Synthesis and Biological Activities of a 3'-Azido Analogue of Doxorubicin Against Drug-Resistant Cancer Cells


Abstract
:1. Introduction
2. Results and Discussion
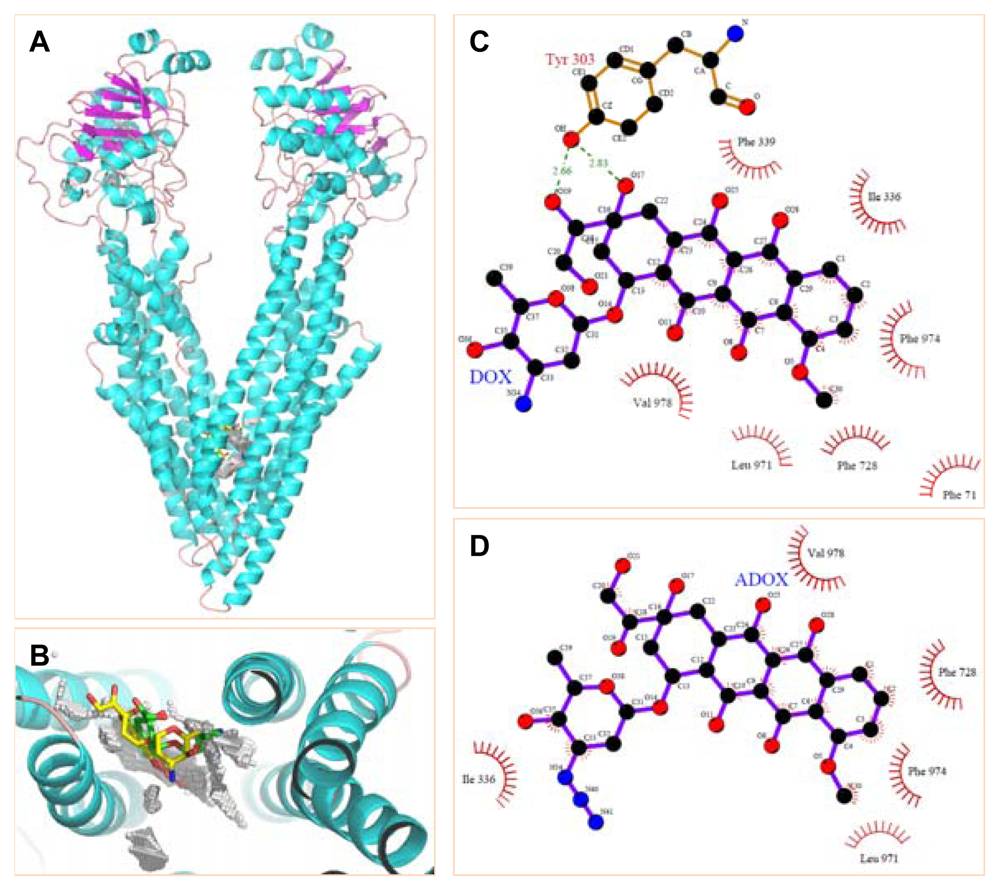
2.1. Molecular Modeling
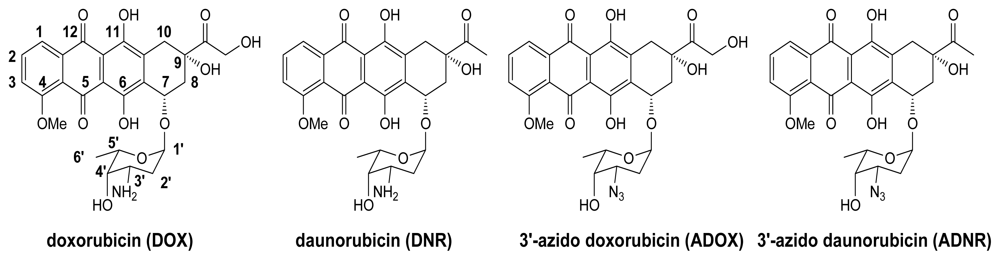
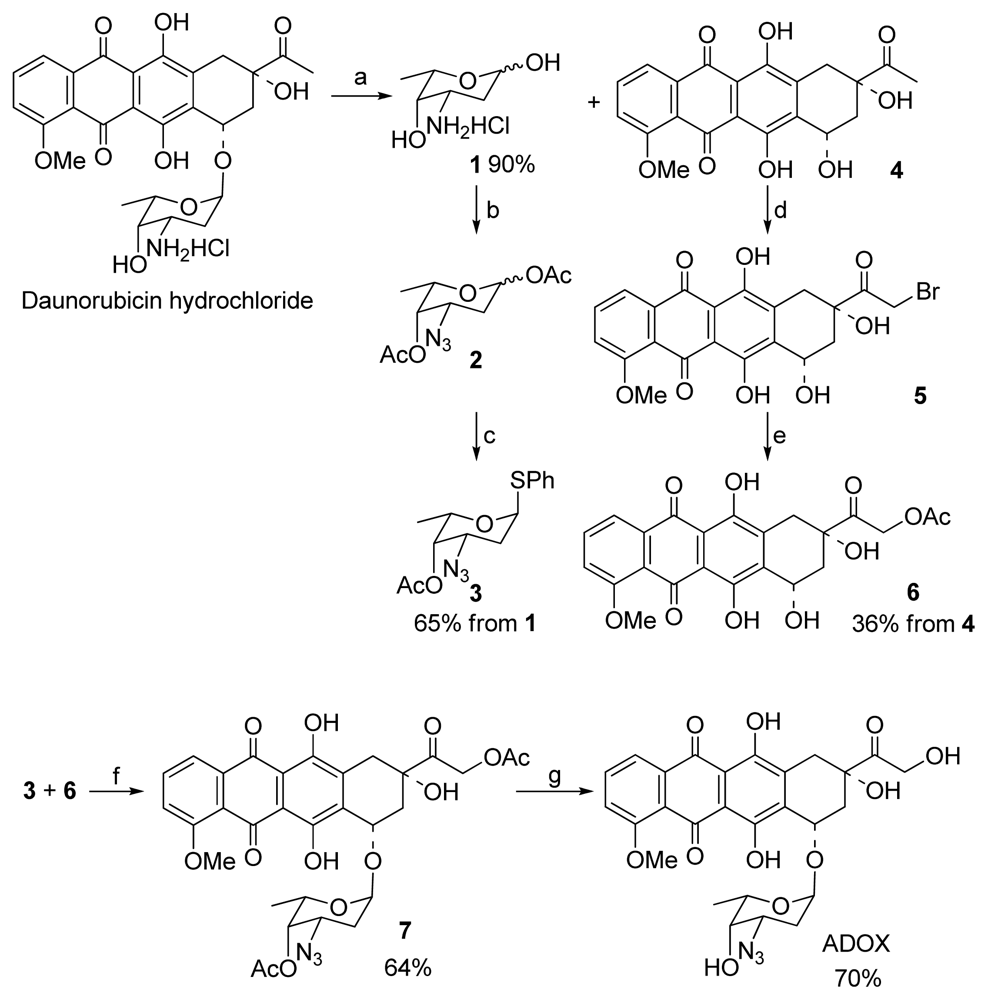
2.2. Synthesis of ADOX
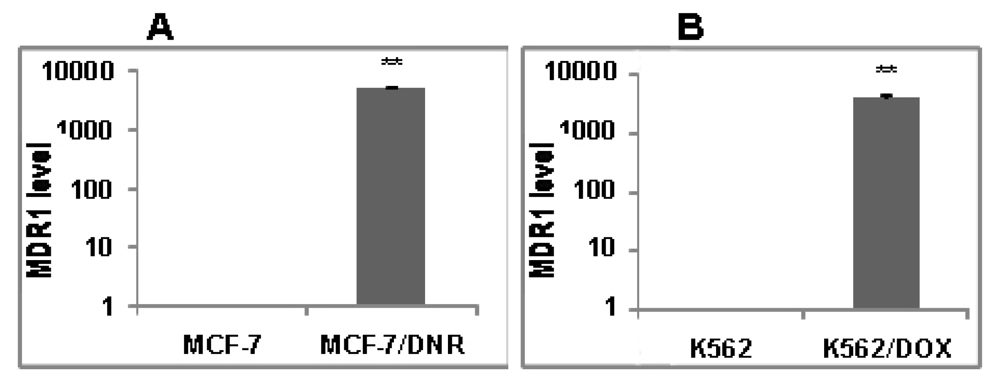
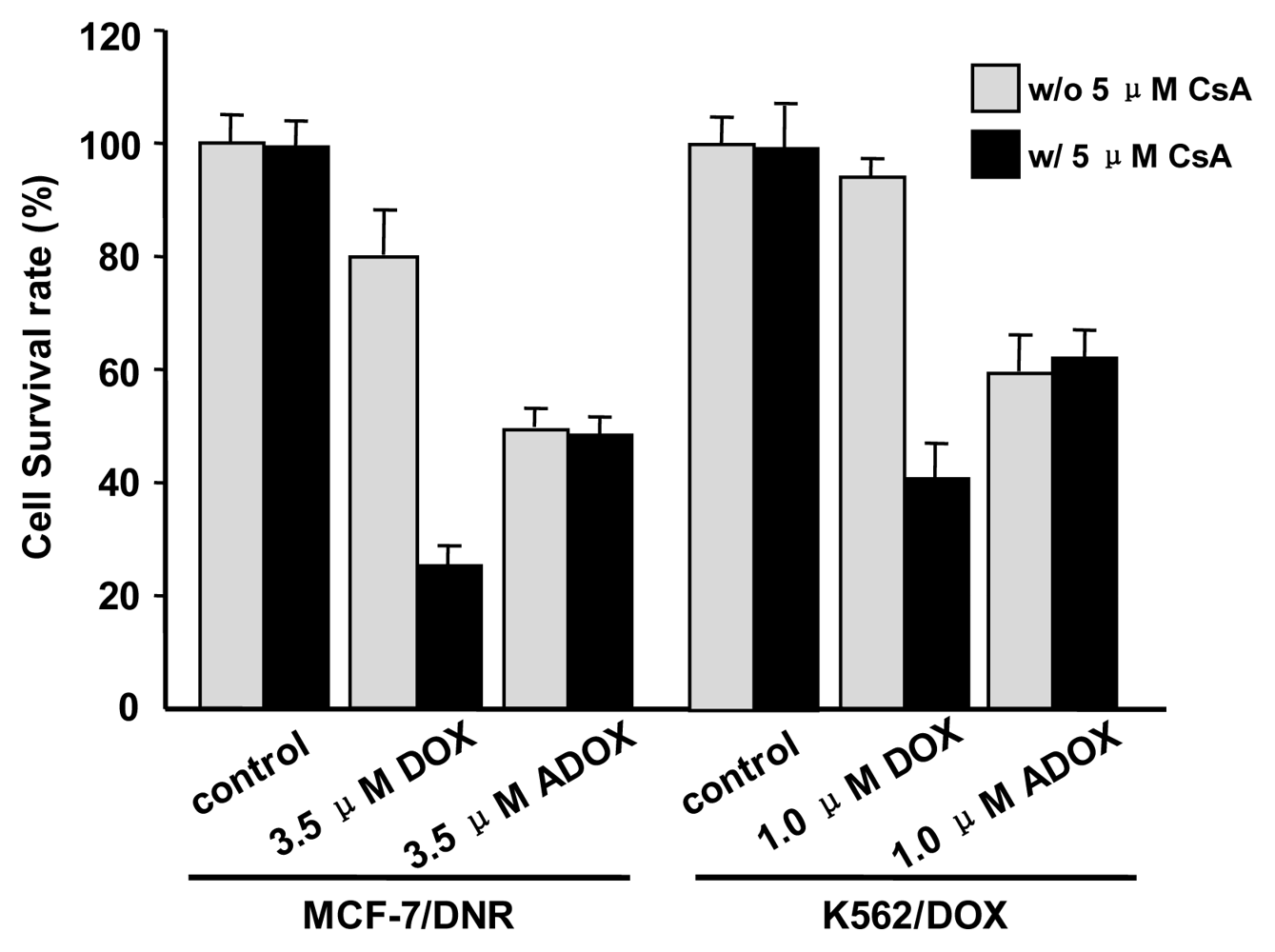
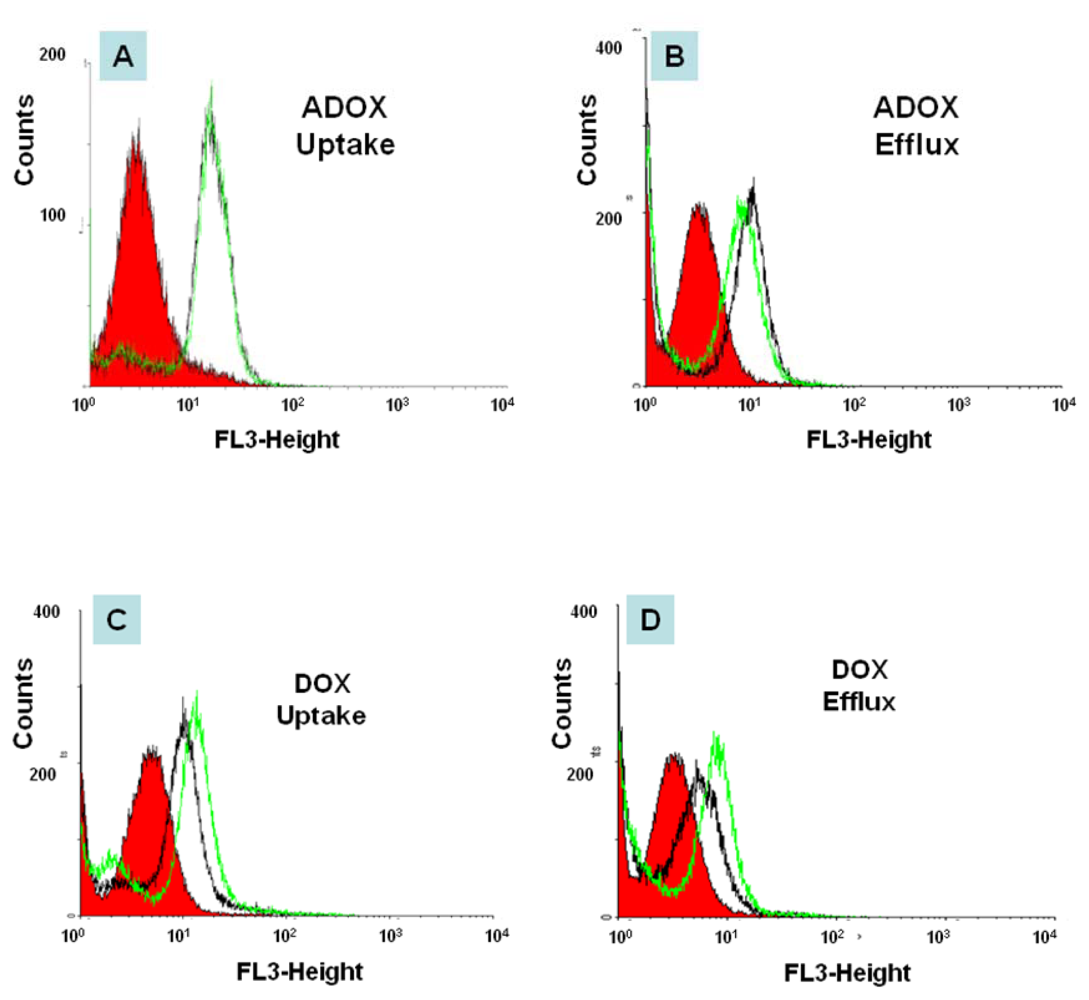
2.3. In Vitro Biological Tests of ADOX
3. Experimental Section
3.1. Synthesis
3.1.1. Glycosylation for Preparation of 1′,14-O-Diacetyl-3′-azido-DOX(7) (Scheme 1)
3.1.2. Deprotection of 7 to Title Compound ADOX (Scheme 1)
3.2. Biology
3.2.1. Cell Culture
3.2.2. MTS Assay of ADOX
3.2.3. RNA Extraction and Real-Time PCR Analysis
3.2.4. Flow Cytometry (FACS)
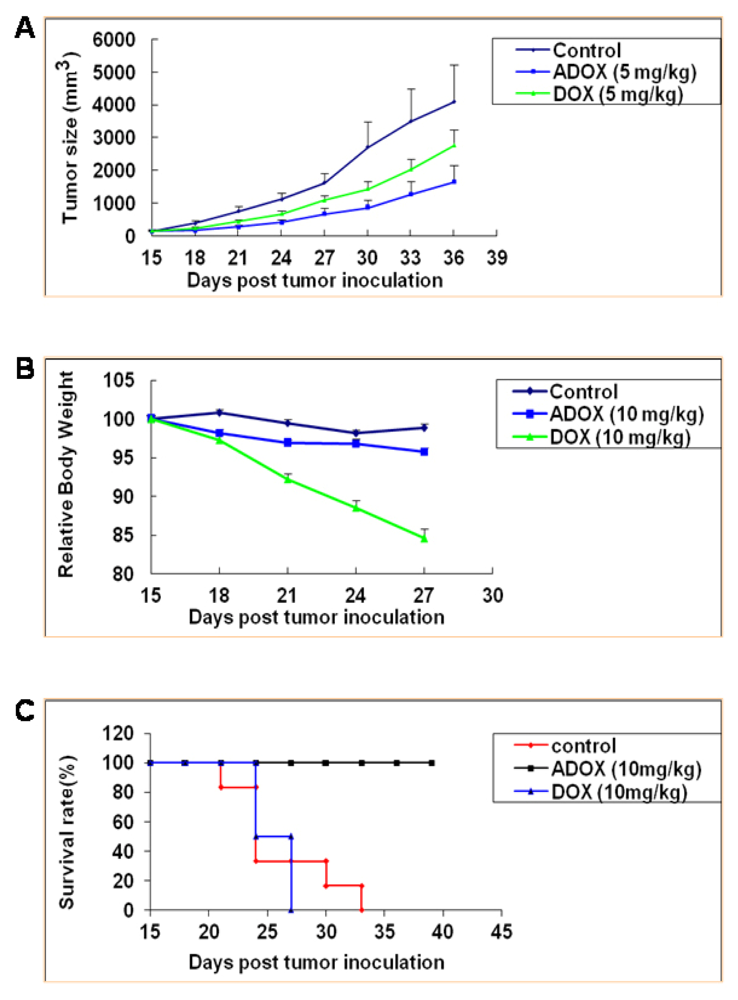
