Abstract
Copper amine oxidases (CAOs) are a ubiquitous group of enzymes that catalyze the conversion of primary amines to aldehydes coupled to the reduction of O2 to H2O2. These enzymes utilize a wide range of substrates from methylamine to polypeptides. Changes in CAO activity are correlated with a variety of human diseases, including diabetes mellitus, Alzheimer’s disease, and inflammatory disorders. CAOs contain a cofactor, 2,4,5-trihydroxyphenylalanine quinone (TPQ), that is required for catalytic activity and synthesized through the post-translational modification of a tyrosine residue within the CAO polypeptide. TPQ generation is a self-processing event only requiring the addition of oxygen and Cu(II) to the apoCAO. Thus, the CAO active site supports two very different reactions: TPQ synthesis, and the two electron oxidation of primary amines. Crystal structures are available from bacterial through to human sources, and have given insight into substrate preference, stereospecificity, and structural changes during biogenesis and catalysis. In particular both these processes have been studied in crystallo through the addition of native substrates. These latter studies enable intermediates during physiological turnover to be directly visualized, and demonstrate the power of this relatively recent development in protein crystallography.
1. Introduction
Amine oxidases are responsible for the oxidative deamination of amines to their corresponding aldehydes sequentially coupled to the reduction of O2 to H2O2. Members of this ubiquitous group of enzymes can be further classified based on the chemical identity of the organic cofactor used during catalysis, into either the flavin adenine dinucleotide-containing amine oxidases (monoamine oxidase (MAO) (E.C. 1.4.3.4) and polyamine oxidase (E.C. 1.5.3.17)) or the copper/quinocofactor-containing amine oxidases. MAOs are found exclusively in the outer mitochondrial membrane of mammalian cells, and catalyze the deamination of primary, secondary, or tertiary amines. These flavoproteins have a firmly established role in the metabolism of aminergic neurotransmitters such as serotonin, norepinephrine, epinephrine, and dopamine [1]. Polyamine oxidases preferentially oxidize polyamines such as spermine and spermidine, and play a role in the regulation of cellular growth [2].
In contrast, the copper/quinocofactor-containing amine oxidases are active against only primary amines, and possess one of two quinone-containing cofactors: 2,4,5-trihydroxyphenylalanine quinone (topaquinone or TPQ) or lysyl tyrosylquinone (LTQ). Although the TPQ- and LTQ-containing amine oxidases both oxidatively deaminate primary amines through the use of an organic quinone-containing cofactor and a copper ion, these two enzyme families are non-homologous. LTQ is found in the lysyl oxidase (LOX)-like family of proteins (LOXL, E.C. 1.4.3.13), named for their ability to convert the ɛ-amino group of peptidyl lysine residues to an aldehyde. This peptidyl aldehyde product can condense with a neighboring lysine residue or react with a second lysine-derived aldehyde to form crosslinks important in the formation and stabilization of collagen and elastin [3]. Amine oxidases containing TPQ (CAOs for copper-containing amine oxidases, E.C. 1.4.3.21) were the first to be characterized in a growing number of bifunctional enzymes that contain “home-made” cofactors produced in situ by the post-translational modification of an endogenous amino acid side chain [4]. This review focuses on cofactor biogenesis and catalysis in the TPQ-containing CAOs, with a particular emphasis on the role X-ray crystallography has played in understanding these two processes.
2. Physiological Significance of CAOs
In the decades since the first discovery of amine oxidase activity in the blood plasma of sheep in 1929 [5], CAOs have been found to exist almost ubiquitously in aerobic organisms, taking on a myriad of functional roles depending on enzyme source, cellular location, and physiological substrate. In bacteria and yeast, CAOs are thought to play mainly a catabolic role, allowing for the use of primary amines as a sole carbon and/or nitrogen source for cellular growth [6,7].
In eukaryotes these enzymes have more poorly defined roles but are known to contribute to a variety of complex cellular activities. CAO activity helps regulate several processes in plants due to the compartment-specific production of product H2O2, which affects plant germination, seedling establishment, and root growth [8]. Hydrogen peroxide production is also associated with cell wall maturation and lignification during growth, as well as wound-healing and the reinforcement of cell walls during cellular stress due to pathogenic attack [9,10]. Moreover, amine catabolism is known to influence the plant stress response upon exposure to cadmium or excess salt [11].
Several CAO homologs are expressed in mammalian tissues. In order to study their distribution amongst mammalian species, four complete genes (AOC1–4, for “amine oxidase, copper-containing”) were cloned and characterized from porcine genomic DNA. All of these encode a bona fide CAO which displays catalytic activity toward primary amines [12]. These were used to identify orthologs in other mammalian genomes; all four AOC genes have been identified in cows, horses, dogs, mice, rats, chimpanzees, macaques, and humans [12]. AOC1 encodes a soluble secretory enzyme known as diamine oxidase (DAO) [13], AOC2 encodes a retina-specific amine oxidase [14], and AOC3 encodes an endothelial vascular adhesion protein (known as VAP-1 in humans) [15]. AOC4 was found to encode an additional serum/plasma CAO homologous to VAP-1, which lacks the N-terminal transmembrane domain found in VAP-1. However, the AOC4 gene in humans is truncated and nonfunctional due to an internal stop codon, and Aoc4 encoded by rodent genomes is present only in small fragments [12].
In mammals, nearly all biogenic primary amines can act as CAO substrates, including the neurotransmitters serotonin, norepinephrine, epinephrine, and dopamine. A large number of additional amines have been identified as CAO substrates, including methylamine, ethylamine, aminoacetone, benzylamine, phenylethylamine, agmatine, spermine, spermidine, histamine, tyramine, mescaline, putrescine, and cadaverine [16–18].
Members of the CAO protein family contribute to the regulation of a myriad of complex processes in mammals due to the diversity in their substrates, their wide distribution throughout mammalian tissues (including the brain, blood plasma, kidneys, placenta, and throughout the cardiovascular and gastrointestinal systems), and changes in expression and activity during disease and throughout pregnancy [19]. For example, histamine, a pro-inflammatory amine produced by mast cells upon allergic reaction or cellular damage, is cleared in human tissues by DAO [20]. Tumor cells are known to contain higher concentrations of amines than cells from normally proliferating tissues, and the oxidation of spermine or spermidine by CAOs produces acrolein, a known cytotoxin which can induce cellular death in cancerous tissues [21]. Agmatine (produced from the decarboxylation of arginine) binds to the α2 adrenergic receptor and imidazoline binding sites; its regulation through CAO activity has been shown to modulate withdrawal anxiety in rats [22].
Changes in CAO activity are correlated with a variety of human diseases, including diabetes mellitus, Alzheimer’s disease, and additional inflammatory disorders [23]. A truncated soluble form of membrane-bound VAP-1 known as semicarbazide-sensitive amine oxidase (SSAO) is known to mediate glucose uptake in adipocytes via the recruitment of the glucose transporter GLUT4 from vesicles within the cellular interior to the cell surface [24]. The soluble SSAO may also compensate for the absence of the Aoc4 product in humans. VAP-1 also controls leukocyte extravasation to sites of inflammation through its activity against amine groups from solvent-exposed lysine residues of membrane-associated counter-receptors, including sialic acid binding Ig-like lectins (siglecs) [15,25]. Mice deficient in Aoc3 expression exhibit impaired immune responses, but notably present no other detectable phenotypic changes [26]. The highly reactive aldehydes produced from the oxidation of methylamine and aminoacetone in human tissues contribute to protein cross-linking, β-amyloid aggregation, and advanced glycation end-product formation in patients with Alzheimer’s disease or diabetes mellitus [27,28]. With CAO activity playing a critical role in such numerous and diverse physiological processes, ranging from basic metabolism in bacteria and yeast to roles in multifaceted disease states in humans, an understanding of TPQ production and catalysis in these enzymes is critical to the development of any potential pharmaceutical therapies which may regulate their activity [23].
3. Post-Translationally Modified Amino Acid Cofactors
A large number of enzymes require low-molecular weight cofactors for catalytic activity, some of which are covalently attached to the protein component. Most cofactors are non-proteinaceous and independently synthesized before their association with the protein component of the relevant enzyme. However, it is now well-established that some enzymes utilize the post-translational modification of endogenous amino acid side chains to form cofactors in situ [4]. This approach enables an enzyme to avoid the energetic costs of cofactor import both to and within the protein, and furthermore may sequester potentially deleterious intermediates produced during cofactor formation. For example, TPQ as a free molecule is a known neurotoxin, however its production from an endogenous tyrosine residue in the deeply buried CAO active site circumvents this problem [29]. Most importantly, this strategy increases the diversity of chemical properties available within an enzyme active site beyond that afforded by the twenty canonical amino acids.
Variations in both the physical properties of these modified amino acid cofactors as well as the mechanisms employed for their formation are widespread. Many modifications involve the crosslinking of two amino acid side chains, for example cytochrome c oxidase and catalase (Tyr-His); tyrosinase, hemocyanin, and catechol oxidase (His-Cys); and galactose oxidase (Tyr-Cys) [30–36]. Cofactor biosynthesis can also involve the modification of a single side chain: sulfatases (cysteine converted to formylglycine); rubisco, urease, and phosphotriesterase (carbamylation of a lysine residue); NADH peroxidase, NADH oxidase, and nitrile hydratase (cysteine converted to cysteinesulfenic acid) [37–43].
