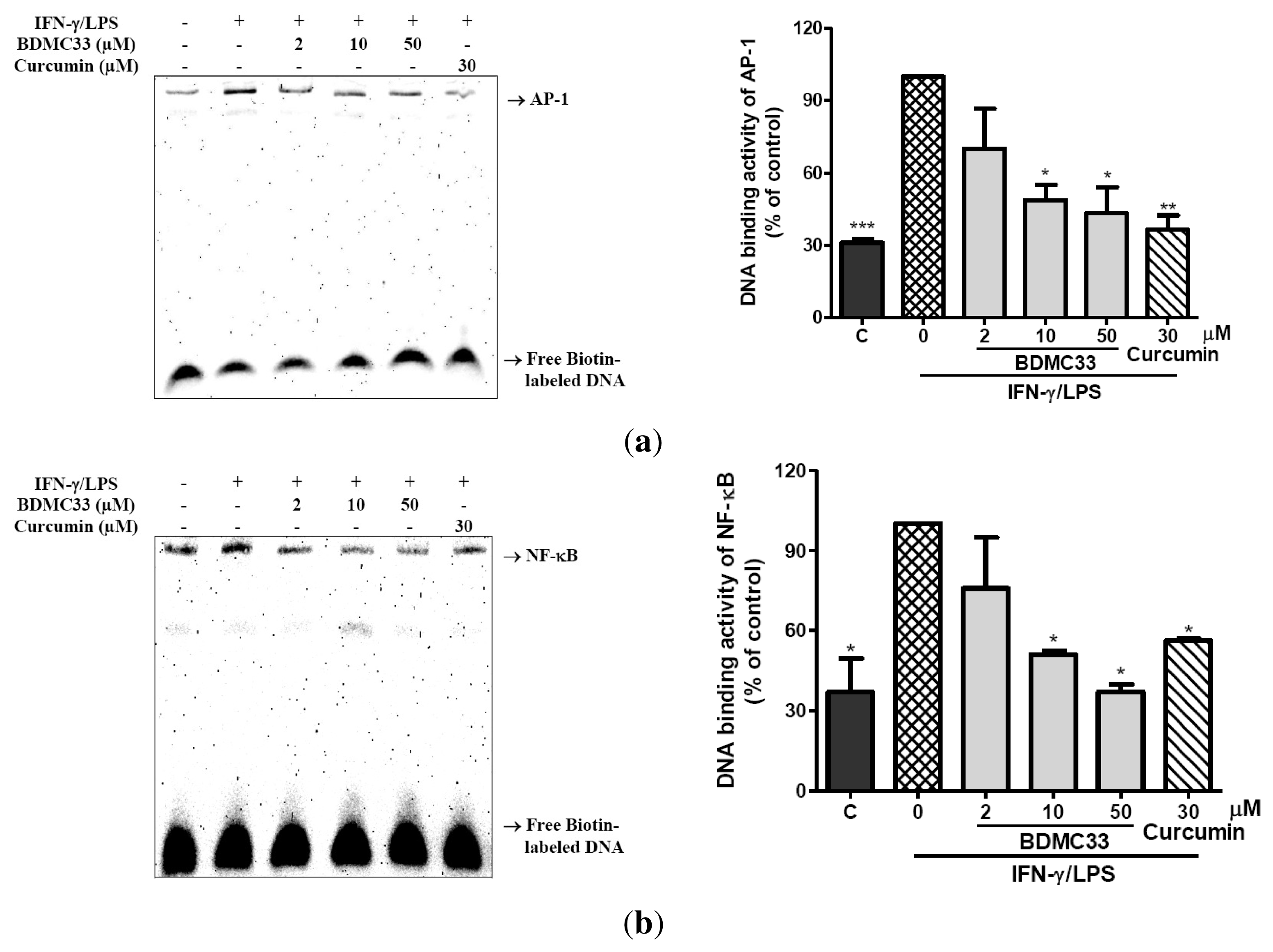
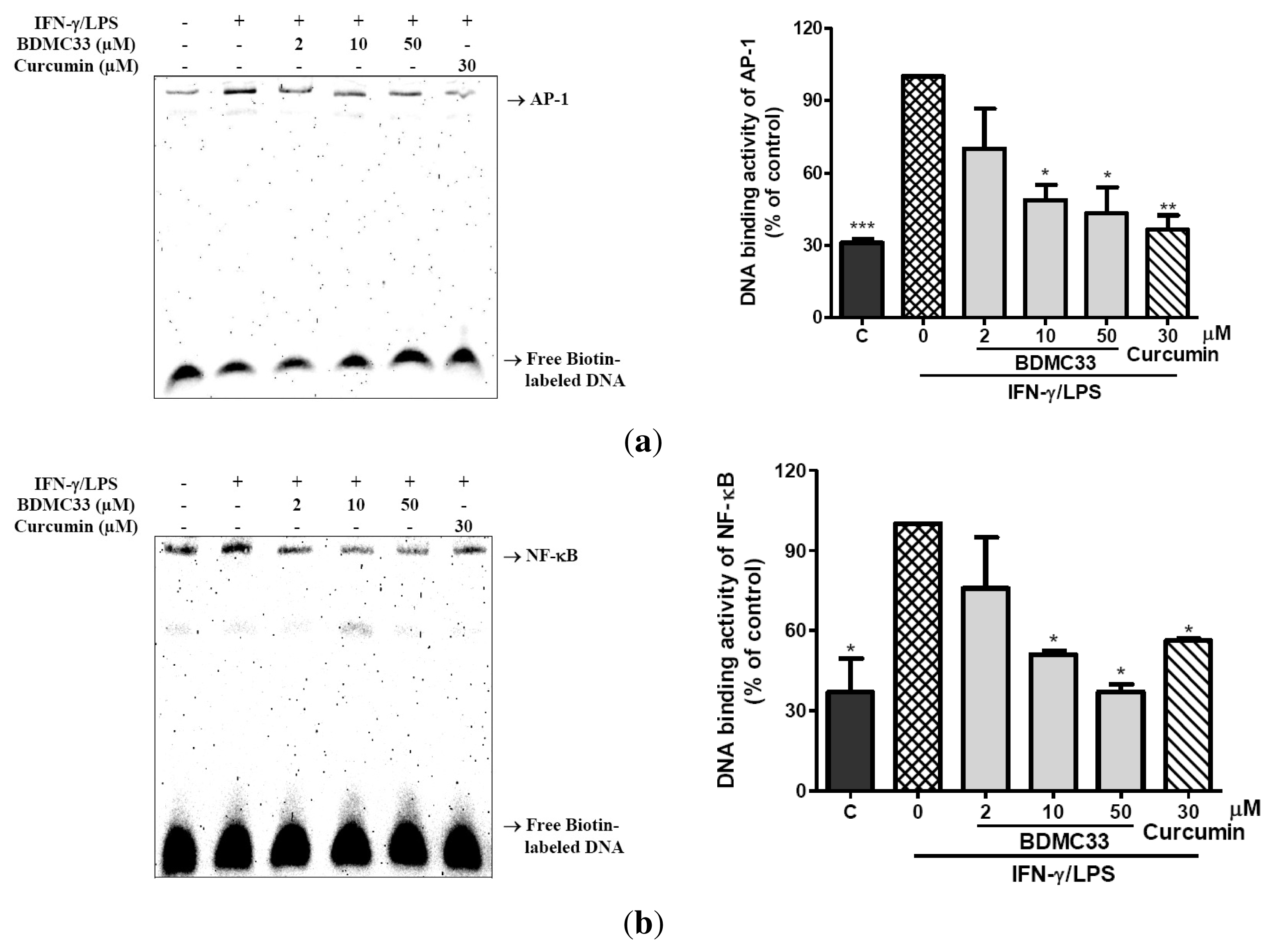
2.3. Attenuation of NF-κB and AP-1 DNA Binding Activity
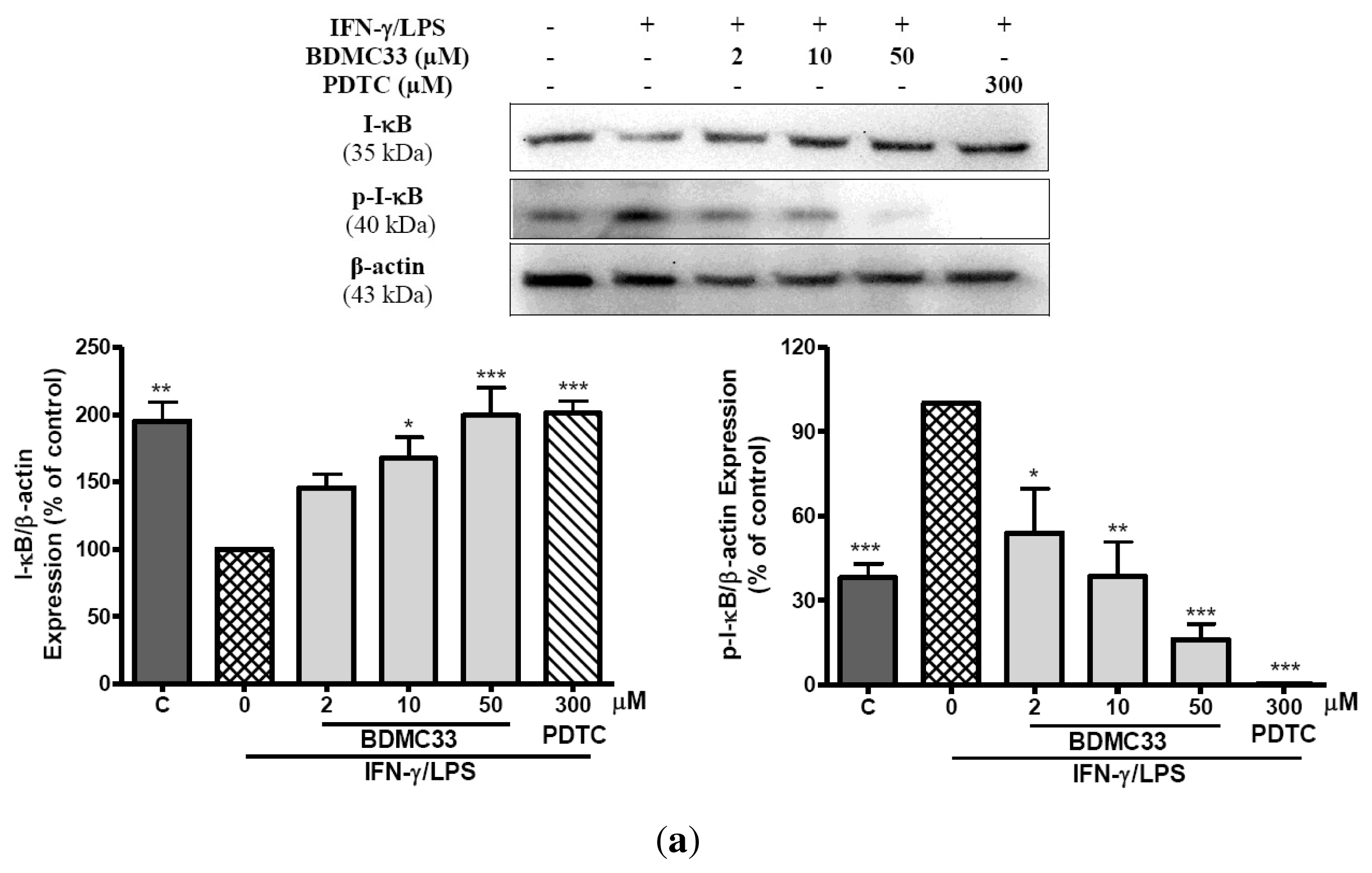
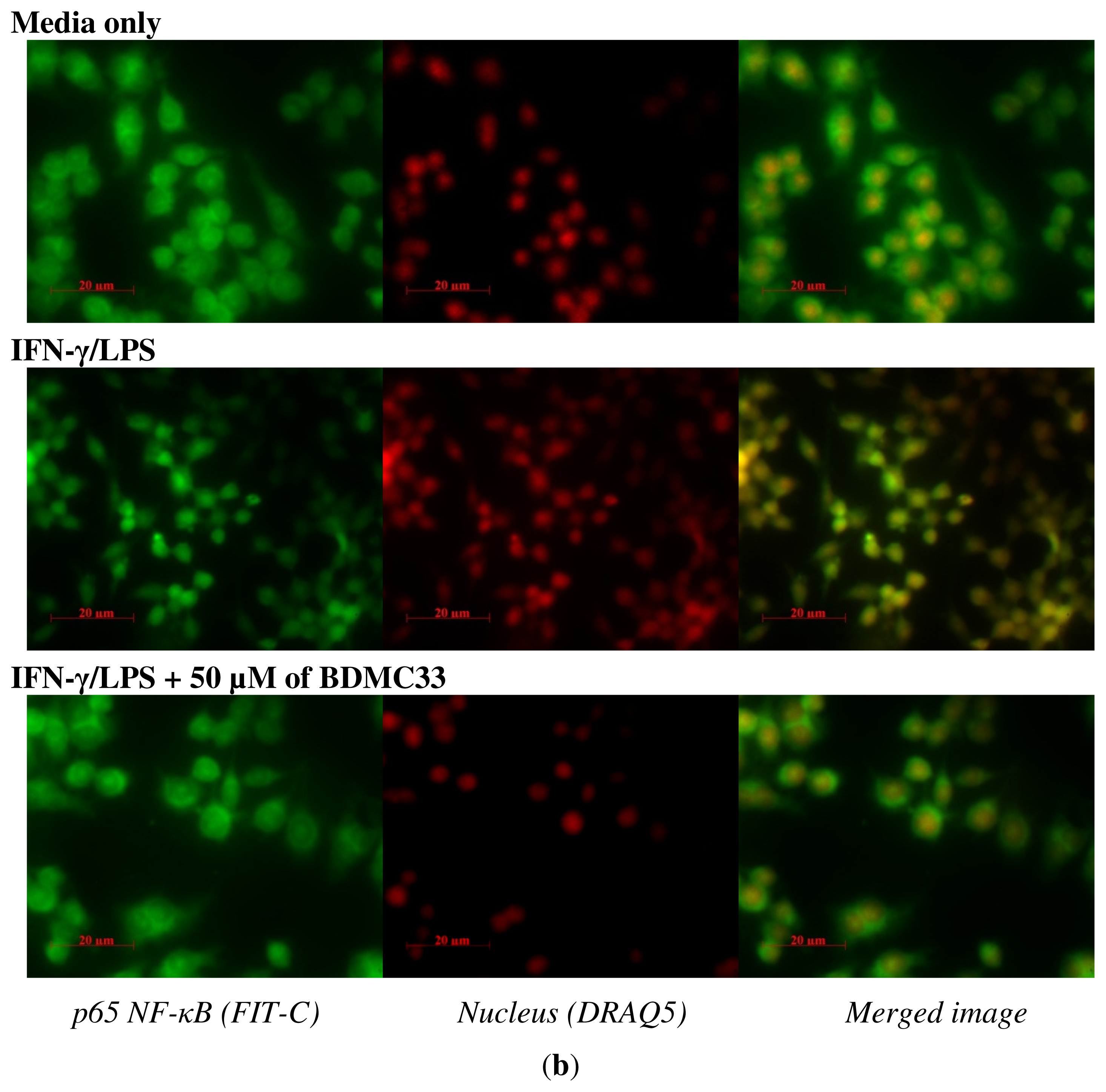
2.4. Effects of BDMC33 on NF-κB Activation
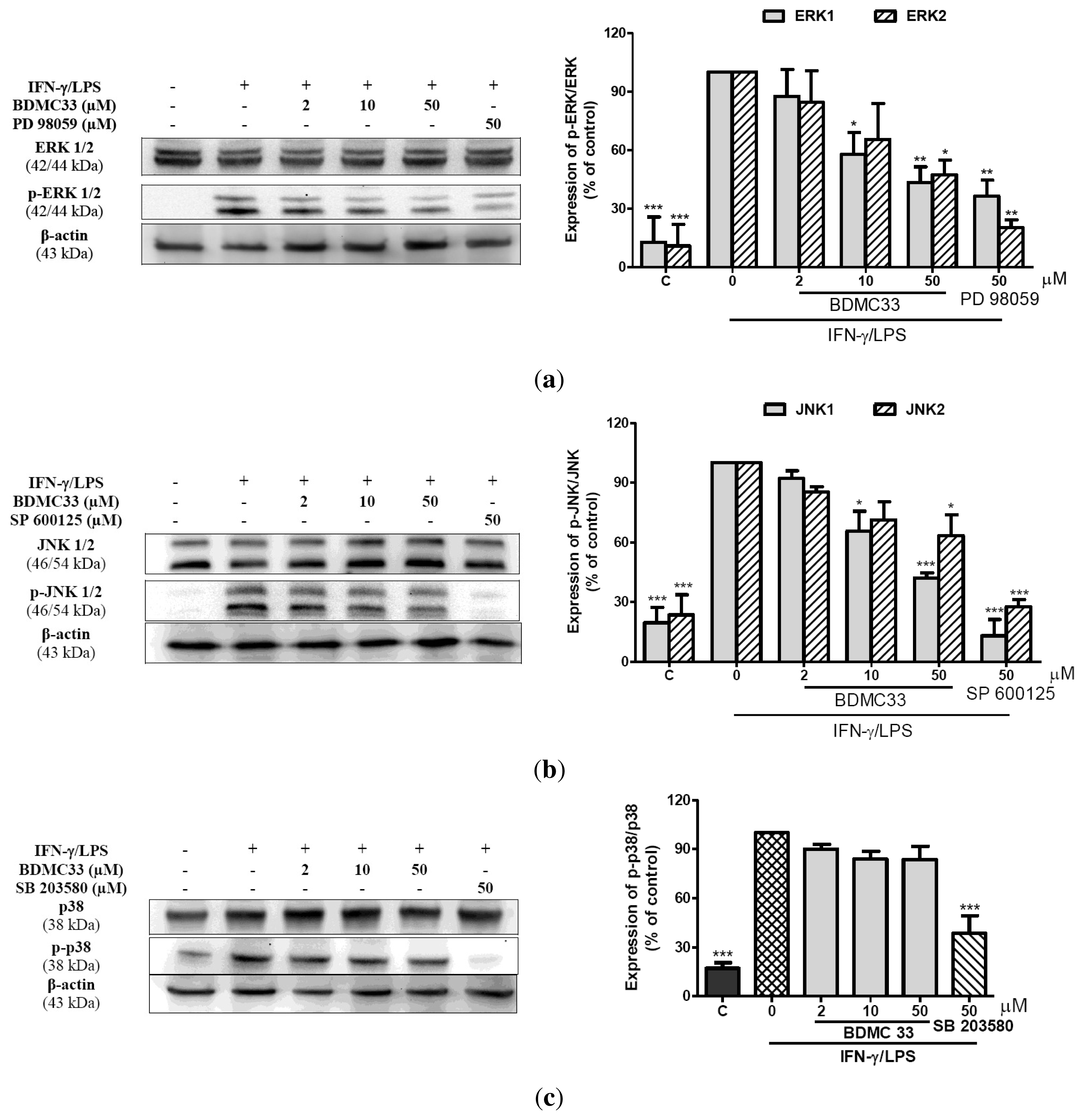
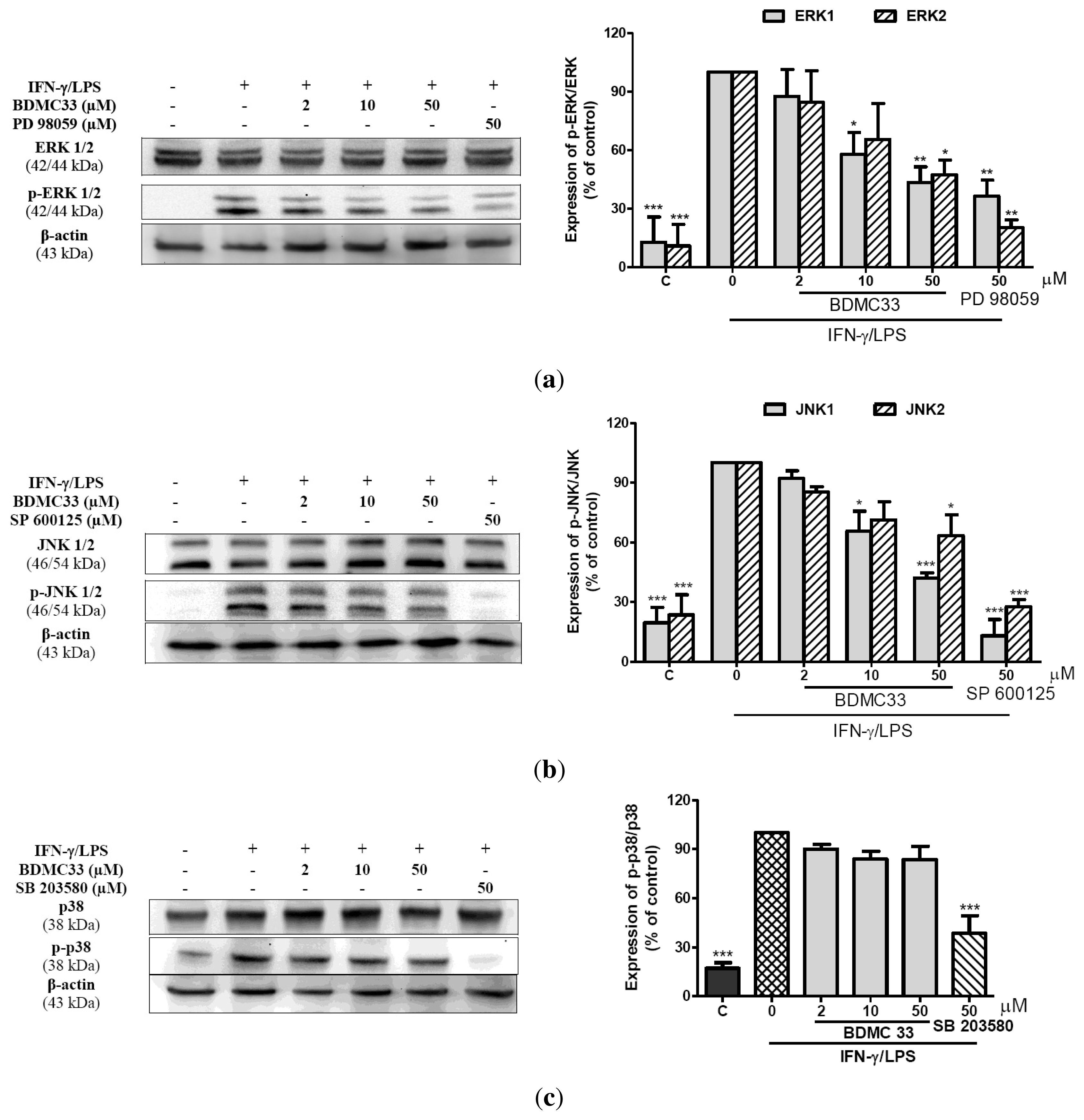
2.5. Effects of BDMC33 on MAPKs Phosphorylation
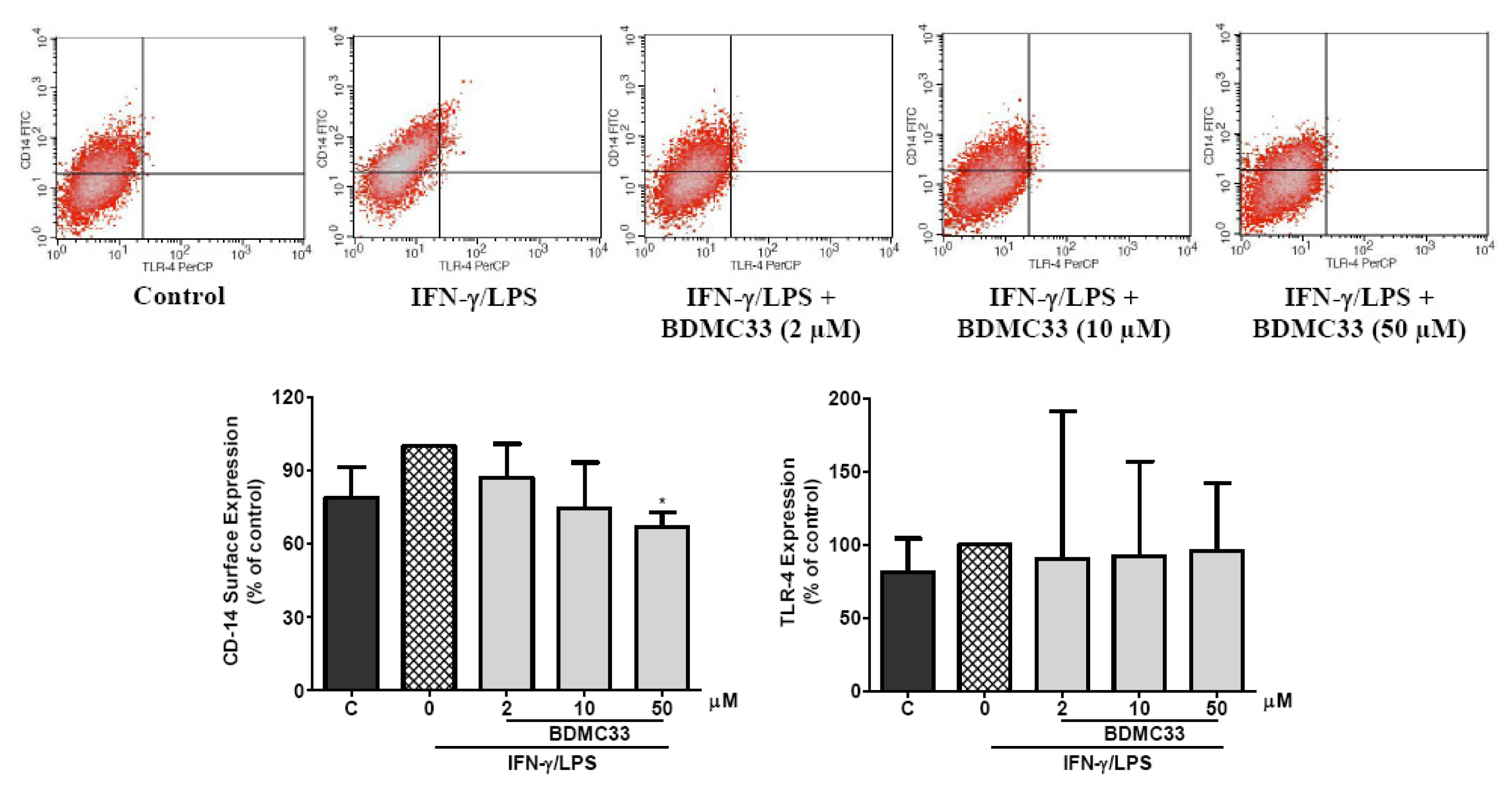
2.7. Effects of BDMC33 on CD-14 and TLR-4 Expression
2.8. Effects of BDMC33 on NO and TNF-α Production from Microglial BV-2 Cells
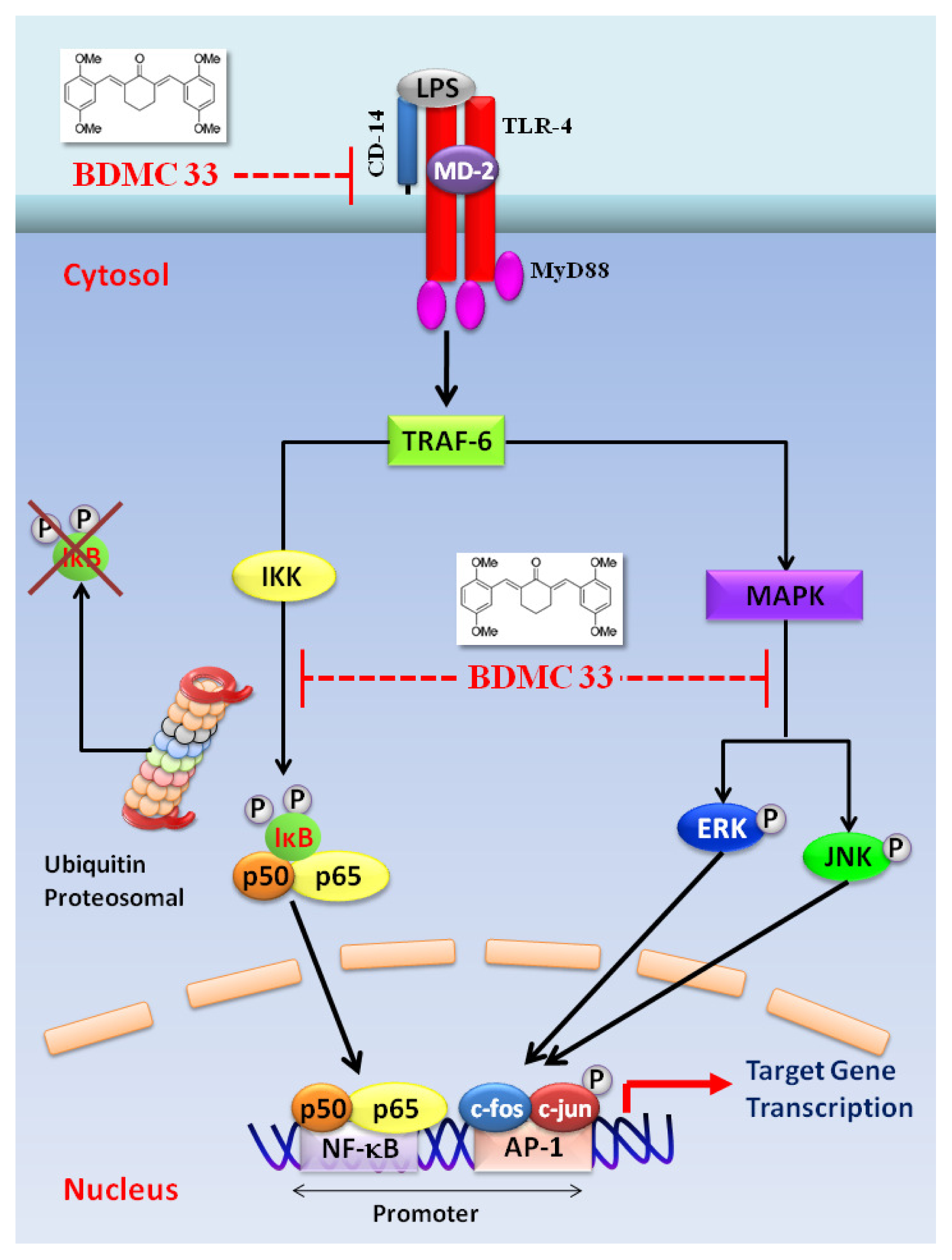
3. Discussion
4. Experimental
4.1. Materials
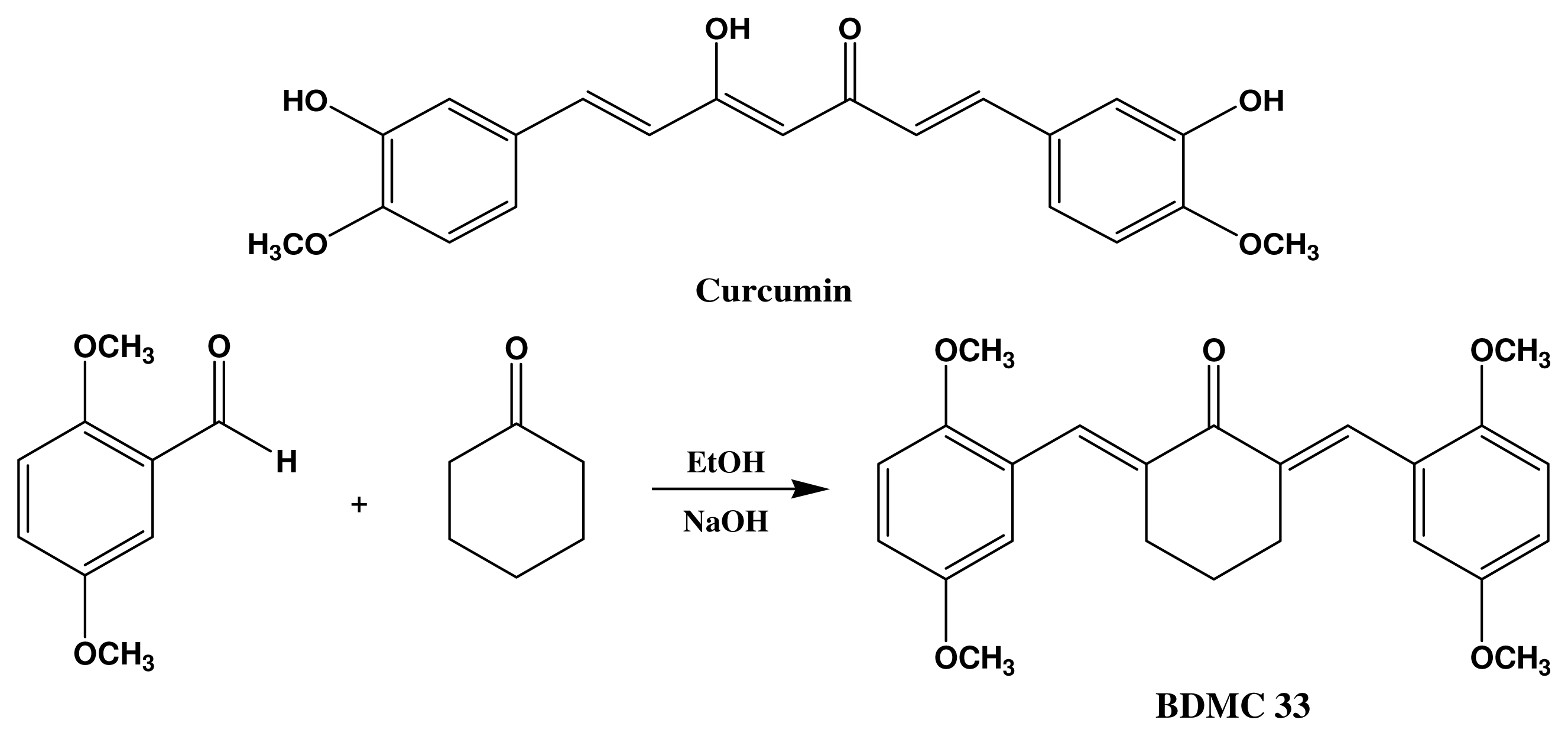
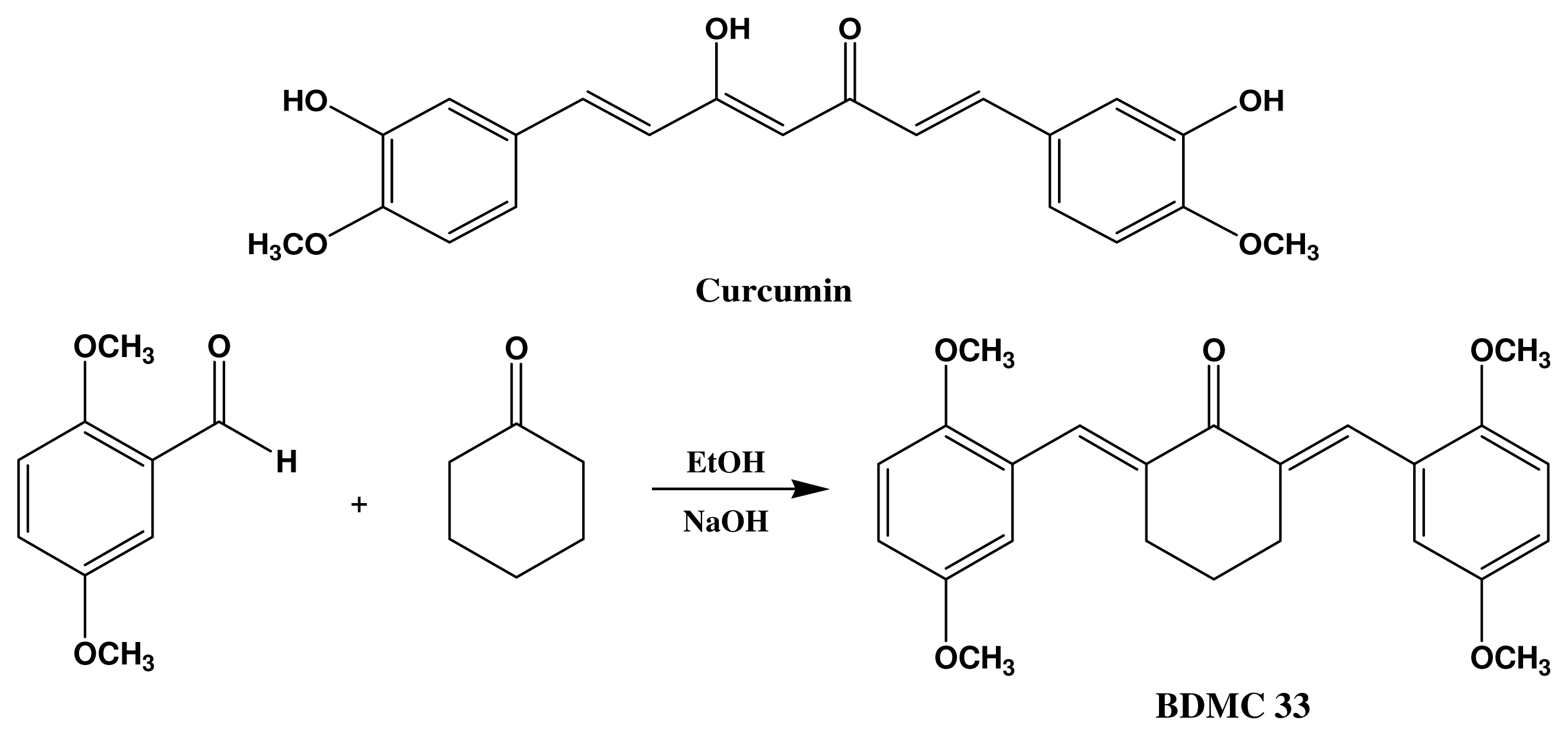
4.2. Synthesis of 2,6-Bis(2,5-dimethoxybenzylidene)Cyclohexanone (BDMC33)
4.3. Cell Culture
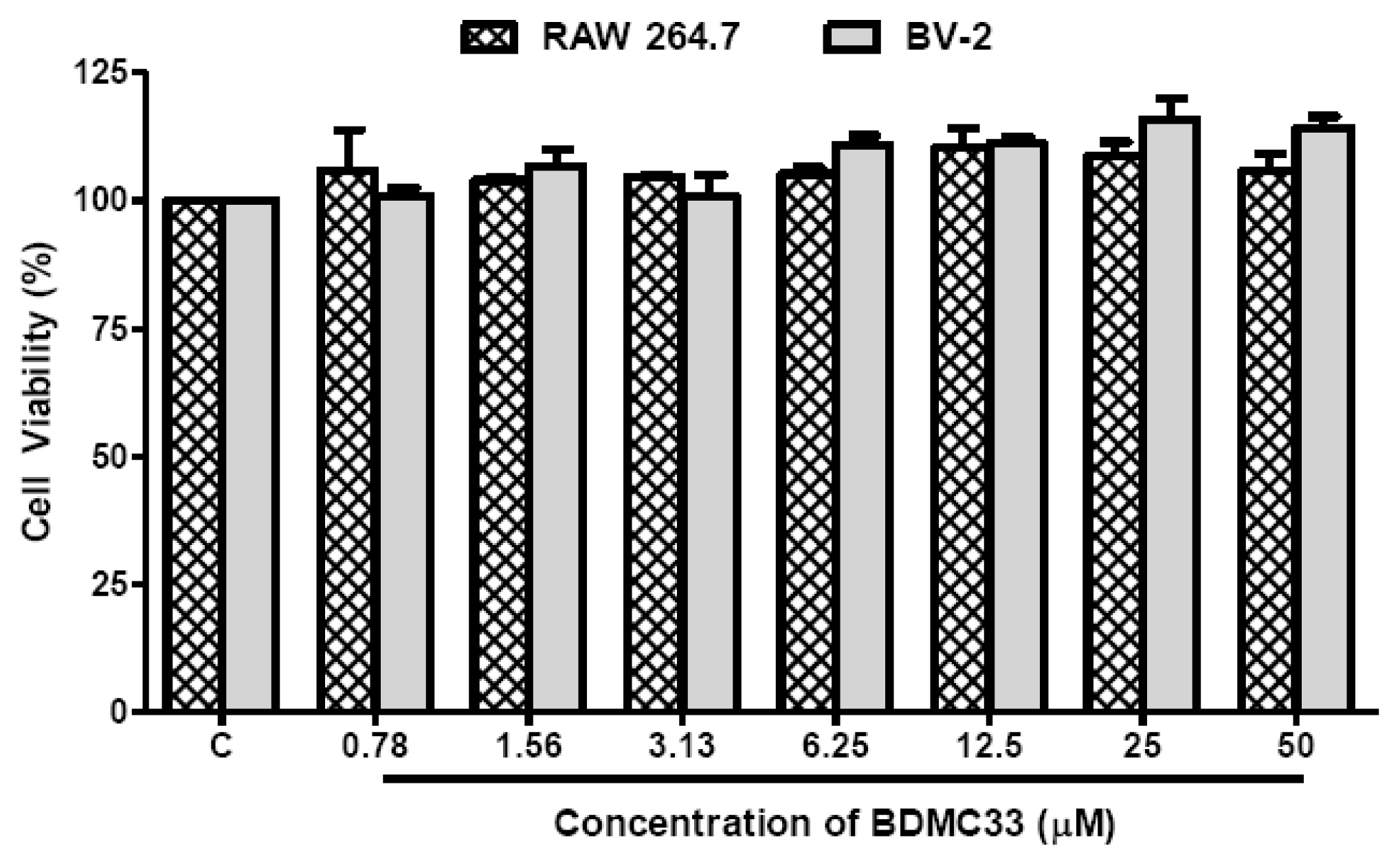
4.4. Cell Viability
4.5. Measurement of Nitric Oxide Production
4.6. Nitrite-Scavenging Activity
4.7. Indirect Determination of iNOS Activity
4.8. Cytokine Immunoassay
4.9. Semi-Quantitative RT-PCR
4.10. Western Blotting Analysis
4.11. Electrophoretic Mobility Shift Assay (EMSA)
4.12. Fluorescence Microscopic Analysis of NF-κB Translocation
4.13. Flow Cytometry Analysis of TLR-4 and CD-14
4.14. Statistical Analysis
5. Conclusion
Acknowledgments
References
- Rodriguez-Vita, J.; Lawrence, T. The resolution of inflammation and cancer. Cytokine Growth Factor Rev 2010, 2, 61–65. [Google Scholar]
- Schwacha, M.G. Macrophages and post-burn immune dysfunction. Burns 2003, 29, 1–14. [Google Scholar]
- Bowdish, D.M.E.; Loffredo, M.S.; Mukhopadhyay, S.; Mantovani, A.; Gordon, S. Macrophage receptors implicated in the “adaptive” form of innate immunity. Microbes Infect 2007, 9, 1680–1687. [Google Scholar]
- Chan, E.D.; Riches, D.W.H. IFN-γ + LPS induction of iNOS is modulated by ERK, JNK/SAPK, and p38mapk in a mouse macrophage cell line. Am. J. Physiol. Cell Physiol 2001, 280, 441–450. [Google Scholar]
