Curcumin: Updated Molecular Mechanisms and Intervention Targets in Human Lung Cancer

Abstract
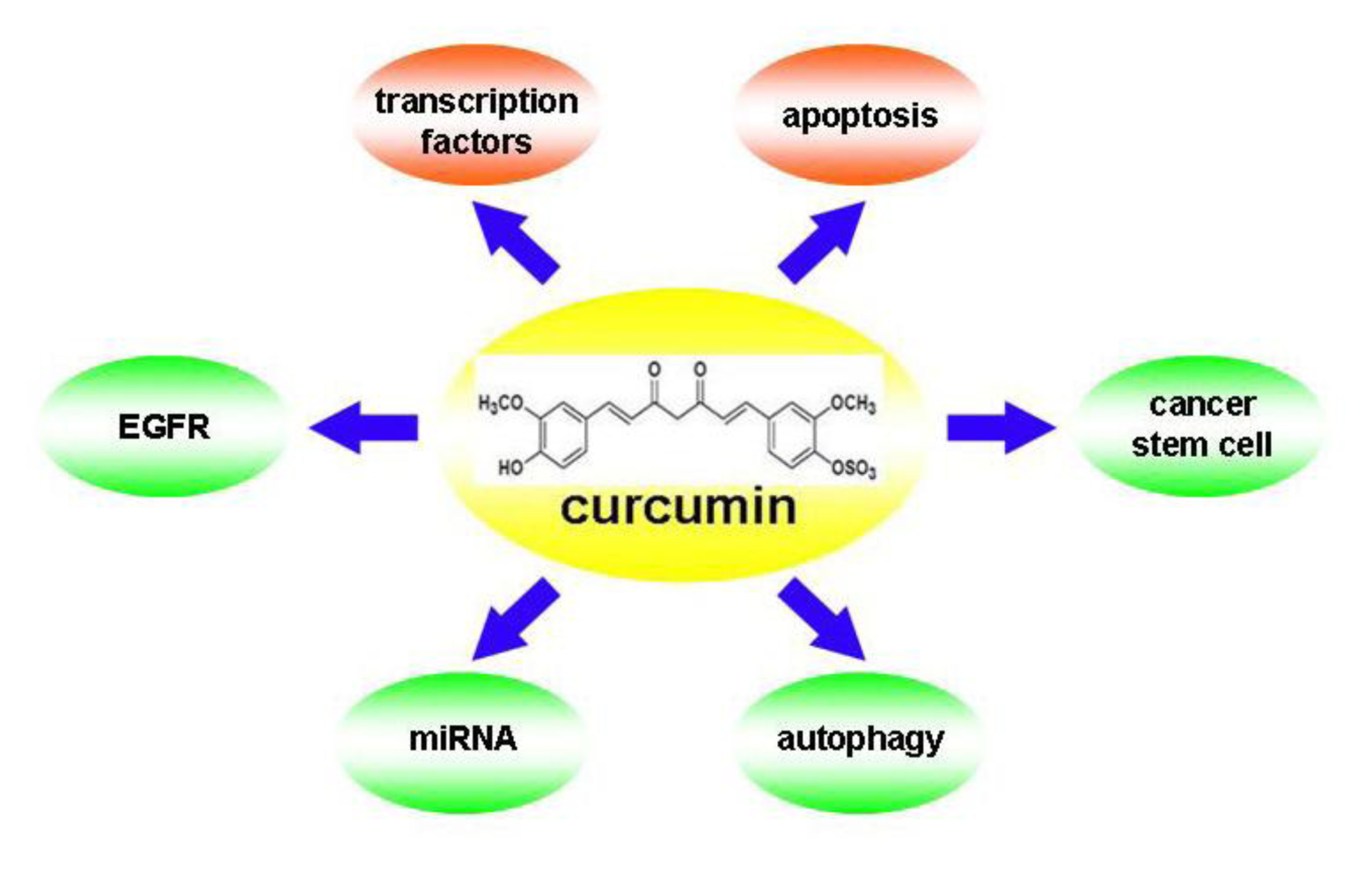
:1. Introduction
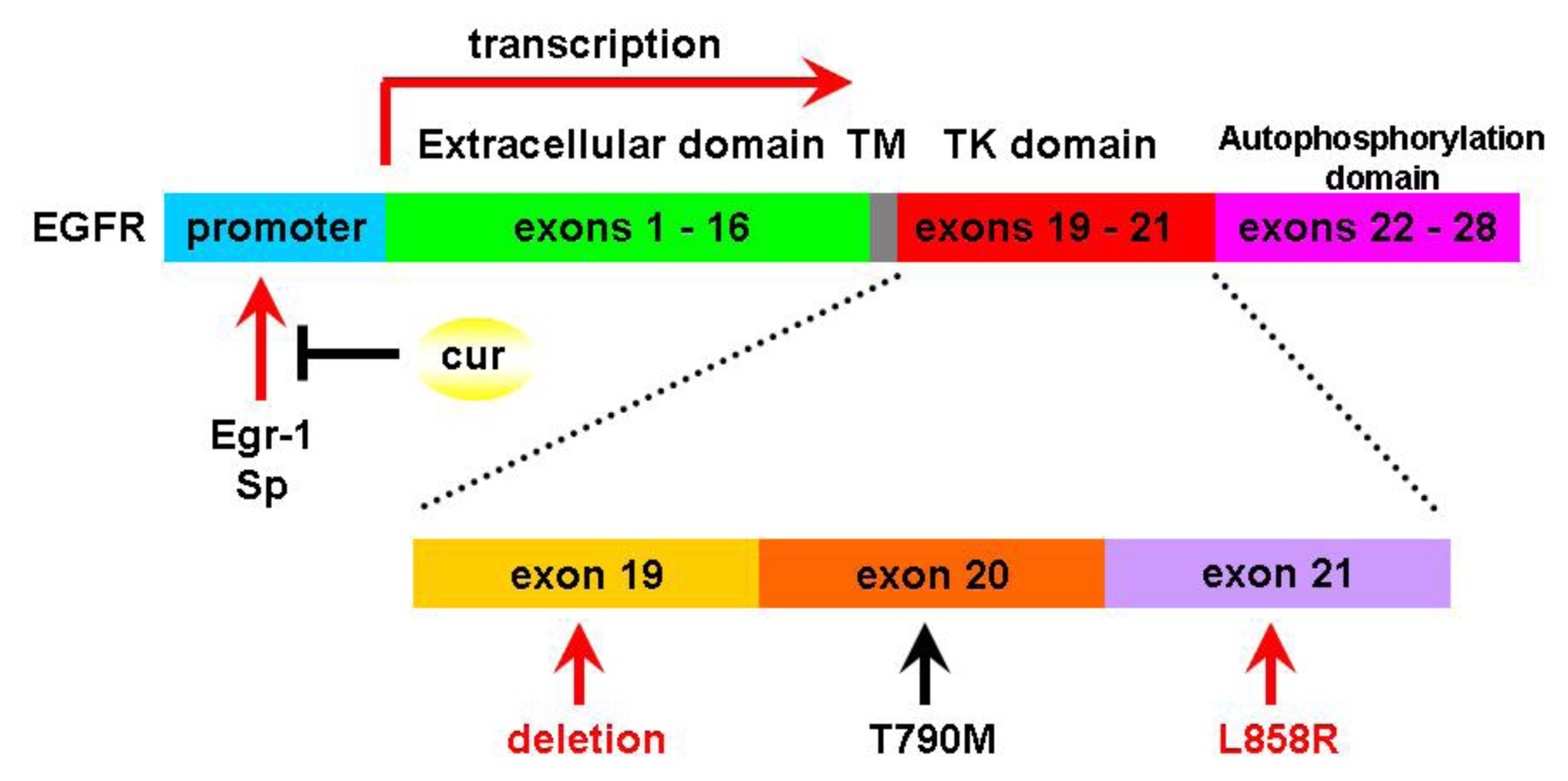
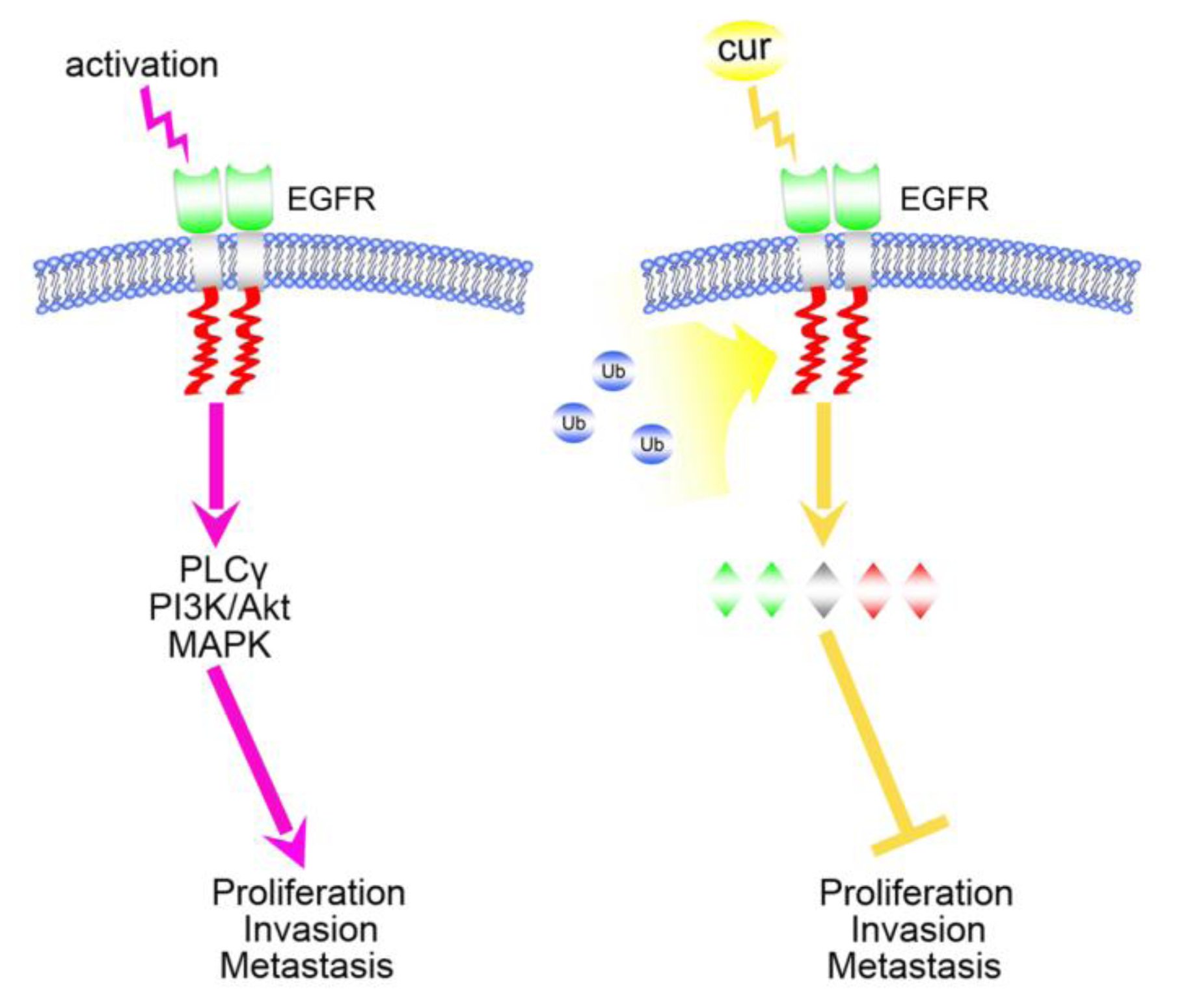
2. Curcumin and Acquired Epidermal Growth Factor Receptor (EGFR)-Tyrosine Kinase (TKIs) Resistance
3. Curcumin and Its Epigenetic Actions on MicroRNAs
4. Curcumin Induces Autophagy: A Double-Edged Sword in Cancer Therapy
5. Targeting Cancer Stem Cell
6. Perspectives and Directions
Acknowledgments
References
- Ramalingam, S.S.; Owonikoko, T.K.; Khuri, F.R. Lung cancer: New biological insights and recent therapeutic advances. CA Cancer J. Clin 2011, 61, 91–112. [Google Scholar]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin 2009, 59, 225–249. [Google Scholar]
- Cheng, A.L.; Hsu, C.H.; Lin, J.K.; Hsu, M.M.; Ho, Y.F.; Shen, T.S.; Ko, J.Y.; Lin, J.T.; Lin, B.R.; Ming-Shiang, W.; et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res 2001, 21, 2895–2900. [Google Scholar]
- Dhillon, N.; Aggarwal, B.B.; Newman, R.A.; Wolff, R.A.; Kunnumakkara, A.B.; Abbruzzese, J.L.; Ng, C.S.; Badmaev, V.; Kurzrock, R. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin. Cancer Res 2008, 14, 4491–4499. [Google Scholar]
- Cruz-Correa, M.; Shoskes, D.A.; Sanchez, P.; Zhao, R.; Hylind, L.M.; Wexner, S.D.; Giardiello, F.M. Combination treatment with curcumin and quercetin of adenomas in familial adenomatous polyposis. Clin. Gastroenterol. Hepatol 2006, 4, 1035–1038. [Google Scholar]
- Chen, J.; Tang, X.Q.; Zhi, J.L.; Cui, Y.; Yu, H.M.; Tang, E.H.; Sun, S.N.; Feng, J.Q.; Chen, P.X. Curcumin protects PC12 cells against 1-methyl-4-phenylpyridinium ion-induced apoptosis by bcl-2-mitochondria-ROS-iNOS pathway. Apoptosis 2006, 11, 943–953. [Google Scholar]
- Divya, C.S.; Pillai, M.R. Antitumor action of curcumin in human papillomavirus associated cells involves downregulation of viral oncogenes, prevention of NFκB and AP-1 translocation, and modulation of apoptosis. Mol. Carcinog 2006, 45, 320–332. [Google Scholar]
- Chanvorachote, P.; Pongrakhananon, V.; Wannachaiyasit, S.; Luanpitpong, S.; Rojanasakul, Y.; Nimmannit, U. Curcumin sensitizes lung cancer cells to cisplatin-induced apoptosis through superoxide anion-mediated Bcl-2 degradation. Cancer Invest 2009, 27, 624–635. [Google Scholar]
- Bava, S.V.; Puliappadamba, V.T.; Deepti, A.; Nair, A.; Karunagaran, D.; Anto, R.J. Sensitization of taxol-induced apoptosis by curcumin involves down-regulation of nuclear factor-kappaB and the serine/threonine kinase Akt and is independent of tubulin polymerization. J. Biol. Chem 2005, 280, 6301–6308. [Google Scholar]
- Yang, C.L.; Ma, Y.G.; Xue, Y.X.; Liu, Y.Y.; Xie, H.; Qiu, G.R. Curcumin induces small cell lung cancer NCI-H446 cell apoptosis via the reactive oxygen species-mediated mitochondrial pathway and not the cell death receptor pathway. DNA Cell Biol 2012, 31, 139–150. [Google Scholar]
- Wu, S.H.; Hang, L.W.; Yang, J.S.; Chen, H.Y.; Lin, H.Y.; Chiang, J.H.; Lu, C.C.; Yang, J.L.; Lai, T.Y.; Ko, Y.C.; et al. Curcumin induces apoptosis in human non-small cell lung cancer NCI-H460 cells through ER stress and caspase cascade- and mitochondria-dependent pathways. Anticancer Res 2010, 30, 2125–2133. [Google Scholar]