Several additional enzymes undergo post-translational modification of an endogenous tyrosine or tryptophan residue to form cofactors which contain a quinone moiety. To date, these include TPQ in CAO (Figure 1A) [29], LTQ in LOXL (Figure 1B) [44], tryptophan tryptophylquinone (TTQ) in bacterial amine dehydrogenase (Figure 1C) [45], and cysteine tryptophylquinone (CTQ) in bacterial quinohemoprotein amine dehydrogenase (Figure 1D) [46].
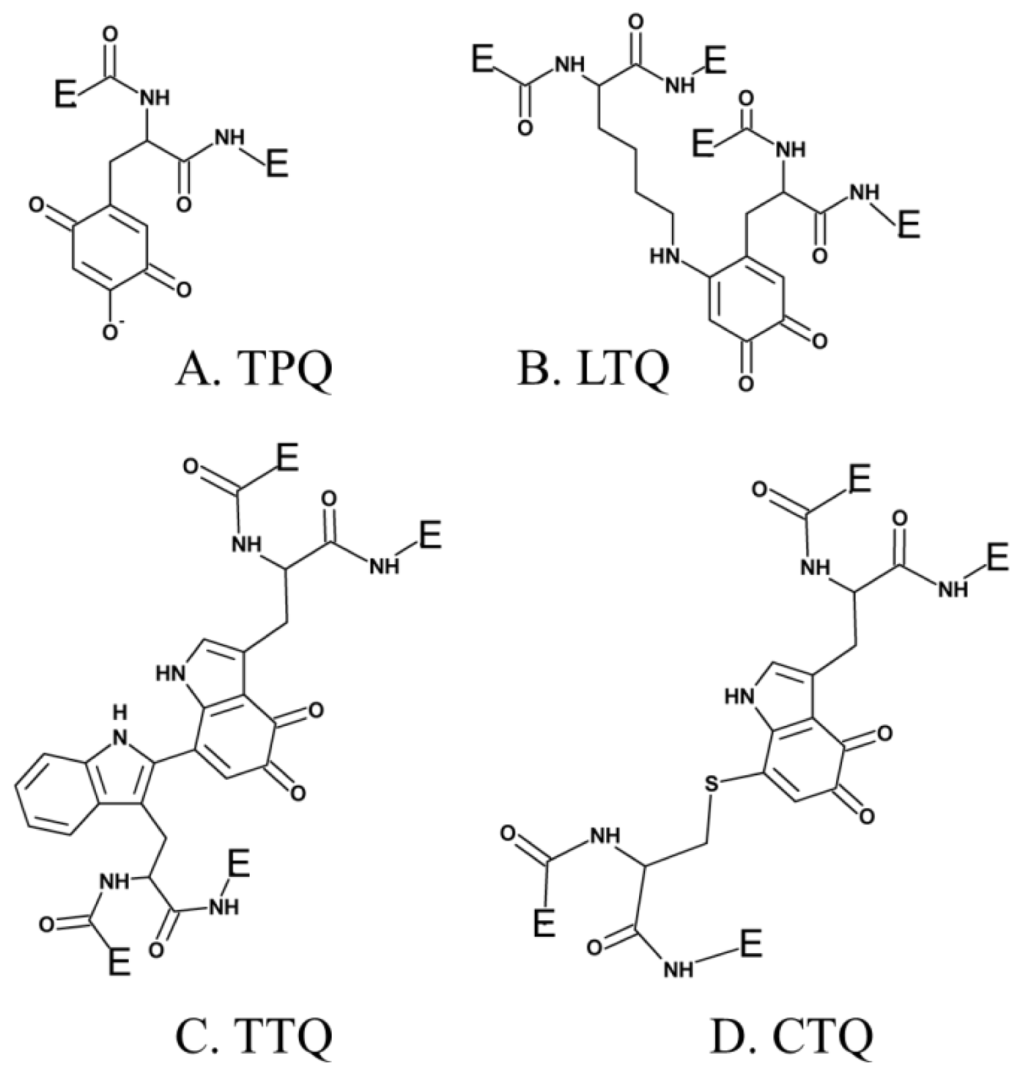
Figure 1.
Quinone-containing cofactors formed by the post-translational modification of tyrosine or tryptophan side chains.
The first of these four quinocofactors to be identified was TPQ, which is located in the deeply-buried CAO active site. The formation of a chromophoric complex between CAO proteins and hydrazine derivatives had originally suggested pyridoxal phosphate (PLP) as a likely candidate for the CAO cofactor (Figure 2A) [47]. Two independent studies later reported the cofactor in Bos taurus serum amine oxidase (BSAO) to be covalently-bound pyrroloquinoline quinone (PQQ) [48,49] (Figure 2B), which was strengthened by resonance Raman experiments conclusively eliminating PLP as a possible CAO cofactor [50,51]. The true identity of the CAO cofactor was unequivocally confirmed to be topaquinone (Figure 1A) by Janes et al. through the use of mass spectrometry, ultraviolet-visible spectroscopy, and proton NMR studies of a pentapeptide isolated from proteolyzed BSAO in comparison to a peptide analog of 6-hydroxydopa [29].
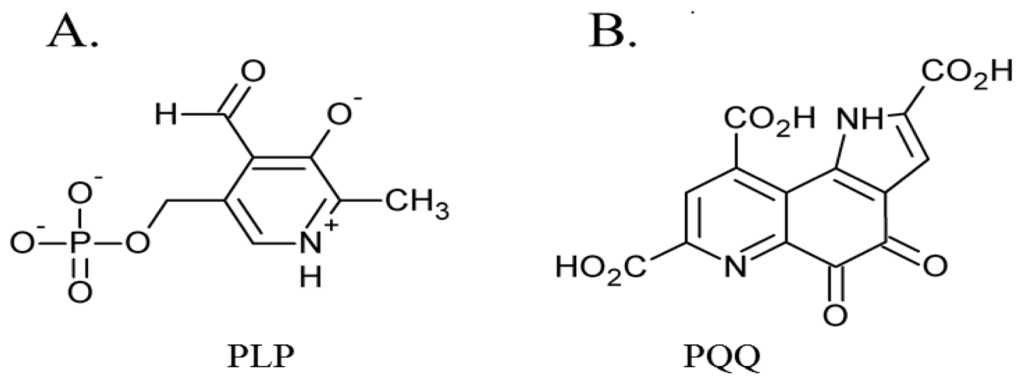
Figure 2.
Structures of initially proposed CAO cofactors (A) PLP and (B) PQQ.
4. Overall CAO Fold
The structures of CAOs isolated from a variety of organisms have been solved by X-ray crystallography (Table 1). Despite sharing only 20–40% sequence homology, they all adopt an archetypal fold which brings together a number of conserved residues to form the CAO active site where both cofactor biogenesis and catalysis take place. There are currently no crystal structures of LTQ-containing LOXL proteins, which are non-homologous to the TPQ-containing CAOs.

Table 1.
TPQ-containing CAOs with available X-ray crystal structures.
All CAOs structurally characterized thus far are homodimeric, with individual protein subunit masses ranging from ~70–80 kDa and eukaryotic CAOs having additional mass through glycosylation. The CAO monomer can be divided into four domains (D1–D4) organized along its primary sequence. The shape of the CAO dimer is similar to that of a mushroom, with the amino-terminal domain D1 (present only in the ECAO crystal structure) acting as a “stalk” (not present in HPAO-1, Figure 3) [59]. D2 and D3 are small α/β domains composed of ~100 residues each, and are thought to have arisen from a gene duplication event given their sequence homology and near identical topology (in HPAO-1, alignment of equivalent D2 and D3 main chain atoms gives a root-mean-square deviation (rmsd) of ~1.1 Å [55]) (purple (D2) and green (D3) in Figure 3).
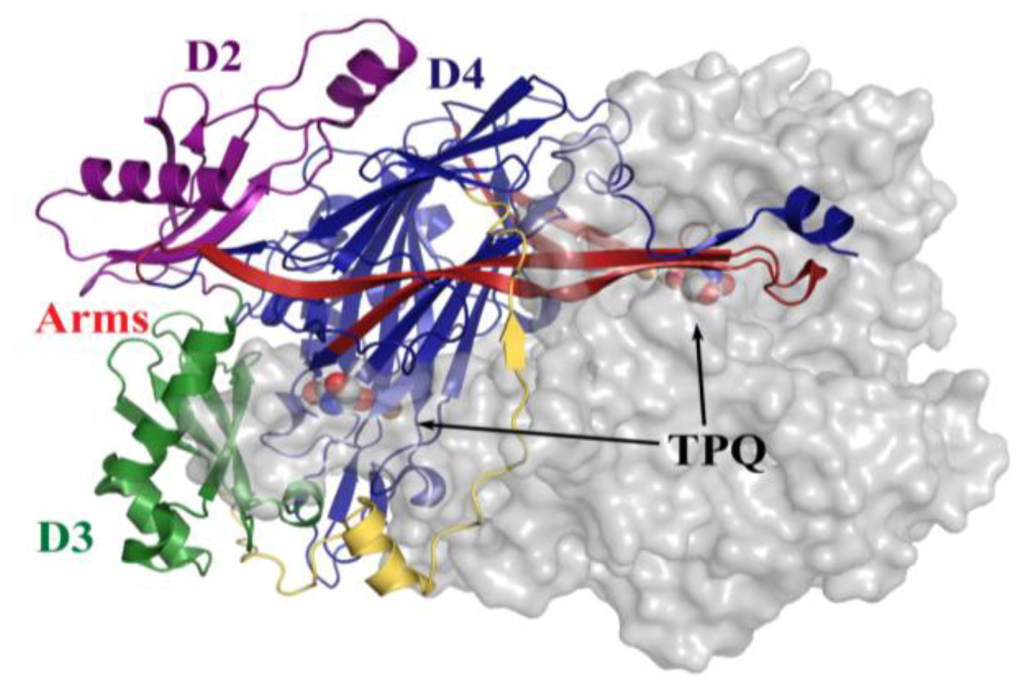
Figure 3.