3.2.5. Anticancer Activity of ADOX Against Drug-Resistant Cancers in Xenograft Model
3.2.6. Acute Toxicity of ADOX Determined by Mouse LD50 Test
3.3. Computation
3.3.1. Molecular Docking of DOX and ADOX to P-gp
3.3.2. Statistics
4. Conclusions
Acknowledgments
References
- Gottesman, M.M. How cancer cells evade chemotherapy: Sixteenth Richard and Hinda Rosenthal foundation award lecture. Cancer Res 1993, 53, 747–754. [Google Scholar]
- Kaye, S.B. The multidrug resistance phenotype. Br. J. Cancer 1988, 58, 691–694. [Google Scholar]
- Lee, V.H. Membrane transporters. Eur. J. Pharm. Sci 2000, 11, S41–S50. [Google Scholar]
- Hosoya, K.I.; Kim, K.J.; Lee, V.H. Age-dependent expression of P-glycoprotein gp170 in Caco-2 cell monolayers. Pharm. Res 1996, 13, 885–890. [Google Scholar]
- Efferth, T. The human ATP-binding cassette transporter genes: From the bench to the bedside. Curr. Mol. Med 2001, 1, 45–65. [Google Scholar]
- Lepage, P.; Gros, P. Structural and functional aspects of P-glycoproteins and related transport proteins. Curr. Opin. Nephrol. Hypertens 1993, 2, 735–743. [Google Scholar]
- Germann, U.A. Molecular analysis of the multidrug transporter. Cytotechnology 1993, 12, 33–62. [Google Scholar]
- Arias, I.M.; Gatmaitan, Z.; Mazzanti, R.; Shu, H.; Kumamoto, Y. Structure and function of P-glycoprotein in the normal liver and intestine. Princess Takamatsu Symp 1990, 21, 229–239. [Google Scholar]
- Gate, L.; Couvreur, P.; Nguyen-Ba, G.; Tapiero, H. N-methylation of anthracyclines modulates their cytotoxicity and pharmacokinetic in wild type and multidrug resistant cells. Biomed. Pharmacother 2003, 57, 301–308. [Google Scholar]