- Sun, Y.; Ren, Y.; Fang, Z.; Li, C.; Fang, R.; Gao, B.; Han, X.; Tian, W.; Pao, W.; Chen, H.; Ji, H. Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J. Clin. Oncol 2010, 28, 4616–4620. [Google Scholar]
- Rosell, R.; Moran, T.; Queralt, C.; Porta, R.; Cardenal, F.; Camps, C.; Majem, M.; Lopez-Vivanco, G.; Isla, D.; Provencio, M.; et al. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med 2009, 361, 958–967. [Google Scholar]
- Sequist, L.V.; Martins, R.G.; Spigel, D.; Grunberg, S.M.; Spira, A.; Jänne, P.A.; Joshi, V.A.; McCollum, D.; Evans, T.L.; Muzikansky, A.; et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J. Clin. Oncol 2008, 26, 2442–2449. [Google Scholar]
- Schiller, J.H.; Harrington, D.; Belani, C.P.; Langer, C.; Sandler, A.; Krook, J.; Zhu, J.; Johnson, D.H. Eastern Cooperative Oncology Group. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N. Engl. J. Med. 2002, 346, 92–98. [Google Scholar]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005, 2. [Google Scholar] [CrossRef] [Green Version]
- Kosaka, T.; Yatabe, Y.; Endoh, H.; Yoshida, K.; Hida, T.; Tsuboi, M.; Tada, H.; Kuwano, H.; Mitsudomi, T. Analysis of epidermal growth factor receptor gene mutation in patients with nonsmall cell lung cancer and acquired resistance to gefitinib. Clin. Cancer Res 2006, 12, 5764–5769. [Google Scholar]
- Uramoto, H.; Sugio, K.; Oyama, T.; Sugaya, M.; Hanagiri, T.; Yasumoto, K. A resistance to gefitinib. Int. J. Clin. Oncol 2006, 11, 487–491. [Google Scholar]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med 2005, 352, 786–792. [Google Scholar]
- Engelman, J.A.; Zejnullahu, K.; Gale, C.M.; Gonzales, A.J.; Shimamura, T.; Zhao, F.; Vincent, P.W.; Naumov, G.N.; Bradner, J.E.; Althaus, I.W.; et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res 2007, 67, 11924–11932. [Google Scholar]
- Kwak, E.L.; Sordella, R.; Bell, D.W.; Godin-Heymann, N.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Driscoll, D.R.; Fidias, P.; Lynch, T.J.; et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. USA 2005, 102, 7665–7670. [Google Scholar]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.R.; Padera, R.F.; Shapiro, G.I.; Baum, A.; Himmelsbach, F.; et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. [Google Scholar]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol 2003, 4, 915–925. [Google Scholar]
- Matsumoto, K.; Nakamura, T. Hepatocyte growth factor and the Met system as a mediator of tumor-stromal interactions. Int. J. Cancer 2006, 119, 477–483. [Google Scholar]
- Matsubara, D.; Ishikawa, S.; Oguni, S.; Aburatani, H.; Fukayama, M.; Niki, T. Molecular predictors of sensitivity to the MET inhibitor PHA665752 in lung carcinoma cells. J. Thorac. Oncol 2010, 5, 1317–1324. [Google Scholar]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar]
- Yano, S.; Wang, W.; Li, Q.; Matsumoto, K.; Sakurama, H.; Nakamura, T.; Ogino, H.; Kakiuchi, S.; Hanibuchi, M.; Nishioka, Y.; et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res 2008, 68, 9479–9487. [Google Scholar]
- Engelman, J.A.; Zejnullahu, K.; Mitsuddomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar]