Overall fold of HPAO-1 viewed along the molecular dyad axis. One monomer is drawn in cartoon, and colored by domain (D2: purple; D3: green; D4: blue; connecting loop: yellow; β-hairpin arms: red). The second monomer is drawn as a semi-transparent molecular surface colored grey. TPQ from both monomers are drawn as space-filling spheres and colored by atom type (carbon, grey).
The large carboxy-terminal domain D4 is composed of ~500 residues and contributes the majority of the conserved amino acids which form the enzyme active site (blue in Figure 3). The topology of D4 is that of a complex antiparallel β-sheet sandwich structure. Two β-hairpin arms protrude from the D4 domains of each monomer, which contain amino acid residues that interact with its partner in the protein dimer and form a portion of the back wall of the other monomer’s active site (red in Figure 3). These arms are thought to be important for the maintenance of dimer stability as well as the regulation of substrate specificity in CAOs. The region between monomers encloses a solvent-filled cavity known as the “inland lake,” and the dimer interface is extensive with a buried surface area comprising ~20% of the total surface area [58,59] (Figure 3).
All structurally characterized CAOs besides HPAO-1 and −2 contain two metal binding sites distinct from the active site copper, one of which is solvent exposed and one of which is less accessible (~36 Å and ~30 Å from the active site in ECAO, respectively) [62]. These metal sites primarily bind Ca(II), with the plant CAOs binding Ca(II) or Mn(II) at the less accessible site dependent on the availability of divalent metal ions [60,63]. The X-ray crystal structures of HPAO-1 and −2 indicate a salt bridge at the corresponding position (HPAO-1: Glu69 and Arg467, HPAO-2: Lys42 and Asp445) [55,56]. A study conducted using EDTA-treated ECAO found that calcium binding at these two sites is not essential for activity, but its removal decreases catalytic efficiency by ~60–90%, which can be partially recovered by the addition of an exogenous divalent cation [62]. This effect has been attributed to long-range structural changes which either alter the conformation of TPQ, or affect the dynamics of a hydrophobic channel important for oxygen transport to the active site.
5. Structure of the CAO Active Site
The deeply buried CAO active site contains a mononuclear type 2 Cu(II) ion bound in a distorted square pyramidal geometry by the imidazole groups of three conserved histidine residues at a distance of ~2.0 Å, a well-ordered axial water ligand at a distance of ~2.4 Å (Wa), and a more labile equatorial water ligand visualized in some CAO structures at a distance of ~2.0 Å (We) (Figure 4A). TPQ has been visualized in two conformations that are named based on the relationship between TPQ and the bound copper ion. The “off-copper” TPQ conformers do not act as copper ligands; instead, the O2 atom interacts with the metal center via the conserved axial water molecule (Figure 4A). The oxygen atom at position 4 of the TPQ ring is involved in a short hydrogen bond (~2.3 Å) with the phenolic hydroxyl of a conserved active site tyrosine residue (Tyr305 in HPAO-1). As this distance is less than 2.5 Å, it indicates a shared proton between Tyr305 and TPQ. The side chain of Tyr305 also interacts with the axial water ligand (Wa) via an intervening water molecule (W1 in Figure 4A). The carbonyl group at position 5 of the TPQ ring points away from the copper center and toward the amine substrate channel. This off-copper species represents a catalytically productive CAO active site, as the C5 atom is the site of nucleophilic attack by amine substrates. A conserved aspartate residue, which acts as a general base during catalysis, sits near the substrate binding pocket (Asp319 in HPAO-1), with an intervening water molecule between it and the cofactor (W2 in Figure 4A). TPQ can also adopt an “on-copper” conformation, in which the ring has rotated about its Cα-Cβ and Cβ-Cγ bonds. In this conformation, TPQ displaces the axial water molecule which is coincident with the off-copper TPQ conformer (Wa) and instead interacts directly with copper via its O4 atom. This represents an unproductive state because the C5 atom of TPQ is inaccessible for nucleophilic attack by substrate (Figure 4B).
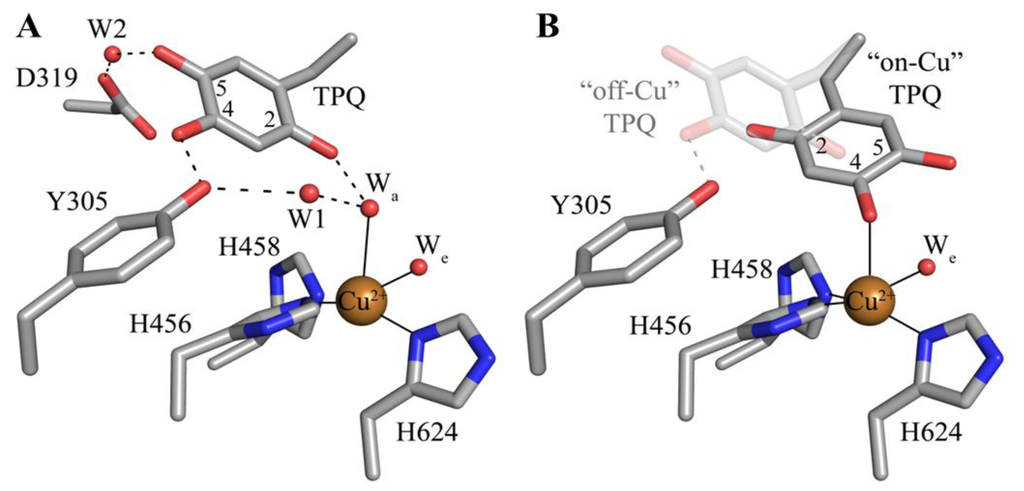
Figure 4.
Crystal structures of the HPAO-1 active site (PDB code 2oov) with TPQ in an (A) “off-copper” or (B) “on-copper” conformation [55,64]. Residues are drawn in stick and colored by atom type (carbon, grey). Copper ions are drawn as gold spheres, and water molecules are drawn as small red spheres. Hydrogen bonds are indicated by dashed lines, and metal-ligand interactions are indicated by solid lines. In (B), the “off-copper” TPQ conformer from panel (A) is drawn in semi-transparent stick.
6. Biogenesis of TPQ
The mechanism by which fully-folded CAOs synthesize a quinone-containing cofactor in situ has been an intriguing question in the field. A number of biochemical studies first established the process to be autocatalytic, requiring copper and oxygen but no auxiliary enzymatic activity or reducing equivalents [65,66]. Two moles of O2 are consumed for every mole of TPQ and H2O2 produced (Scheme 1) [67].
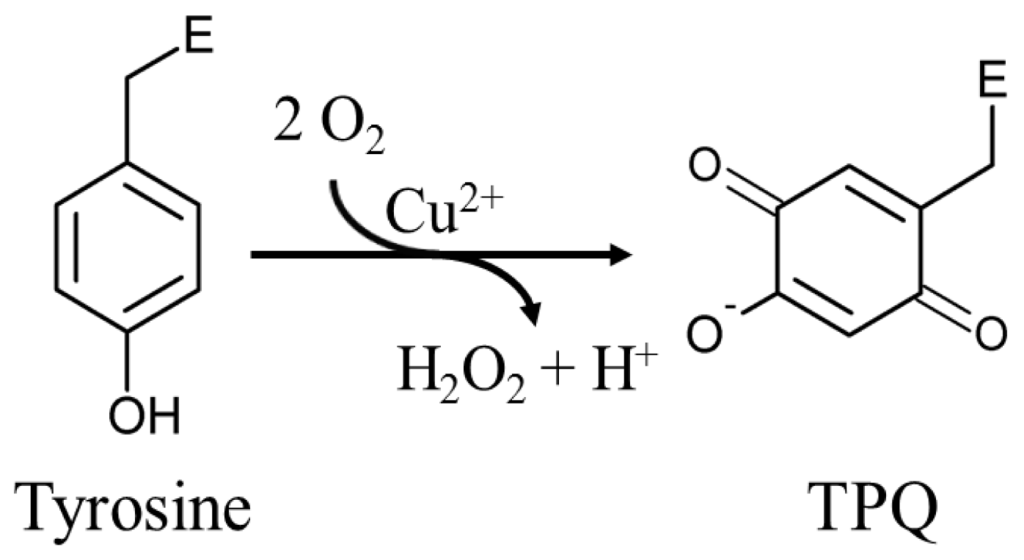
Scheme 1.