- Held, T.K.; WeiHua, X.; Yuan, L.; Kalvakolanu, D.V.; Cross, A.S. Gamma interferon uugments macrophage activation by lipopolysaccharide by two distinct mechanisms, at the signal transduction level and via an autocrine mechanism involving tumor necrosis factor alpha and interleukin-1. Infect. Immun 1999, 67, 206–212. [Google Scholar]
- Dobrovolskaia, M.A.; Vogel, S.N. Toll receptors, CD14, and macrophage activation and deactivation by LPS. Microbes Infect 2002, 4, 903–914. [Google Scholar]
- Schroder, K.; Sweeta, M.J.; Hume, D.A. Signal integration between IFN-γ and TLR signaling pathways in macrophages. Immunobiology 2006, 211, 511–524. [Google Scholar]
- Takeda, K.; Akira, S. TLR signaling pathways. Semin. Immunol 2004, 16, 3–9. [Google Scholar]
- Aktan, F. iNOS-mediated nitric oxide production and its regulation. Life Sci 2004, 75, 639–653. [Google Scholar]
- Hofseth, L.J. Nitric oxide as a target of complementary and alternative medicines to prevent and treat inflammation and cancer. Cancer Lett 2008, 268, 10–30. [Google Scholar]
- Murtaugh, M.P.; Foss, D.L. Inflammatory cytokines and antigen presenting cell activation. Vet. Immun. Immunop 2002, 87, 109–121. [Google Scholar]
- Christ-Crain, M.; Opal, S.M. The role of biomarkers in the diagnosis and management of community-acquired pneumonia. Crit. Care 2010, 14, 203–214. [Google Scholar]
- Makarov, S.S. NF-kB as a therapeutic target in chronic inflammation: Recent advances. Mol. Med. Today 2000, 6, 441–448. [Google Scholar]
- Li, Q.; Verma, I.M. NF-κB Regulation in the immune system. Nat. Rev. Immunol 2002, 2, 725–734. [Google Scholar]
- Beauparlant, P.; John, H. Biological and Biochemical Inhibitors of the NF-KB/Rel Proteins and Cytokine Synthesis. Cytokine Growth Factor Rev 1996, 7, 175–190. [Google Scholar]