- Engelman, J.A.; Jänne, P.A. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin. Cancer Res 2008, 14, 2895–2899. [Google Scholar]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.V.; et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar]
- Shishodia, S.; Potdar, P.; Gairola, C.G.; Aggarwal, B.B. Curcumin (diferuloylmethane) down-regulates cigarette smoke-induced NF-kappaB activation through inhibition of IkappaBalpha kinase in human lung epithelial cells: Correlation with suppression of COX-2, MMP-9 and cyclin D1. Carcinogenesis 2003, 24, 1269–1279. [Google Scholar]
- Lin, S.S.; Laai, K.C.; Hsu, S.C.; Yang, J.S.; Kuo, C.L.; Lin, J.P.; Ma, Y.S.; Wu, C.C.; Chung, J.G. Curcumin inhibits the migration and invasion of human A549 lung cancer cells through the inhibition of matrix metalloproteinase-2 and −9 and Vascular Endothelial Growth Factor (VEGF). Cancer Lett 2009, 285, 127–133. [Google Scholar]
- Chen, L.; Tian, G.; Shao, C.; Cobos, E.; Gao, W. Curcumin modulates eukaryotic initiation factors in human lung adenocarcinoma epithelial cells. Mol. Biol. Rep 2010, 37, 3105–3110. [Google Scholar]
- Fu, S.; Kurzrock, R. Development of curcumin as an epigenetic agent. Cancer 2010, 116, 4670–4676. [Google Scholar]
- Lev-Ari, S.; Vexler, A.; Starr, A.; Ashkenazy-Voghera, M.; Greif, J.; Aderka, D.; Ben-Yosef, R. Curcumin augments gemcitabine cytotoxic effect on pancreatic addenocarcinoma cell lines. Cancer Invest 2007, 25, 411–418. [Google Scholar]
- Kunnumakkara, A.B.; Guha, S.; Krishnan, S.; Diagaradjane, P.; Gelovani, J.; Aggarwal, B.B. Curcumin potentiates antitumor activity of gemcitabin in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor-kappaB-regulated gene products. Cancer Res 2007, 67, 3853–3861. [Google Scholar]
- Seol, D.W.; Chen, Q.; Zarnegar, R. Transcriptional activation of the hepatocyte growth factor receptor (c-met) gene by its ligand (hepatocyte growth factor) is mediated through AP-1. Oncogene 2000, 19, 1132–1137. [Google Scholar]
- Chen, A.; Xu, J.; Johnson, A.C. Curcumin inhibits human colon cancer cell growth by suppressing gene expressions of epidermal growth factor receptor through reducing the activity of the transcription factor Egr-1. Oncogene 2006, 25, 278–287. [Google Scholar]
- Chadalapaka, G.; Jutooru, I.; Burghardt, R.; Safe, S. Drugs that target specificity proteins downregulate epidermal growth factor receptor in bladder cancer cells. Mol. Cancer Res 2010, 8, 739–750. [Google Scholar]
- Lee, J.Y.; Lee, Y.M.; Chang, G.C.; Yu, S.L.; Hsieh, W.Y.; Chen, J.J.; Chen, H.W.; Yang, P.C. Curcumin induces EGFR degradation in lung adenocarcinoma and modulates p38 activation in intestine: The versatile adjuvant for gefitinib therapy. PLoS One 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Croce, C.M. Causes and consequence of miRNA dysregulation in cancer. Nat. Rev. Genet 2009, 10, 704–714. [Google Scholar]
- Valencia-Sanchez, M.A.; Liu, J.; Hannon, G.J.; Parker, R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev 2006, 20, 515–524. [Google Scholar]
- Chen, C.Z. MicroRNAs as oncogenes and tumor suppressors. N. Engl. J. Med 2005, 353, 1768–1771. [Google Scholar]