An endogenous tyrosine residue is converted to TPQ in an autocatalytic oxygen- and copper-dependent process. E represents the enzyme polypeptide.
Kinetic and structural investigations using metal-free precursor (apo) protein from Hansenula polymorpha (apoHPAO-1) and Arthrobacter globiformis (apoAGAO), the only metal-free precursor CAO proteins currently available, have led to a proposed mechanism for TPQ biogenesis. This process in HPAO-1 is outlined in Scheme 2. ApoCAO is defined as amine oxidase protein which lacks both active site cofactors: a mononuclear copper ion and TPQ. The tyrosine residue which undergoes modification is contained within the conserved active site consensus sequence Thr-X-X-Asn-Tyr-Asp/Glu (precursor tyrosine residue underlined) [68]. TPQ biogenesis begins with apoCAO, in which the precursor tyrosine side chain is unmodified (A in Scheme 2) [58,69]. Copper binds at the active site, and is ligated by three strictly conserved histidine residues (A→B in Scheme 2). A kinetic study conducted using apoHPAO-1 with and without pre-bound copper reported a rate of TPQ formation which was unchanged with the pre-binding of Cu(II), indicating that copper binding is a fast process relative to the overall rate of biogenesis [70].
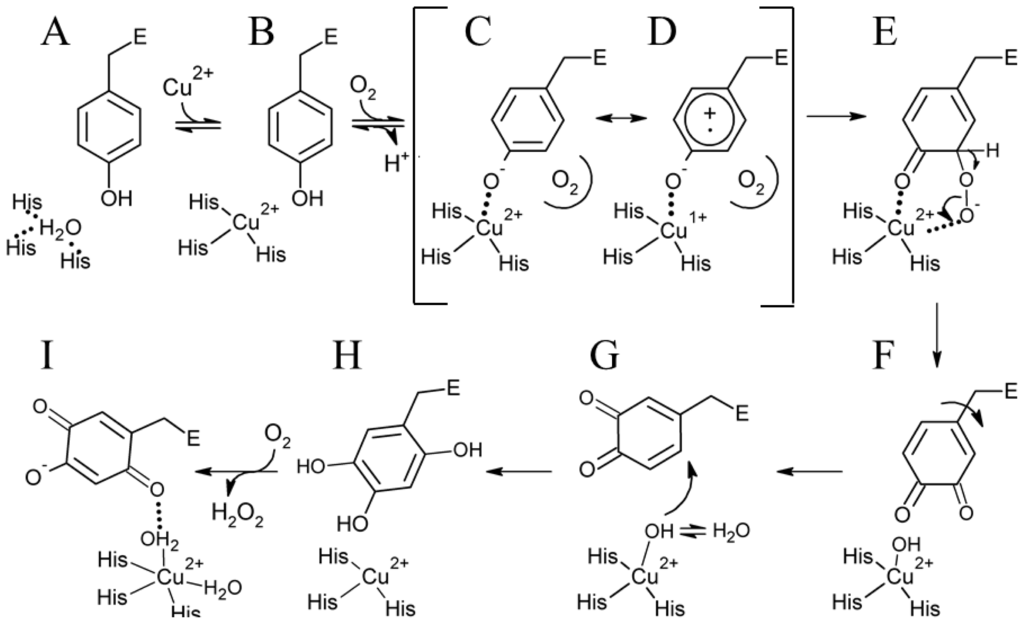
Scheme 2.
Proposed TPQ biogenesis mechanism in HPAO-1. Figure adapted from [70].
In HPAO-1, molecular oxygen then binds in a nearby off-copper hydrophobic pocket, which induces a conformational change in the precursor tyrosine side chain such that its hydroxyl group becomes oriented toward the copper (B→C in Scheme 2). Structural work suggests that at this point, the tyrosine residue is present in its protonated form, as indicated by the long distance between the phenolic oxygen and bound copper in crystal structures of anaerobic Cu(I)-apoHPAO-1 and Cu(II)-apoAGAO complexes (~2.8 Å and ~2.5 Å, respectively) [69,71].
Support for the oxygen-dependent formation of a tyrosine/copper complex comes from spectroscopic studies carried out using apoHPAO-1 [72]. The pre-incubation of apoHPAO-1 with copper followed by exposure to molecular oxygen resulted in a feature which absorbs at λmax = 350 nm. This species decays isosbestically with the formation of TPQ, as indicated by a broad feature absorbing at 480 nm. Importantly, oxygen was found to be required but not consumed during this process, suggesting that oxygen binding results in a conformational change in the side chain of the precursor tyrosine residue. This movement, followed by deprotonation of the tyrosine, results in the formation of a ligand-metal charge transfer (LMCT) species which gives rise to the feature at 350 nm (C in Scheme 2). A putative oxygen binding pocket comprised of residues Met634, Tyr407, and Leu425 has been identified in HPAO-1, and the rate of biogenesis is linked to the volume of the hydrophobic residue at position 634 [73,74].
The Cu(II)-tyrosine complex, along with its Cu(I)-tyrosyl radical resonance form (D in Scheme 2), is activated for monooxygenation at position 3 of the tyrosine ring by the pre-bound O2, which forms a bridged peroxo adduct (C/D→E in Scheme 2). This species rapidly collapses to produce dopaquinone (DPQ) (E→F in Scheme 2). The DPQ ring flips ~180° about its Cα-Cβ bond by an unknown mechanism (F→G in Scheme 2), which leaves it positioned for the incorporation of a second oxygen atom at position 6 of the ring by a copper-activated water/hydroxyl (G→H in Scheme 2). This produces TPQred (2,4,5-trihydroxyphenylalanine = 2-electron reduced TPQ) (H in Scheme 2), which is then oxidized to mature TPQ with the concomitant reduction of molecular oxygen to hydrogen peroxide (H→I in Scheme 2). Lastly, TPQ moves to the catalytically productive off-copper position seen in structures of native CAO (I in Scheme 2).
The preceding mechanism is strongly supported by a crystallographic investigation of TPQ biogenesis in apoAGAO [69]. The crystal structure of apoAGAO has been solved to a resolution of 2.2 Å, and unambiguously shows the precursor tyrosine residue (Y382 in AGAO) in its unmodified form with its hydroxyl group pointed toward the vacant metal binding site [58] (Figure 5A). This residue and the three conserved active site histidine residues that ligate copper in the native enzyme (His431, His433, and His592) are arranged around the empty metal binding site as if in a tetrahedral geometry. His431 and His433 adopt the conformers seen in the native enzyme, but His592 is present as two conformers, indicating some positional flexibility at this site. The two water molecules normally observed in the native CAO active site which ligate the copper (Wa and We in Figure 4A) are absent in the apoAGAO structure.
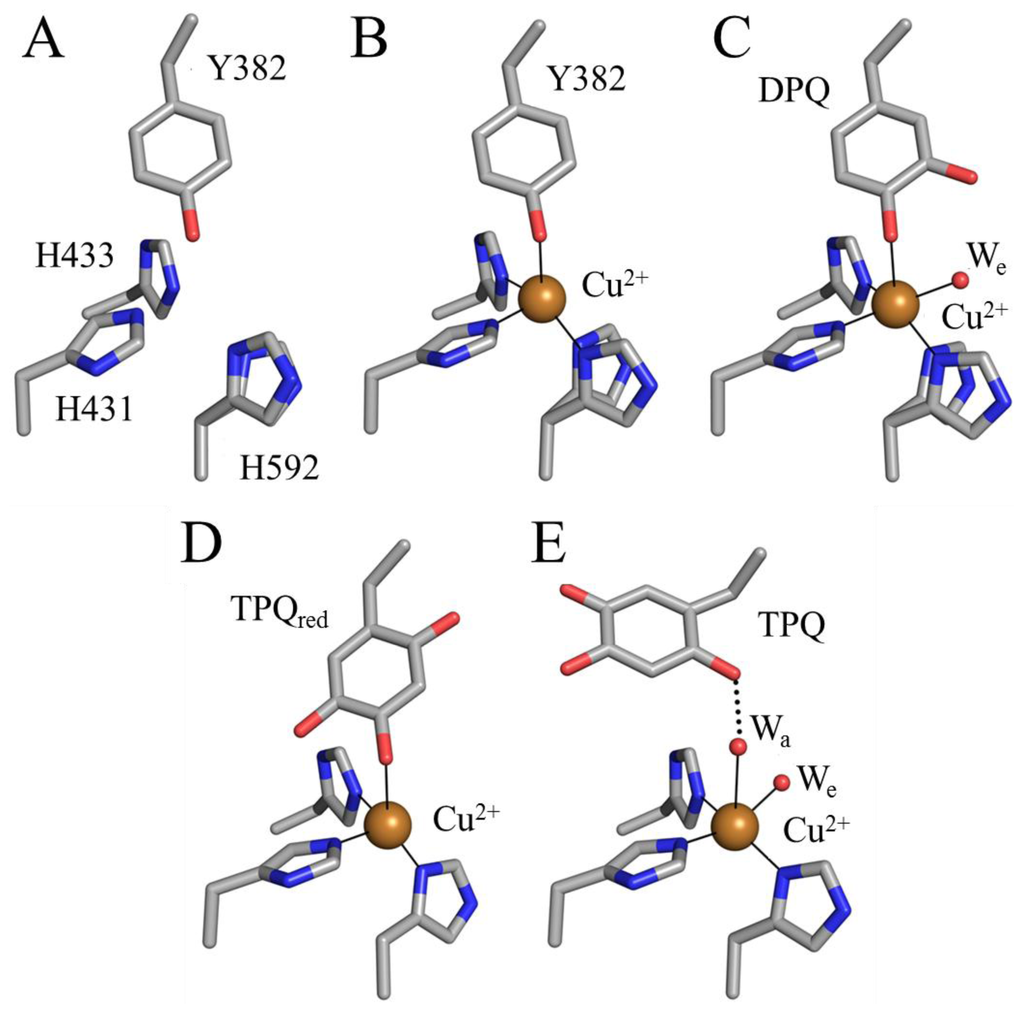
Figure 5.