- May, M.J.; Ghosh, S. Signal transduction through NF-kB. Immunol. Today 1998, 19, 80–88. [Google Scholar]
- Roshak, A.K.; Callahan, J.F.; Blake, S.M. Small-molecule inhibitors of NF-κB for the treatment of inflammatory joint disease. Curr. Opin. Pharmacol 2002, 2, 316–321. [Google Scholar]
- Gabby Krens, S.F.; Spaink, H.P.; Snaar-Jagalska, B.E. Functions of the MAPK family in vertebrate-development. FEBS Lett 2006, 580, 4984–4990. [Google Scholar]
- English, J.M.; Cobb, M.H. Pharmacological inhibitors of MAPK pathways. Trends Pharmacol. Scis 2002, 23, 40–45. [Google Scholar]
- Kim, E.K.; Choi, E. Pathological roles of MAPK signaling pathways in human diseases. Biochem. Biophys. Acta 2010, 1802, 396–405. [Google Scholar]
- Anand, P.; Thomas, S.G.; Kunnumakkara, A.B.; Sundaram, C.; Harikumar, K.B.; Sung, B.; Tharakan, S.T.; Misra, K.; Priyadarsini, I.K.; Rajasekharan, K.N.; et al. Biological activities of curcumin and its analogues (Congeners) made by man and Mother Nature. Biochem. Pharmacol 2008, 76, 1590–1611. [Google Scholar]
- Aggarwal, B.B.; Harikumar, B.B. Potential therapeutic effects of curcumin, the anti-inflammatory agent, against neurodegenerative, cardiovascular, pulmonary, metabolic, autoimmune and neoplastic diseases. Int. J. Biochem. Cell B 2009, 41, 40–59. [Google Scholar]
- Chathoth, S.; Thayyullathil, F.; Galadari, S. Curcumin cell signaling: A possible target for chemotherapy. Curr. Trends Biotechnol. Pharm 2008, 2, 226–238. [Google Scholar]
- Liang, G.; Li, X.; Chen, L.; Yang, S.; Wu, X.; Studer, E.; Gurley, E.; Hylemon, P.B.; Ye, F.; Li, Y.; Zhou, H. Synthesis and anti-inflammatory activities of mono-carbonyl analogues of curcumin. Bioorg. Med. Chem. Lett 2008, 18, 1525–1529. [Google Scholar]
- Lee, K.H.; Abd Aziz, F.H.; Syahida, A.; Abas, F.; Shaari, K.; Israf, D.A.; Lajis, N.H. Synthesis and biological evaluation of curcumin-like diarylpentanoid analogues for anti-inflammatory, antioxidant and anti-tyrosinase activities. Eur. J. Med. Chem 2009, 44, 3195–3200. [Google Scholar]