- Ambros, V. MicroRNA pathways in flies and worms: Growth, death, fat, stress, and timing. Cell 2003, 113, 673–676. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—MicroRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar]
- Calin, G.A.; Croce, C.M. MicroRNA signature in human cancer. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar]
- Kumar, M.S.; Lu, J.; Mercer, K.L.; Golub, T.R.; Jacks, T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat. Genet 2007, 39, 673–677. [Google Scholar]
- Takamizawa, J.; Konishi, H.; Yanagisawa, K.; Tomida, S.; Osada, H.; Endoh, H.; Harano, T.; Yatabe, Y.; Nagino, M.; Nimura, Y.; et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res 2004, 64, 3753–3756. [Google Scholar]
- Hayashita, Y.; Osada, H.; Tatematsu, Y.; Yamada, H.; Yanagisawa, K.; Tomida, S.; Yatabe, Y.; Kawahara, K.; Sekido, Y.; Takahashi, T. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res 2005, 65, 9628–9632. [Google Scholar]
- Osada, H.; Takahashi, T. let-7 and miR-17-92: Small-sized major players in lung cancer development. Cancer Sci 2011, 102, 9–17. [Google Scholar]
- Yu, L.; Todd, N.W.; Xing, L.; Xie, Y.; Zhang, H.; Liu, Z.; Fang, H.; Zhang, J.; Katz, R.L.; Jiang, F. Early detection of lung adenocarcinoma in sputum by a panel of microRNA markers. Int. J. Cancer 2010, 127, 2870–2878. [Google Scholar]
- Foss, K.M.; Sima, C.; Ugolini, D.; Neri, M.; Allen, K.E.; Weiss, G.J. miR-1254 and miR-574-5p: Serum-based microRNA biomarkers for early-stage non-small cell lung cancer. J. Thorac. Oncol 2011, 6, 482–488. [Google Scholar]
- Yu, S.L.; Chen, H.Y.; Chang, G.C.; Chen, C.Y.; Chen, H.W.; Singh, S.; Cheng, C.L.; Yu, C.J.; Lee, Y.C.; Chen, H.S.; et al. MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell 2008, 13, 48–57. [Google Scholar]
- Patnaik, S.; Kannisto, E.; Knudsen, S.; Yendamuri, S. Evaluation of microRNA expression profiles that may predict recurrence of localized stage I non-small cell lung cancer after surgical resection. Cancer Res 2010, 70, 36–45. [Google Scholar]
- Matsubara, H.; Takeuchi, T.; Nishikawa, E.; Yanagisawa, K.; Hayashita, Y.; Ebi, H.; Yamada, H.; Suzuki, M.; Nagino, M.; Nimura, Y.; et al. Apoptosis induction by antisense oligonucleotides against miR-17-5p and miR-20a in lung cancers overexpressing miR-17-92. Oncogene 2007, 26, 6099–6105. [Google Scholar]
- Sun, M.; Estrov, Z.; Ji, Y.; Coombes, K.R.; Harris, D.H.; Kurzrock, R. Curcumin (diferuloylmethane) alters the expression profiles of microRNAs in human pancreatic cancer cells. Mol. Cancer Ther 2008, 7, 464–473. [Google Scholar]
- Zhang, J.; Du, Y.; Wu, C.; Ren, X.; Ti, X.; Shi, J.; Zhao, F.; Yin, H. Curcumin promotes apoptosis in human adenocarcinoma cells through miR-186* signaling pathway. Oncol. Rep 2010, 24, 1217–1223. [Google Scholar]
- Mudduluru, G.; George-William, J.N.; Muppala, S.; Asangani, I.A.; Kumarswamy, R.; Nelson, L.D.; Allgayer, H. Curcumin regulates miR-21 expression and inhibits invasion and metastasis in colorectal cancer. Biosci. Rep 2011, 31, 185–197. [Google Scholar]
- Yang, J.; Cao, Y.; Sun, J.; Zhang, Y. Curcumin reduces the expression of Bcl-2 by upregulating miR-15a and miR-16 in MCF-7 cells. Med. Oncol 2010, 27, 1114–1118. [Google Scholar]