Structurally characterized TPQ biogenesis intermediates in AGAO [58,69]: (A) apoAGAO (PDB code 1avk); (B) apoAGAO/Cu(II) complex (PDB code 1ivu); (C) dopaquinone (DPQ)-containing early intermediate (PDB code 1ivv); (D) 2-electron reduced TPQ (TPQred)-containing late intermediate (PDB code 1ivw) and (E) holoAGAO generated in the crystal (PDB code 1ivx). Residues are drawn in stick and colored by atom type (carbon, grey). Copper ions are drawn as gold spheres, and water molecules as small red spheres. Solid lines indicate metal-ligand interactions, and dashed lines indicate hydrogen bonds.
A series of crystal structures containing AGAO biogenesis intermediates were solved by exposing anaerobic apoAGAO crystals, which had been pre-bound with Cu(II), to O2 followed by freezing after different amounts of time [69]. This study produced structures of an anaerobic complex between apoAGAO and Cu(II) (flash-frozen after 0 min of O2 exposure), an early intermediate (flash-frozen after 10 min of O2 exposure), and a late intermediate (flash-frozen after 100 min of O2 exposure) formed during biogenesis (Figure 5B,C,D), which are consistent with the mechanism presented in Scheme 2. Finally, exposure of apoAGAO to O2 for a week before flash-freezing resulted in the structure of a species identical to holoAGAO, confirming that biogenesis can go to completion in the crystal (Figure 5E).
The structure of the anaerobic complex between apoAGAO and Cu(II) was solved to a resolution of 1.9 Å (Figure 5B). The electron density clearly shows the unmodified side chain of Tyr382, consistent with the requirement of molecular oxygen for TPQ biogenesis. The phenolic group of Tyr382 is thought to be protonated based on the distance between its hydroxyl group and the bound copper (~2.5 Å). Aside from the bound copper ion, this structure is essentially identical to that of apoAGAO (Figure 5A), and represents the first step of TPQ biogenesis, activating the precursor tyrosine side chain for monooxygenation at position 3 of the phenyl ring.
The structure of apoHPAO-1 in complex with Zn(II) (PDB code 1ekm) solved to 2.5 Å resolution in the presence of oxygen, reinforces the importance of the tyrosyl/copper complex in activating the precursor tyrosine ring for the initial oxygenation reaction [75]. Zinc is known to bind tightly at the CAO active site and resist displacement by copper [76]. Because zinc cannot support TPQ production, the structure of the Zn(II)-apoHPAO-1 complex mimics the copper bound species (B in Scheme 2). Similar to the anaerobic Cu(II)-apoAGAO complex, zinc was found to bind at the copper binding site and was ligated by the three conserved histidine residues and the unmodified precursor tyrosine residue in a tetrahedral geometry [75].
A crystal structure containing the dopaquinone intermediate formed during TPQ biogenesis (F in Scheme 2) was solved to a resolution of 2.1 Å (Figure 5C) [69]. The electron density confirmed that in this intermediate one oxygen atom had been inserted into the ring of Tyr382 to form dopaquinone. An additional feature of this structure is the presence of an equatorial water ligand at a distance ~2.1 Å from the copper center (We in Figure 5C) that is absent in the anaerobic complex between apoAGAO and Cu(II) (Figure 5B).
Freezing apoAGAO crystals after exposure to O2 for 100 minutes trapped TPQred (H in Scheme 2) solved to a resolution of 1.9 Å (Figure 5D) [69]. As indicated by the electron density, two oxygen atoms had been inserted into the Tyr382 ring, forming either TPQred or TPQ. This intermediate is formed only after rotation of the dopaquinone ring by ~180°, resulting in a species in which the O4 atom acts as a direct ligand to the bound copper. Single crystal microspectrophotometry experiments as well as the presence of a hydrogen bond between the O2 atom of the ring with the backbone carbonyl of Thr403 (indicating that the O2 atom is protonated), confirmed that this species is TPQred and not TPQ [69].
It was previously thought that only copper, the physiological relevant metal in CAOs, could support TPQ biogenesis. Studies conducted with apoHPAO-1 and apoAGAO, however, have revealed that biogenesis is supported by alternate metals in vitro, although at decreased rates [77,78]. In apoHPAO-1, biogenesis is supported by Cu(II), Cu(I), or Ni(II) bound at the active site, but does not occur upon Co(II) binding [78,79]. In contrast, biogenesis in apoAGAO is supported by Co(II) in addition to Cu(II) and Ni(II) (Cu(I) has not been tested) [77]. In both apoCAOs, Zn(II) binding at the active site renders the enzyme inert, as it does not support biogenesis and furthermore resists displacement by Cu(II) [75,77]. Several properties are thought to contribute to a metal’s ability to initiate and support biogenesis in apoCAOs. The low reduction potentials for the Ni(II)/Ni(I) and Co(II)/Co(I) couples disfavor a mechanism requiring the reduction of the metal for biogenesis [80,81]. Lewis acidity has been proposed to be important for biogenesis; however Zn(II), an effective Lewis acid, is unable to support TPQ synthesis, suggesting that reduction potential does play some role in determining which metals can initiate biogenesis [75,77].
The crystal structures of apoHPAO-1, Cu(I)-apoHPAO-1 (prepared anaerobically) and Co(II)-apoHPAO-1 have also been reported [71]. The active sites of apoHPAO-1 and Cu(I)-apoHPAO-1 are nearly superimposable with those of apoAGAO and Cu(II)-apoAGAO, respectively [58,69]. The structure of Co(II)-apoHPAO-1, however, reveals that cobalt binds in apoHPAO-1 and apoAGAO with different geometries (Figure 6). In apoHPAO-1, the cobalt is 5-coordinate, bound in a distorted square pyramidal geometry by the imidazole groups of three histidine residues, the precursor tyrosine residue, and an equatorial water molecule (Figure 6A) [71]. In contrast, cobalt in the apoAGAO structure is bound tetrahedrally by the three conserved histidine residues and the precursor tyrosine residue (Figure 6B) [77]. Given that Co(II) supports biogenesis in apoAGAO but not apoHPAO-1, it is likely that differences in metal coordination influence the ability of Co(II) to support biogenesis.
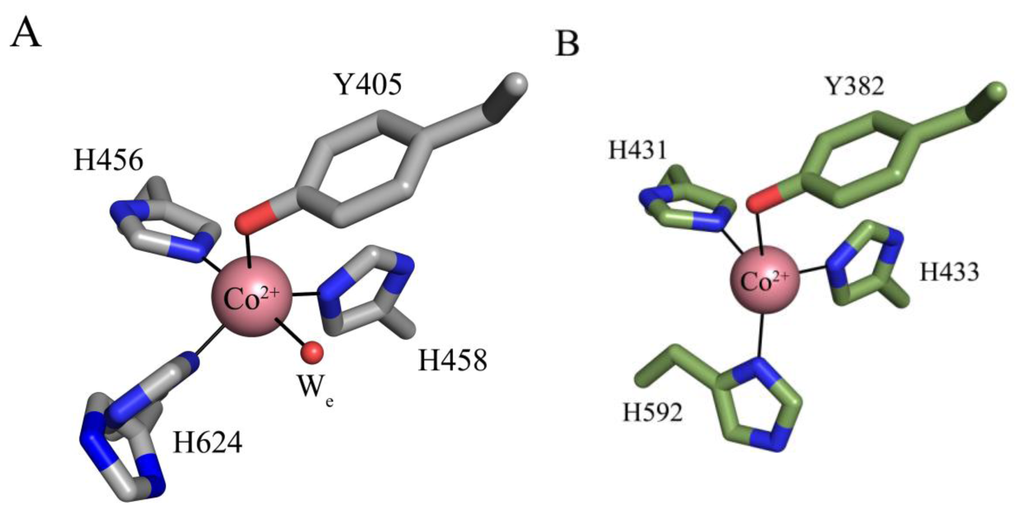
Figure 6.