- Pan, M.H.; Lin-Shiau, S.Y.; Lin, J.K. Comparative studies on the suppression of nitric oxide synthase by curcumin and its hydrogenated metabolites through down-regulation of IκB kinase and NFκB activation in macrophages. Biochem. Pharmacol 2000, 60, 1665–1676. [Google Scholar]
- Camacho-Barquero, L.; Villegas, I.; Sánchez-Calvo, J.M.; Talero, E.; Sánchez-Fidalgo, S.; Motilva, V.; Alarcón de la Lastra, C. Curcumin, a Curcuma longa constituent, acts on MAPK p38 pathways modulating COX-2 and iNOS expression in chronic experimental colitis. Int. J. Immunopharm 2007, 7, 333–342. [Google Scholar]
- Gafner, S.; Lee, S.K.; Cuendet, M.; Barthelemy, S.; Vergnes, L.; Labidalle, S.; Mehta, R.; Boone, D.W.; Pezzuto, J.M. Biologic evaluation of curcumin and structural derivatives in cancer chemoprevention model systems. Phytochemistry 2004, 65, 2849–2859. [Google Scholar]
- Shealy, D.J.; Wooley, P.H.; Emmell, E.; Volk, A.; Rosenberg, A.; Treacy, G.; Wagner, C.L.; Mayton, L.; Griswold, D.E.; Song, X.R. Anti-TNF-α antibody allows healing of joint damage in polyarthritic transgenic mice. Arthritis Res 2002, 4, R7–R14. [Google Scholar]
- Alten, R.; Gram, H.; Joosten, L.A.; van der Berg, W.B.; Sieper, J.; Wassenberg, S.; Burmester, G.; Riel, P.V.; Diaz-Lorente, M.; Bruin, G.L.M.; et al. The human anti-IL-1β monoclonal antibody ACZ885 is effective in joint inflammation models in mice and in a proof-of-concept study in patients with rheumatoid arthritis. Arthritis Res. Ther 2008, 10, R67–R76. [Google Scholar]
- Moon, D.O.; Kim, M.O.; Choi, Y.H.; Park, Y.M.; Kim, G.Y. Cucurmin attenuates inflammatory response in IL-1β-induced human synovial fibroblast and collagen-induced arthritis in mouse model. Int. J. Immunopharm 2010, 10, 605–610. [Google Scholar]