- Fang, L.; Zhang, G.; Li, C.; Zheng, X.; Zhu, L.; Xiao, J.J.; Szakacs, G.; Nadas, J.; Chan, K.K.; Wang, P.G.; et al. Discovery of a daunorubicin analogue that exhibits potent antitumor activity and overcomes P-gp-mediated drug resistance. J. Med. Chem 2006, 49, 932–941. [Google Scholar]
- Battisti, R.F.; Zhong, Y.; Fang, L.; Gibbs, S.; Shen, J.; Nadas, J.; Zhang, G.; Sun, D. Modifying the sugar moieties of daunorubicin overcomes P-gp-mediated multidrug resistance. Mol. Pharm 2007, 4, 140–153. [Google Scholar]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [Google Scholar]
- Zhu, L.; Cao, X.; Chen, W.; Zhang, G.; Sun, D.; Wang, P.G. Syntheses and biological activities of daunorubicin analogs with uncommon sugars. Bioorg. Med. Chem 2005, 13, 6381–6387. [Google Scholar]
- Alper, P.B.; Hung, S.-C.; Wong, C.-H. Metal catalyzed diazo transfer for the synthesis of azides from amines. Tetrahedron Lett 1996, 37, 6029–6032. [Google Scholar]
- Rho, Y.S.; Kim, S.Y.; Kim, W.-J.; Yun, Y.K.; Sin, H.S.; Yoo, D.J. Convenient syntheses of daunomycinone-7-d-glucuronides and doxorubicinone-7-d-glucuronides. ChemInform 2005, 36. [Google Scholar] [CrossRef]
- Hou, D.; Lowary, T.L. Recent advances in the synthesis of 2-deoxy-glycosides. Carbohydr. Res 2009, 344, 1911–1940. [Google Scholar]
- Marzabadi, C.H.; Franck, R.W. The synthesis of 2-deoxyglycosides: 1988–1999. Tetrahedron 2000, 56, 8385–8417. [Google Scholar]
- Lear, M.J.; Yoshimura, F.; Hirama, M. A direct and efficient α-selective glycosylation protocol for the kedarcidin sugar, l-mycarose: AgPF6 as a remarkable activator of 2-deoxythioglycosides. Angew. Chem. Int. Edit 2001, 40, 946–949. [Google Scholar]
- Zhang, G.; Fang, L.; Zhu, L.; Aimiuwu, J.E.; Shen, J.; Cheng, H.; Muller, M.T.; Lee, G.E.; Sun, D.; Wang, P.G. Syntheses and biological activities of disaccharide daunorubicins. J. Med. Chem 2005, 48, 5269–5278. [Google Scholar]
- Fang, L.; Zhang, G.; Li, C.; Zheng, X.; Zhu, L.; Xiao, J.J.; Szakacs, G.; Nadas, J.; Chan, K.K.; Wang, P.G.; et al. Discovery of a daunorubicin analogue that exhibits potent antitumor activity and overcomes P-gp-mediated drug resistance. J. Med. Chem 2006, 49, 932–941. [Google Scholar]
- Deprez-De Campeneere, D.; Baurain, R.; Huybrechts, M.; Trouet, A. Comparative study in mice of the toxicity, pharmacology, and therapeutic activity of daunorubicin-DNA and doxorubicin-DNA complexes. Cancer Chemother. Pharmacol 1979, 2, 25–30. [Google Scholar]
- WinNonlin Software, version 4.1; Pharsight Corp.: Cary NC, USA, 1998.