Active sites of (A) Co(II)-apoHPAO-1 (PDB code 3sxx) and (B) Co(II)-apoAGAO (PDB code 1wmp) [71,77]. Residues are drawn in stick and colored by atom type (apoHPAO1: carbon, grey; apoAGAO: carbon, green). Cobalt ions are drawn as pink spheres, and a water molecule as a small red sphere. Ligand-metal interactions are indicated by solid lines. Figure adapted from [71].
Biogenesis requires precise control in terms of positioning the precursor tyrosine residue and the intermediates formed during biogenesis within the active site. These species undergo rotations about their Cα-Cβ and Cβ-Cγ bonds to occupy both on- and off-copper positions. A number of active site residues, including some within the consensus sequence which contains the precursor tyrosine residue, contribute to this high level of conformational regulation by stabilizing the emerging cofactor in conformations accessed during biogenesis (Figure 7).
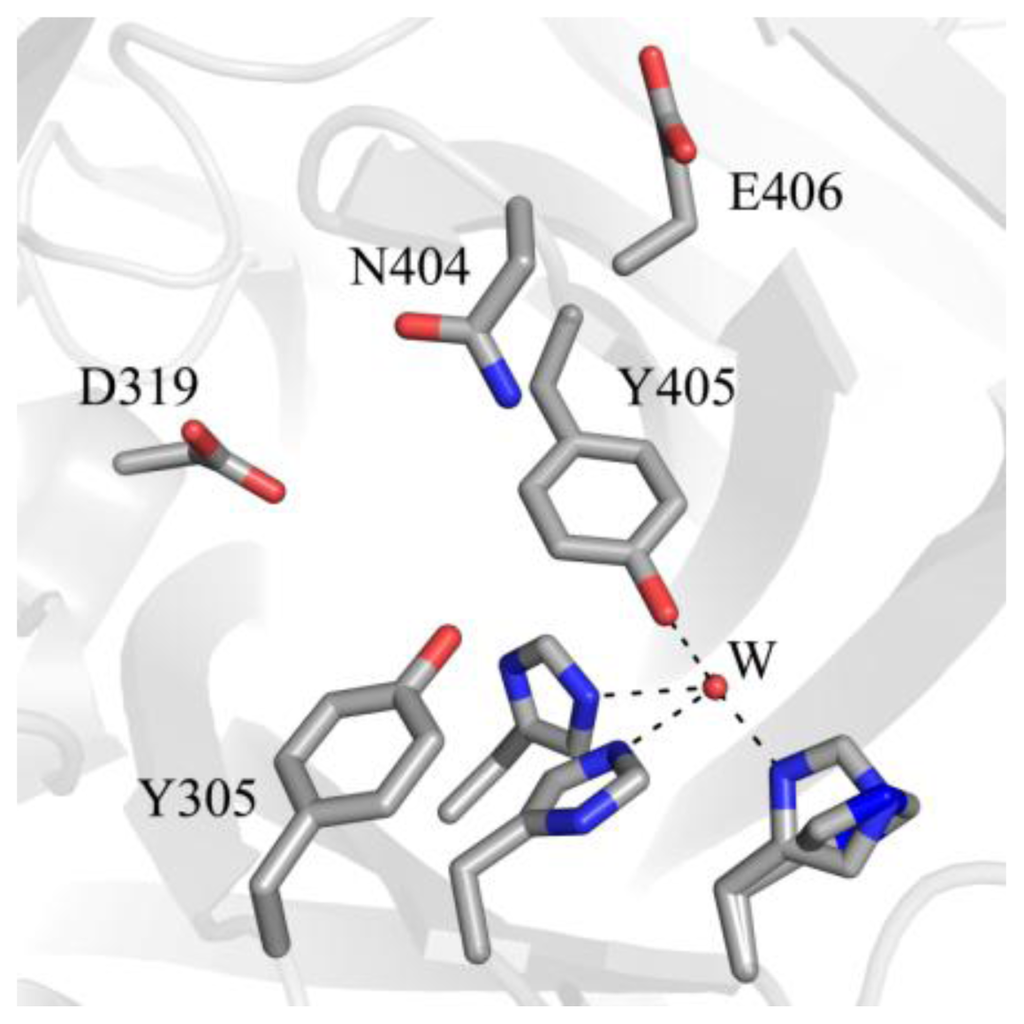
Figure 7.
Amino acid residues in apoHPAO-1 shown to be involved in stabilizing biogenesis intermediates (PDB code 3sx1) [71]. Residues are drawn in stick and colored by atom type (carbon, grey). A water molecule is drawn as a small red sphere. Dashed lines indicate hydrogen bonds.
A strictly conserved active site tyrosine residue (Tyr305 in HPAO-1) is involved in a short hydrogen bond with the O4 atom of TPQ, helping to stabilize it in the appropriate position for catalysis of amine oxidation. The mutation of Tyr305 to a phenylalanine residue interferes with normal O–O bond cleavage during biogenesis and results in indiscriminant oxidative damage, indicating that this strictly conserved residue plays a role in conformational stabilization of the precursor tyrosine residue during biogenesis as well [82]. In HPAO-1, the mutation of the strictly conserved residue N-terminal to the precursor tyrosine (Asn404) to an alanine results in only 5–10% TPQ formation relative to the native enzyme, while the mutation of Asn404 to an aspartate results in a 2-fold decrease in kTPQ [72,83]. The rate of TPQ biogenesis is also decreased by an order of magnitude when the residue C-terminal to the precursor tyrosine in HPAO-1 (Glu406) is mutated to a glutamine [72]. These effects illustrate the importance of the chemical properties of active site residues for biogenesis, such that mobility of the precursor tyrosine side chain is enabled during certain biogenesis steps, whereas a particular conformer is stabilized during others.
The TPQ cofactor provides a chromophoric handle that enables changes in the electronic properties of TPQ to be monitored. ApoCAO protein is colorless with no near-UV/visible absorption peak, whereas the mature protein is yellow-pink in color (λmax = 480 nm) due to electronic transitions within the cofactor [84]. When apoHPAO-1 is aerobically reconstituted with Cu(II), an intermediate absorbing at 380 nm forms and decays before the formation of the species at 480 nm which indicates mature TPQ (Figure 8). The rates of formation and decay of the 380 nm species are unaffected by pre-incubation with zinc or the addition of Cu(II) to the mature TPQ-containing HPAO-1. Thus, this species is thought to arise from an off-pathway LMCT interaction [72].
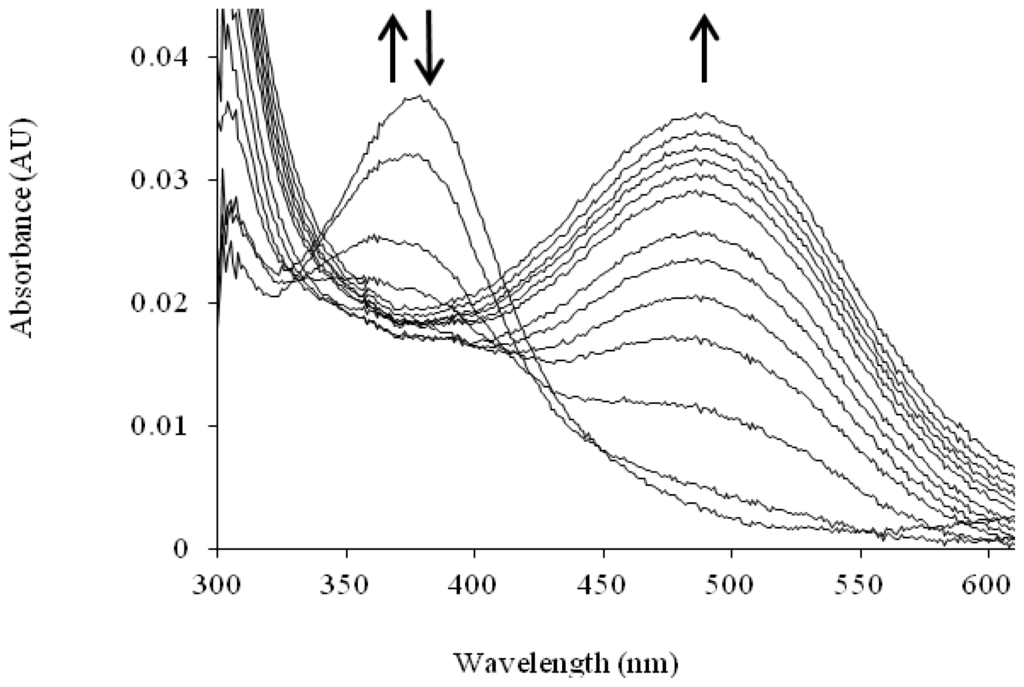
Figure 8.
Solution UV/visible spectra showing the time course of the aerobic reconstitution of apoHPAO-1 with Cu(II) at pH 7.0. The directions of change for near-UV/visible absorbance features over time are indicated by arrows. Figure from [71].
The copper bound at the CAO active site is ligated by three N and one/two O ligands and thus is type 2 or “non-blue” copper. Consequently, the intense spectral feature absorbing at ~600 nm associated with type 1 “blue” copper sites (ɛ = ~5000 M−1·cm−1) due to the presence of a cysteinic sulfur ligand is absent in CAOs [85]. The coupling of X-ray crystallography with single crystal spectroscopy is a powerful tool, allowing chromophoric species formed during enzymatic reactions to be identified before, during, and after X-ray data collection.