- Kleinert, H.; Pautz, A.; Linker, K.; Schwarz, P.M. Regulation of the expression of inducible nitric oxide synthase. Eur. J. Pharmacol 2004, 500, 255–266. [Google Scholar]
- Liu, H.; Sidiropoulos, P.; Song, G.; Pagliari, L.J.; Birrer, M.J.; Stein, B.; Anrather, J.; Pope, R.M. TNF-α gene expression in macrophages: Regulation by NF-κB is independent of c-Jun or C/EBPβ. J. Immunol 2000, 164, 4277–4285. [Google Scholar]
- Zhang, Y.; Saccani, S.; Shin, H.; Nikolajczyk, B.S. Dynamic protein associations define two phases of IL-1β transcriptional activation. J. Immunol 2008, 181, 503–512. [Google Scholar]
- Jung, K.K.; Lee, H.S.; Cho, J.Y.; Shin, W.C.; Rhee, M.H.; Kim, T.G.; Kang, J.H.; Kim, S.H.; Hong, S.; Kang, S.Y. Inhibitory effect of curcumin on nitric oxide production from lipopolysaccharide-activated primary microglia. Life Sci 2006, 79, 2022–2031. [Google Scholar]
- Lim, J.H.; Kwon, T.K. Curcumin inhibits phorbol myristate acetate (PMA)-induced MCP-1 expression by inhibiting ERK and NF-κB transcriptional activity. Food Chem. Toxicol 2010, 48, 47–52. [Google Scholar]
- Kamijo, R.; Harada, H.; Matsuyama, T.; Bosland, M.; Gerecitano, J.; Shapiro, D.; Le, J.; Koh, S.I.; Kimura, T.; Green, S.J. Requirement for transcription factor IRF-1 in NO synthase induction in macrophages. Science 1994, 263, 1612–1615. [Google Scholar]
- Guo, Z.; Shao, L.; Feng, X.; Reid, K.; Marderstein, E.; Nakao, A.; Geller, D.A. A critical role for C/EBPβ binding to the AABS promoter response element in the human iNOS gene. FASEB J 2003, 17, 1718–1720. [Google Scholar]
- Rossi, A.; Kapahi, P.; Natoli, G.; Takahashi, T.; Chen, Y.; Karin, M.; Santoro, M.G. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature 2000, 403, 103–118. [Google Scholar]
- Reynaert, N.L.; Ckless, K.; Korn, S.H.; Vos, N.; Guala, A.S.; Wouters, E.F.M.; van der Vliet, A.; Janssen-Heininger, Y.M. Nitric oxide represses inhibitory kappaB kinase through S-nitrosylation. Proc. Natl. Acad. Sci. USA 2004, 101, 8945–8950. [Google Scholar]