- Guo, L.; Liu, Y.; Bai, Y.; Sun, Y.; Xiao, F.; Guo, Y. Gene expression profiling of drug-resistant small cell lung cancer cells by combining microRNA and cDNA expression analysis. Eur. J. Cancer 2010, 46, 1692–1702. [Google Scholar]
- Garofalo, M.; Quintavalle, C.; di Leva, G.; Zanca, C.; Romano, G.; Taccioli, C.; Liu, C.G.; Croce, C.M.; Condorelli, G. MicroRNA signatures of TRAIL resistance in human non-small cell lung cancer. Oncogene 2008, 27, 3845–3855. [Google Scholar]
- Zhu, W.; Shan, X.; Wang, T.; Shu, Y.; Liu, P. MiR-181b modulates multidrug resistance by targeting BCL2 in human cancer cell lines. Int. J. Cancer 2010, 127, 2520–2529. [Google Scholar]
- Zhang, J.; Zhang, T.; Ti, X.; Shi, J.; Wu, C.; Ren, X.; Yin, H. Curcumin promotes apoptosis in A549/DDP multidrug-resistant human lung adenocarcinoma through an miRNA signaling pathway. Biochem. Biophys. Res. Commun 2010, 399, 1–6. [Google Scholar]
- Ali, S.; Ahmad, A.; Banerjee, S.; Padhye, S.; Dominiak, K.; Schaffert, J.M.; Wang, Z.; Philip, P.A.; Sarkar, F.H. Gemcitabine sensitivity can be induced in pancreatic cancer cells through modulation of miR-200 and miR-21 expression by curcumin or its analogue CDF. Cancer Res 2010, 70, 3606–3617. [Google Scholar]
- Baehrecke, E.H. How death shapes life during development. Nat. Rev. Mol. Cell Biol 2002, 3, 779–787. [Google Scholar]
- Jaboin, J.J.; Hwang, M.; Lu, B. Autophagy in lung cancer. Method Enzymol 2009, 453, 287–304. [Google Scholar]
- Canuto, R.A.; Tessitore, L.; Muzio, G.; Autelli, R.; Baccino, F.M. Tissue protein turnover during liver carcinogenesis. Carcinogenesis 1993, 14, 2581–2587. [Google Scholar]
- Schwartz, L.M.; Smith, S.W.; Jones, M.E.; Osborne, B.A. Do all programmed cell deaths occur via apoptosis? Proc. Natl. Acad. Sci. USA 1993, 90, 980–984. [Google Scholar]
- Schwarze, P.E.; Seglen, P.O. Reduced autophagic activity, improved protein balance and enhanced in vitro survival of hepatocytes isolated from carcinogen-treated rats. Exp. Cell Res 1985, 157, 15–28. [Google Scholar]
- Ryter, S.W.; Choi, A.M.K. Autophagy in the lung. Proc. Am. Thorac. Soc 2010, 7, 13–21. [Google Scholar]
- Levine, B. Unraveling the role of autophagy in cancer. Autophagy 2006, 2, 65–66. [Google Scholar]
- Kondo, Y.; Kondo, S. Autophagy and cancer therapy. Autophagy 2006, 2, 85–90. [Google Scholar]
- Gozuacik, D.; Kimchi, A. Autophagy and cell death. Curr. Top. Dev. Biol 2007, 78, 217–245. [Google Scholar]
- Kim, K.W.; Hwang, M.; Moretti, L.; Jaboin, J.J.; Cha, Y.I.; Lu, B. Autophagy upregulation by inhibitors of caspase-3 and mTOR enhances radiotherapy in a mouse model of lung cancer. Autophagy 2008, 4, 659–668. [Google Scholar]
- Ding, X.L.; Zhang, H.Y.; Qi, L.; Zhao, B.X.; Lian, S.; Lv, H.S.; Miao, J.Y. Synthesis of novel pyrazole carboxamide derivatives and discovery of modulators for apoptosis or autophagy in A549 lung cancer cells. Bioorg. Med. Chem. Lett 2009, 19, 5325–5328. [Google Scholar]
- Zheng, L.W.; Li, Y.; Ge, D.; Zhao, B.X.; Liu, Y.R.; Lv, H.S.; Ding, J.; Miao, J.Y. Synthesis of novel oxime-containing pyrazole derivatives and discovery of regulators for apoptosis and autophagy in A549 lung cancer cells. Bioorg. Med. Chem. Lett 2010, 20, 4766–4770. [Google Scholar]