7. Catalysis in CAOs
Catalysis in CAOs utilizes a ping-pong mechanism and can be thought of as two distinct half-reactions: (1) the oxidation of a primary amine substrate, generating product aldehyde and the 2-electron reduced aminoquinol form of the cofactor in which O5 is displaced by a substrate-derived amine group (known as the reductive half-reaction) and (2) the re-oxidation of TPQ with concomitant reduction of O2 to H2O2 and the release of NH4 + (known as the oxidative half-reaction) (Scheme 3).

Scheme 3.
The catalytic half-reactions of CAOs. TPQamq, aminoquinol. E, enzyme.
The active sites of resting native CAOs contain oxidized TPQ and Cu(II) (A in Scheme 4). Catalysis is initiated through the nucleophilic attack by a primary amine on the C5 atom of TPQ forming a covalent substrate Schiff base complex (A→B in Scheme 4) [86]. Proton abstraction by a conserved aspartate residue from the C1 atom of the substrate, which is expected to have a decreased pKa, forms the corresponding product Schiff base species (D in Scheme 4) via the rapid rearrangement of a carbanionic intermediate (C→D in Scheme 4) [87]. Hydrolysis of the product Schiff base releases the corresponding aldehyde product and leaves the cofactor as a 2-electron reduced aminoquinol (D→E in Scheme 4) [88].
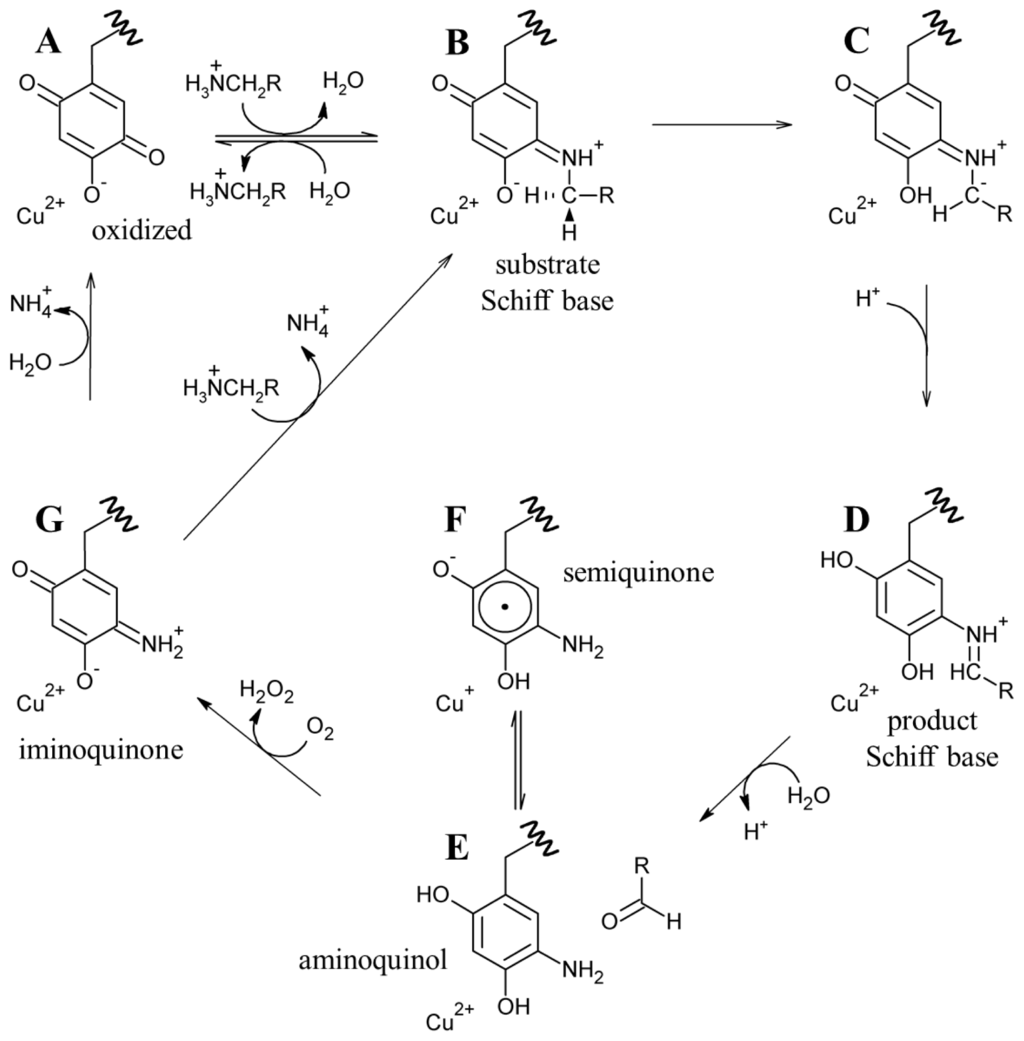
Scheme 4.
Proposed CAO catalytic mechanism. RCH3NH3 + is representative of all primary amine substrates.
The aminoquinol represents the conclusion of the reductive half-reaction, and acts as the initial species in the oxidative half-reaction. While the study of CAOs from several sources has led to a general consensus regarding the reductive half-reaction, details concerning the oxidative half-reaction have remained unclear, particularly concerning the nature of the first electron transfer from reduced cofactor to O2 [89]. Reduced TPQ exists in an equilibrium between the aminoquinol/Cu(II) couple and a semiquinone radical/Cu(I) form (E and F, respectively, in Scheme 4) [90]. The distribution of the aminoquinol/semiquinone equilibrium is source-, pH-, and temperature-dependent [91–93]. At pH 7, CAOs derived from plant sources form as much as 40% semiquinone when anaerobically reduced with substrate, while other non-plant eukaryotic CAOs contain very low levels of semiquinone nearing the limit of detection [91]. Bacterial CAOs contain semiquinone at levels somewhere in between those of the two eukaryotic groups.
Two proposals to describe the first electron transfer to O2 have been put forth. The first utilizes an inner sphere electron transfer mechanism, in which O2 binds to the reduced copper in the semiquinone/Cu(I) couple. This is followed by electron transfer from Cu(I) to O2 to form copper(II)/superoxide. The transfer of another electron from the semiquinone and two protons to the superoxide yields the iminoquinone form of cofactor (G in Scheme 4) and hydrogen peroxide [94]. Support for an inner sphere electron transfer mechanism derives from the detection of the semiquinone radical form of TPQ [90] and the demonstration of a catalytically competent electron transfer rate from the aminoquinol to Cu(II) [93,95]. Additionally, azide, which is expected to ligate copper, was found to exhibit competitive inhibition with respect to O2 in PSAO and a CAO isolated from pig plasma, as well as partially competitive inhibition in DAO [96,97].
An outer sphere electron transfer mechanism has also been proposed, in which an electron from aminoquinol is transferred directly to O2 bound in a nearby hydrophobic pocket, forming superoxide and the semiquinone [98,99]. This mechanism does not involve an obligate change in the oxidation state of the copper, with its primary role being to stabilize the resultant superoxide. A kinetic study using BSAO demonstrated that the single electron reduction of O2 is rate-limiting during the oxidative half-reaction [98]. Chemical intuition suggests that this should be a fast process if the electron is derived from reduced Cu(I), thus an outer sphere electron transfer mechanism consistent with the kinetic data was proposed. Major support for an outer sphere electron transfer mechanism derives from work done with HPAO-1 [99,100]. The removal of copper from the HPAO-1 active site followed by reconstitution with Co(II) resulted in fully-functional protein with kinetic parameters under O2-saturating conditions indistinguishable from those of the native enzyme [100]. In addition, copper-depleted lentil seedling amine oxidase (LSAO) which was reconstituted with Co(II) regained partial catalytic competency [101]. The reduction potential of the Co(II)/Co(I) couple is very low, for example −400 to −500 mV vs. SHE in methionine synthase [80]. Co(II) is thus unlikely to be reduced during Co(II)-mediated catalysis, suggesting that a redox role for copper is unnecessary in the native enzyme. The use of a hydrophobic oxygen binding site near the copper is supported by kinetic data indicating that oxygen binding in HPAO-1 is noncompetitive with azide predicted to bind at the active site copper [102]. In addition, the oxidation of a model compound for the reduced cofactor has been shown to occur in the absence of metal [99]. Finally, the semiquinone intermediate in some anaerobically reduced CAOs is virtually undetectable, which lends additional support to an outer sphere electron transfer mechanism [98].
The issue of the first electron transfer from cofactor to O2 is being actively pursued, and it has been suggested that CAOs may be capable of using more than one mechanism to reduce O2 but that each different CAO has a clear preference for either inner or outer sphere electron transfer [89,103,104]. Regardless, O2 ultimately accepts two electrons and two protons from the cofactor, yielding H2O2 and the iminoquinone (G in Scheme 4). Hydrolysis of the iminoquinone releases product ammonium and regenerates oxidized TPQ (G→A in Scheme 4). Alternatively, when substrate levels are high the iminoquinone can react with a second amine substrate, releasing product ammonium and generating the substrate Schiff base (G→B in Scheme 4).