- Kapahi, P.; Takahashi, T.; Natoli, G.; Adams, S.R.; Chen, Y.; Tsien, R.Y.; Karin, M. Inhibition of NF-κB activation by arsenite through reaction with a critical cysteine in the activation loop of IkB kinase. J. Biol. Chem 2000, 275, 36062–36066. [Google Scholar]
- Jeon, K.I.; Byun, M.S.; Jue, D.M. Gold compound auranofin inhibits IκB kinase (IKK) by modifying Cys-179 of IKKβ subunit. Exp. Mol. Med 2003, 35, 61–66. [Google Scholar]
- Conroy, J.L.; Seto, C.T. Demonstration by 13C NMR Studies that tetrahydropyranone-based inhibitors bind to cysteine proteases by reversible formation of a hemithioketal adduct. J. Org. Chem 1998, 63, 2367–2370. [Google Scholar]
- Bonvina, C.; Guillona, A.; van Bemmelena, M.X.; Gerwinsb, P.; Johnsonc, G.L.; Widmann, C. Role of the amino-terminal domains of MEKKs in the activation of NFκB and MAPK pathways and in the regulation of cell proliferation and apoptosis. Cell Signal 2002, 14, 123–131. [Google Scholar]
- Zhao, Q.; Lee, F.S. Mitogen-activated protein kinase/ERK kinase kinases 2 and 3 activate nuclear factor-κB through IκB kinase-α and IκB kinase-β. J. Org. Chem 1999, 274, 8355–8358. [Google Scholar]
- Xue, F.; Seto, C.T. A comparison of cyclohexanone and tetrahydro-4H-thiopyran-4-one 1,1-dioxide as pharmacophores for the design of peptide-based inhibitors of the serine protease plasmin. J. Org. Chem 2005, 70, 8309–8321. [Google Scholar]
- Palusiaka, M.; Grabowski, S.J. Methoxy group as an acceptor of proton in hydrogen bonds. J. Mol. Struct 2002, 642, 97–104. [Google Scholar]
- Pae, H.O.; Jeong, S.O.; Kim, H.S.; Kim, S.H.; Song, Y.S.; Kim, S.K.; Chai, K.Y.; Chung, H.T. Dimethoxycurcumin, a synthetic curcumin analogue with higher metabolic stability, inhibits NO production, inducible NO synthase expression and NF-kappaB activation in RAW264.7 macrophages activated with LPS. Mol. Nutr. Food Res 2008, 52, 1082–1091. [Google Scholar]