- Origin Software, version 7.5; OriginLab Corporation: Northampton, UK, 2004.
- DOCK Program, version 6.5; University of California: San Francisco, CA, USA, 2011.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell lines and DRI a | DOX | ADOX |
|---|---|---|
| MCF-7 cell line | 0.11 | 2.2 |
| MCF-7/DNR cell line b | 20 | 3.5 |
| DRIa | 182 | 1.6 |
| K562 cell line | 0.080 | 0.64 |
| K562/DOX cell line b | 27 | 0.87 |
| DRI a | 338 | 1.4 |
| CV-1 cell line c | 25 | 2.8 |
| 1.5 μM-CPT-CV-1 cell line d | > 10 | 6.6 |
| 4 μM-CPT-CV-1 cell line d | > 10 | 2.6 |
| 3 μM-mAMSA-CV-1 cell line d | > 10 | 2.4 |
| Dose (mg/kg) a | 120 | 84 | 59 | 41 | 29 | 20 |
|---|---|---|---|---|---|---|
| Number of mice in test b | 10 c | 10 | 10 | 10 | 10 | 10 |
| Number of dead mice | 9 | 7 | 6 | 6 | 4 | 0 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yu, S.; Zhang, G.; Zhang, W.; Luo, H.; Qiu, L.; Liu, Q.; Sun, D.; Wang, P.-G.; Wang, F. Synthesis and Biological Activities of a 3'-Azido Analogue of Doxorubicin Against Drug-Resistant Cancer Cells. Int. J. Mol. Sci. 2012, 13, 3671-3684. https://doi.org/10.3390/ijms13033671
Yu S, Zhang G, Zhang W, Luo H, Qiu L, Liu Q, Sun D, Wang P-G, Wang F. Synthesis and Biological Activities of a 3'-Azido Analogue of Doxorubicin Against Drug-Resistant Cancer Cells. International Journal of Molecular Sciences. 2012; 13(3):3671-3684. https://doi.org/10.3390/ijms13033671
Chicago/Turabian StyleYu, Shuwen, Guisheng Zhang, Wenpeng Zhang, Huanhua Luo, Liyun Qiu, Qingfeng Liu, Duxin Sun, Peng-George Wang, and Fengshan Wang. 2012. "Synthesis and Biological Activities of a 3'-Azido Analogue of Doxorubicin Against Drug-Resistant Cancer Cells" International Journal of Molecular Sciences 13, no. 3: 3671-3684. https://doi.org/10.3390/ijms13033671
APA StyleYu, S., Zhang, G., Zhang, W., Luo, H., Qiu, L., Liu, Q., Sun, D., Wang, P.-G., & Wang, F. (2012). Synthesis and Biological Activities of a 3'-Azido Analogue of Doxorubicin Against Drug-Resistant Cancer Cells. International Journal of Molecular Sciences, 13(3), 3671-3684. https://doi.org/10.3390/ijms13033671