- Han, W.; Pan, H.; Chen, Y.; Sun, J.; Wang, Y.; Li, J.; Ge, W.; Feng, L.; Lin, X.; Wang, X.; et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Viola, G.; Bortolozzi, R.; Hamel, E.; Moro, S.; Brun, P.; Castagliuolo, I.; Ferlin, M.G.; Basso, G. MG-2477, a new tubulin inhibitor, induces autophagy through inhibition of the Akt/mTOR pathway and delayed apoptosis in A549 cells. Biochem. Pharmacol 2012, 83, 16–26. [Google Scholar]
- He, Q.; Huang, B.; Zhao, J.; Zhang, Y.; Zhang, S.; Miao, J. Knockdown of integrin β4-induced autophagic cell death associated with P53 in A549 lung adenocarcinoma cells. FEBS J 2008, 275, 5725–5732. [Google Scholar]
- Jia, Y.L.; Li, J.; Qin, Z.H.; Liang, Z.Q. Autophagic and apoptotic mechanisms of curcumin-induced death in K562 cells. J. Asian Nat. Prod. Res 2009, 11, 918–928. [Google Scholar]
- O’Sullivan-Coyne, G.; O’Sullivan, G.C.; O’Donovan, T.R.; Piwocka, K.; McKenna, S.L. Curcumin induces apoptosis-independent death in oesophageal cancer cells. Br. J. Cancer 2009, 101, 1585–1595. [Google Scholar]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000, 19, 5720–5728. [Google Scholar]
- Aoki, H.; Takada, Y.; Kondo, S.; Sawaya, R.; Aggarwal, B.B.; Kondo, Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: Role of Akt and extracellular signal-regulated kinase signaling pathways. Mol. Pharmacol 2007, 72, 29–39. [Google Scholar]
- Shinojima, N.; Yokoyama, T.; Kondo, Y.; Kondo, S. Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in curcumin-induced autophagy. Autophagy 2007, 3, 635–637. [Google Scholar]
- Qian, H.; Yang, Y.; Wang, X. Curcumin enhanced adriamycin-induced human liver-derived Hepatoma G2 cell death through activation of mitochondria-mediated apoptosis and autophagy. Eur. J. Pharm. Sci 2011, 43, 125–131. [Google Scholar]
- Wilken, R.; Veena, M.S.; Wang, M.B.; Srivatsan, E.S. Curcumin: A review of anti-cancer properties and therapeutic activity in head and neck squamous cell carcinoma. Mol. Cancer 2011, 10. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med 1997, 3, 730–737. [Google Scholar]
- Maitland, N.J.; Collins, A.T. Cancer stem cells—A therapeutic target? Curr. Opin. Mol. Ther 2010, 12, 662–673. [Google Scholar]
- Hermann, P.C.; Bhaskar, S.; Cioffi, M.; Heeschen, C. Cancer stem cells in solid tumors. Semin. Cancer Biol 2010, 20, 77–84. [Google Scholar]
- Chen, S.Y.; Huang, Y.C.; Liu, S.P.; Tsai, F.J.; Shyu, W.C.; Lin, S.Z. An overview of concepts for cancer stem cells. Cell Transplant 2011, 20, 113–120. [Google Scholar]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med 2011, 17, 313–319. [Google Scholar]
- Ho, M.M.; Ng, A.V.; Lam, S.; Hung, J.Y. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res 2007, 67, 4827–4833. [Google Scholar]
- Levina, V.; Marrangoni, A.M.; DeMarco, R.; Gorelik, E.; Lokshin, A.E. Drug-selected human lung cancer stem cells: Cytokine network, tumorigenic and metastatic properties. PLoS One 2008, 3. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, H.; Okudela, K.; Yazawa, T.; Sato, H.; Shimoyamada, H. Cancer stem cell: Implications in cancer biology and therapy with special reference to lung cancer. Lung Cancer 2009, 66, 275–281. [Google Scholar]