- Thorgersen, E.B.; Hellerud, B.C.; Nielsen, E.W.; Barratt-Due, A.; Fure, H.; Lindstad, J.K.; Pharo, A.; Fosse, E.; Tønnessen, T.I.; Johansen, H.T.; et al. CD14 inhibition efficiently attenuates early inflammatory and hemostatic responses in Escherichia coli sepsis in pigs. FASEB J 2010, 24, 712–722. [Google Scholar]
- Genth-Zotz, S.; von Haehling, S.; Bolger, A.P.; Kalra, P.R.; Wensel, R.; Coats, A.J.S.; Volk, H.D.; Anker, S.D. The anti-CD14 antibody IC14 suppresses ex vivo endotoxin stimulated tumor necrosis factor-alpha in patients with chronic heart failure. Eur. J. Heart Fail 2006, 8, 366–372. [Google Scholar]
- Verbon, A.; Dekkers, P.E.P.; Hove, T.T.; Hack, C.E.; Pribble, J.P.; Turner, T.; Souza, S.; Axtelle, T.; Hoek, F.J.; van Deventer, S.J.H.; et al. IC14, an Anti-CD14 antibody, inhibits endotoxin-mediated symptoms and inflammatory responses in humans. J. Immunol 2001, 166, 3599–3605. [Google Scholar]
- Mir, M.; Tolosa, L.; Asensio, V.J.; Lladó, J.; Olmos, G. Complementary roles of tumor necrosis alpha and interferon gamma in inducible microglial nitric oxide generation. J. Neuroimmunol 2008, 204, 101–109. [Google Scholar]
- Wang, M.J.; Kuo, J.S.; Lee, W.W.; Huang, H.Y.; Chen, W.F.; Lin, S.Z. Translational event mediates differential production of tumor necrosis factor-alpha in hyaluronan-stimulated microglia and macrophages. J. Neurochem 2006, 97, 857–871. [Google Scholar]
- Watters, J.J.; Sommer, J.A.; Pfeiffer, Z.A.; Prabhu, U.; Guerra, A.N.; Bertics, P.J. A differential role for the mitogen activated protein kinases in lipopolysaccharide signaling: The MEK/ERK pathway is not essential for nitric oxide and interleukin 1β production. J. Biol. Chem 2002, 277, 9077–9087. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Gen Bank No. | Base Pair | Primer | |
|---|---|---|---|---|
| TNF-α | NM_013693 | 324 | Forward | 5′-ACGGCATGGATCTCAAAGAC-3′ |