- Wu, C.; Alman, B.A. Side population cells in human cancers. Cancer Lett 2008, 268, 1–9. [Google Scholar]
- Sophos, N.A.; Vasiliou, V. Aldehyde dehydrogenase gene superfamily: The 2002 update. Chem. Biol. Interact 2003, 143–144, 5–22. [Google Scholar]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar]
- Teng, Y.; Wang, X.; Wang, Y.; Ma, D. Wnt/beta-catenin signaling regulates cancer stem cells in lung cancer A549 cells. Biochem. Biophys. Res. Commun 2010, 392, 373–379. [Google Scholar]
- Bertolini, G.; Roz, L.; Perego, P.; Tortoreto, M.; Fontanella, E.; Gatti, L.; Pratesi, G.; Fabbri, A.; Andriani, F.; Tinelli, S.; et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc. Natl. Acad. Sci. USA 2009, 106, 16281–16286. [Google Scholar]
- Riccioni, R.; Dupuis, M.L.; Bernabei, M.; Petrucci, E.; Pasquini, L.; Mariani, G.; Cianfriglia, M.; Testa, U. The cancer stem cell selective inhibitor salinomycin is a p-glycoprotein inhibitor. Blood Cells Mol. Dis 2010, 45, 86–92. [Google Scholar]
- Yilmaz, O.H.; Valdez, R.; Theisen, B.K.; Guo, W.; Ferguson, D.O.; Wu, H.; Morrison, S.J. PTEN dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006, 441, 475–482. [Google Scholar]
- Lin, L.; Fuchs, J.; Li, C.; Olson, V.; Bekaii-Saab, T.; Li, J. STAT3 signaling pathway is necessary for cell survival and tumorsphere forming capacity in ALDH+/CD133+ stem cell-like human colon cancer cells. Biochem. Biophys. Res. Commun 2011, 416, 246–251. [Google Scholar]
- Kakarala, M.; Brenner, D.E.; Korkaya, H.; Cheng, C.; Tazi, K.; Ginestier, C.; Liu, S.; Dontu, G.; Wicha, M.S. Targeting breast stem cells with the cancer preventive compounds curcumin and piperine. Breast Cancer Res. Treat 2010, 122, 777–785. [Google Scholar]
- Fong, D.; Yeh, A.; Naftalovich, R.; Choi, T.H.; Chan, M.M. Curcumin inhibits the side population (SP) phenotype of the rat C6 glioma cell line: Towards targeting of cancer stem cells with phytochemicals. Cancer Lett 2010, 293, 65–72. [Google Scholar]
- Lim, K.J.; Bisht, S.; Bar, E.E.; Maitra, A.; Eberhart, C.G. A polymeric nanoparticle formulation of curcumin inhibits growth, clonogenicity and stem-like fraction in malignant brain tumors. Cancer Biol. Ther 2011, 5, 464–473. [Google Scholar]
- Lin, L.; Liu, Y.; Li, H.; Li, P.K.; Fuchs, J.; Shibata, H.; Iwabuchi, Y.; Lin, J. Targeting colon cancer stem cells using a new curcumin analogue, GO-Y030. Br. J. Cancer 2011, 105, 212–220. [Google Scholar]
- Bao, B.; Ali, S.; Banerjee, S.; Wang, Z.; Logna, F.; Azmi, A.S.; Kong, D.; Ahmad, A.; Li, Y.; Padhye, S.; et al. Curcumin analogue CDF inhibits pancreatic tumor growth by switching on suppressor microRNAs and attenuating EZH2 expression. Cancer Res 2012, 72, 335–345. [Google Scholar]
- Jordan, C.T. Can we finally target the leukemic stem cells? Best Pract. Res. Clin. Haematol 2008, 21, 615–620. [Google Scholar]
- Neelakantan, S.; Nasim, S.; Guzman, M.L.; Jordan, C.T.; Crooks, P.A. Aminoparthenolides as novel anti-leukemic agents: Discovery of the NF-κB inhibitor, DMAPT (LC-1). Bioorg. Med. Chem. Lett 2009, 19, 4346–4349. [Google Scholar]
- National Cancer Institute. Clinical development plan: Curcumin. J. Cell Biochem. Suppl. 1996, 26, 72–85.
- Sharma, R.A.; Euden, S.A.; Platton, S.L.; Cooke, D.N.; Shafayat, A.; Hewitt, H.R.; Marczylo, T.H.; Morgan, B.; Hemingway, D.; Plummer, S.M.; et al. Phase I clinical trial of oral curcumin: Biomarkers of systemic activity and compliance. Clin. Cancer Res 2004, 10, 6847–6854. [Google Scholar]
- Cheng, A.L.; Hsu, C.H.; Lin, J.K.; Hsu, M.M.; Ho, Y.F.; Shen, T.S.; Ko, J.Y.; Lin, J.T.; Lin, B.R.; Ming-Shiang, W.; et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res 2001, 21, 2895–2900. [Google Scholar]
- Wahlstrom, B.; Blennow, G. A study on the fate of curcumin in the rat. Acta Pharmacol. Toxicol. (Copenh.) 1978, 43, 86–92. [Google Scholar]
- Shoba, G.; Joy, D.; Joseph, T.; Majeed, M.; Rajendran, R.; Srinivas, P.S. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med 1998, 64, 353–356. [Google Scholar]
- Bisht, S.; Feldmann, G.; Soni, S.; Ravi, R.; Karikar, C.; Maitra, A.; Maitra, A. Polymeric nanoparticleencapsulated curcumin (“nanocurcumin”): A novel strategy for human cancer therapy. J. Nanobiotechnol 2007, 5. [Google Scholar] [CrossRef]
- Li, L.; Braiteh, F.S.; Kurzrock, R. Liposome-encapsulated curcumin: In vitro and in vivo effects on proliferation, apoptosis, signaling, and angiogenesis. Cancer 2005, 104, 1322–1231. [Google Scholar]
- Maiti, K.; Mukherjee, K.; Gantait, A.; Saha, B.P.; Mukherjee, P.K. Curcumin-phospholipid complex: Preparation, therapeutic evaluation and pharmacokinetic study in rats. Int. J. Pharm 2007, 330, 155–163. [Google Scholar]
- Marczylo, T.H.; Verschoyle, R.D.; Cooke, D.N.; Morazzoni, P.; Steward, W.P.; Gescher, A.J. Comparison of systemic availability of curcumin with that of curcumin formulated with phosphatidylcholine. Cancer Chemother. Pharmacol 2007, 60, 171–177. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Cancer Origin | Upregulate | Downregulate | Targets | Reference |
|---|---|---|---|---|
| Pancrea | miR-22 | miR-21 | SP1, ESR1 | [58] |
| miR-200 | miR-199a* | PTEN | [66] | |
| Colorectum | miR-21 | AP1, Pdcd4 | [60] | |
| Breast | miR-15a | Bcl-2 | [61] | |
| miR-16 | ||||
| Lung | miR-186* | Casp-10 | [59,65] | |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ye, M.-X.; Li, Y.; Yin, H.; Zhang, J. Curcumin: Updated Molecular Mechanisms and Intervention Targets in Human Lung Cancer. Int. J. Mol. Sci. 2012, 13, 3959-3978. https://doi.org/10.3390/ijms13033959
Ye M-X, Li Y, Yin H, Zhang J. Curcumin: Updated Molecular Mechanisms and Intervention Targets in Human Lung Cancer. International Journal of Molecular Sciences. 2012; 13(3):3959-3978. https://doi.org/10.3390/ijms13033959
Chicago/Turabian StyleYe, Ming-Xiang, Yan Li, Hong Yin, and Jian Zhang. 2012. "Curcumin: Updated Molecular Mechanisms and Intervention Targets in Human Lung Cancer" International Journal of Molecular Sciences 13, no. 3: 3959-3978. https://doi.org/10.3390/ijms13033959
APA StyleYe, M.-X., Li, Y., Yin, H., & Zhang, J. (2012). Curcumin: Updated Molecular Mechanisms and Intervention Targets in Human Lung Cancer. International Journal of Molecular Sciences, 13(3), 3959-3978. https://doi.org/10.3390/ijms13033959