| Reverse | 5′-CGGACTCCGCAAAGTCTAAG-3′ | |||
| IL-1β | NM_008361 | 497 | Forward | 5′-TGACGTTCCCATTAGACAGC-3′ |
| Reverse | 5′-TGGGGAAGGCATTAGAAACA-3′ | |||
| GADPH | M32599 | 269 | Forward | 5′-TGTTCCTACCCCCAATGTGT-3′ |
| Reverse | 5′-CCCTGTTGCTGTAGCCGTAT-3′ |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, K.-H.; Chow, Y.-L.; Sharmili, V.; Abas, F.; Alitheen, N.B.M.; Shaari, K.; Israf, D.A.; Lajis, N.H.; Syahida, A. BDMC33, A Curcumin Derivative Suppresses Inflammatory Responses in Macrophage-Like Cellular System: Role of Inhibition in NF-κB and MAPK Signaling Pathways. Int. J. Mol. Sci. 2012, 13, 2985-3008. https://doi.org/10.3390/ijms13032985
Lee K-H, Chow Y-L, Sharmili V, Abas F, Alitheen NBM, Shaari K, Israf DA, Lajis NH, Syahida A. BDMC33, A Curcumin Derivative Suppresses Inflammatory Responses in Macrophage-Like Cellular System: Role of Inhibition in NF-κB and MAPK Signaling Pathways. International Journal of Molecular Sciences. 2012; 13(3):2985-3008. https://doi.org/10.3390/ijms13032985
Chicago/Turabian StyleLee, Ka-Heng, Yuh-Lit Chow, Vidyadaran Sharmili, Faridah Abas, Noorjahan Banu Mohamed Alitheen, Khozirah Shaari, Daud Ahmad Israf, Nordin Haji Lajis, and Ahmad Syahida. 2012. "BDMC33, A Curcumin Derivative Suppresses Inflammatory Responses in Macrophage-Like Cellular System: Role of Inhibition in NF-κB and MAPK Signaling Pathways" International Journal of Molecular Sciences 13, no. 3: 2985-3008. https://doi.org/10.3390/ijms13032985
APA StyleLee, K.-H., Chow, Y.-L., Sharmili, V., Abas, F., Alitheen, N. B. M., Shaari, K., Israf, D. A., Lajis, N. H., & Syahida, A. (2012). BDMC33, A Curcumin Derivative Suppresses Inflammatory Responses in Macrophage-Like Cellular System: Role of Inhibition in NF-κB and MAPK Signaling Pathways. International Journal of Molecular Sciences, 13(3), 2985-3008. https://doi.org/10.3390/ijms13032985