Catalysis in CAOs can be monitored spectroscopically owing to changes in the electronic form of TPQ, both in solution and in crystallo (Table 2).
Table 2.
λmax of spectroscopic intermediates formed during CAO catalysis.
A number of catalytic intermediates formed during both half-reactions have been structurally characterized. The active sites from structures of native TPQ-containing CAOs from a variety of organisms (Table 1) are nearly identical, and contain oxidized TPQ, copper ligated by the imidazole groups of three strictly conserved histidine residues, an aspartate residue which acts as the catalytic base during catalysis, and several conserved water molecules. The active site structure of HPAO-1 is shown in Figure 9A [64]. As described previously, catalytically productive TPQ in the native enzyme is in an off-copper conformation with its C5 carbonyl pointed toward the substrate amine channel, ideally positioned for attack by a primary amine substrate. This conformation is stabilized by hydrogen bonding between the O2 atom of TPQ and an axial water molecule (Wa in Figure 9A), and between the O4 atom of TPQ and the hydroxyl of a conserved tyrosine residue. Results from resonance Raman spectroscopy using AGAO indicate that underivatized TPQ exhibits significant electron delocalization between the C2 and C4 oxygen atoms, with only the C5 atom possessing significant C=O character [111]. This is consistent with the formation of a covalent cofactor-substrate Schiff base complex following nucleophilic attack at the C5 position during catalysis as opposed to the C2 or C4 positions.
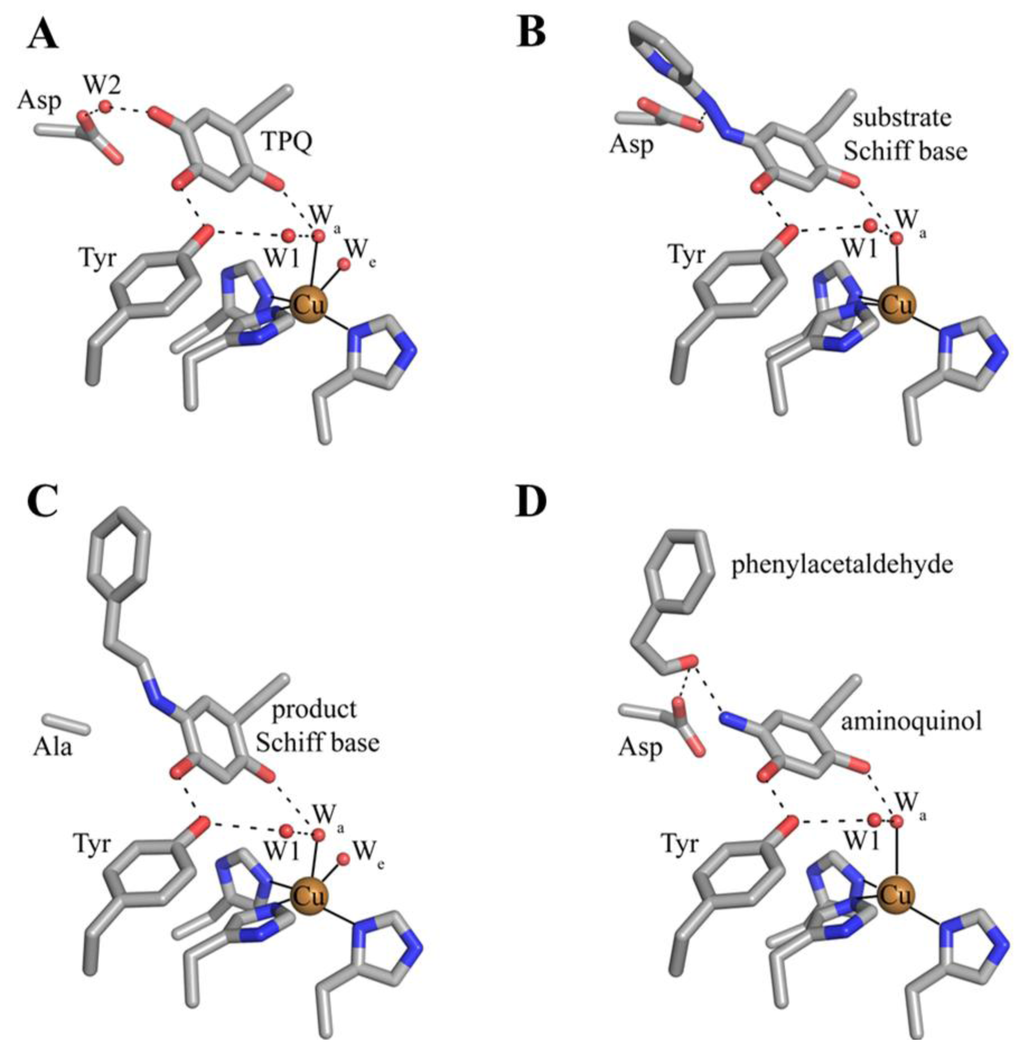
Figure 9.
Structurally characterized intermediates formed during the CAO reductive half-reaction. (A) native HPAO-1 active site (PDB code 2oov) [64]; (B) ECAO containing a substrate Schiff base analog formed with the inhibitor 2-hydrazinopyridine (PDB code 1spu) [86]; (C) D298A AGAO containing product Schiff base (PDB code 2cwv) [113]; (D) substrate-reduced ECAO containing the aminoquinol (PDB code 1d6u) [88]. Residues are drawn in stick and colored by atom type (carbon, grey). Copper ions are drawn as gold spheres, and water molecules as small red spheres. Dashed lines indicate hydrogen bonds, and solid lines indicate metal-ligand interactions.
The structure of ECAO in a covalent complex with the inhibitor 2-hydrazinopyridine has been solved to a resolution of 2.0 Å [86] (Figure 9B). The inhibitor was found to bind to atom C5 of the cofactor, displacing the O5 atom and generating a Schiff base analog (B in Scheme 4). Because 2-hydrazinopyridine contains a nitrogen atom instead of a carbon at the C1 position, the covalent complex formed with the inhibitor cannot be deprotonated and thus accumulates in the crystal. The pyridine and quinone rings are not coplanar, suggesting that the complex is an analog of the substrate Schiff base and not the product Schiff base. A notable feature of this structure is the hydrogen bond between a nitrogen atom (corresponding to C1 in a physiological Schiff base) of the inhibitor/cofactor complex and an active site strictly conserved aspartate residue, which suggested this residue as the general catalytic base in the reductive half-reaction that abstracts a proton from the substrate Schiff base [86]. This was unequivocally confirmed through the study of twelve amino acid variants at this site in ECAO, which established that only glutamic acid had catalytic activity, although its kcat was reduced by a factor of 6.4 × 104 compared to the native aspartic acid [86,87]. Recently, AGAO has also been solved in complex with three different hydrazine inhibitors: benzylhydrazine, 4-hydroxybenzylhydrazine, and phenylhydrazine [112]. As in ECAO, these bind as hydrazone adducts analogous to substrate Schiff base.
The mutation of the active site aspartate residue (D298) to an alanine in AGAO results in a decrease in catalytic efficiency by ~106 orders of magnitude, with the low level of activity possibly due to water acting as a base [113]. The incubation of D298A AGAO crystals with the physiological substrate 2-phenylethylamine for one week before freeze-trapping and structure solution resulted in a 1.85 Å resolution crystal structure containing a product Schiff base intermediate (Figure 9C). This assignment was confirmed by single crystal microspectrophotometry [113]. The product Schiff base has not been observed in solution, presumably due to the fast rate of hydrolysis which produces aldehyde and the aminoquinol (D→E in Scheme 4). In the crystal, however, the reaction is considerably slower and the product Schiff base is able to accumulate. The C2 atom of the product is coplanar with the TPQ ring and the imine double bond in this structure, which is anticipated for the product Schiff base formed with 2-phenylethylamine (Figure 9C).
The 2-electron reduced aminoquinol is the final cofactor intermediate formed during the reductive half-reaction, and marks the initiation of the oxidative half-reaction. Crystals containing the aminoquinol at the active site were obtained by freeze-trapping crystals of ECAO after their anaerobic reduction with 2-phenylethylamine. The structure of this species was solved to a resolution of 2.4 Å (Figure 9D) [88]. Single crystal microspectrophotometry indicated that the species was bleached, which is consistent with the aminoquinol (Table 2) [106]. Present in the off-copper conformation, the aminoquinol interacts with the copper ion through the axial water molecule seen in native CAO structures. A surprising feature in this structure was the presence of phenylacetaldehyde product bound at the back of the active site. Crystal contacts appeared to inhibit interdomain movement such that aldehyde remained trapped in the enzyme at the back of the active site [88].
Intermediates formed during the oxidative half-reaction which have been structurally characterized are shown in Figure 10. The oxidative half-reaction involves the reoxidation of TPQ, and begins with TPQ in an equilibrium between the aminoquinol/Cu(II) couple and a semiquinone radical/Cu(I) species. A structure containing the aminoquinol has been solved, and is described in the preceding paragraph. Currently there is no published crystal structure of a reduced CAO containing semiquinone and Cu(I).
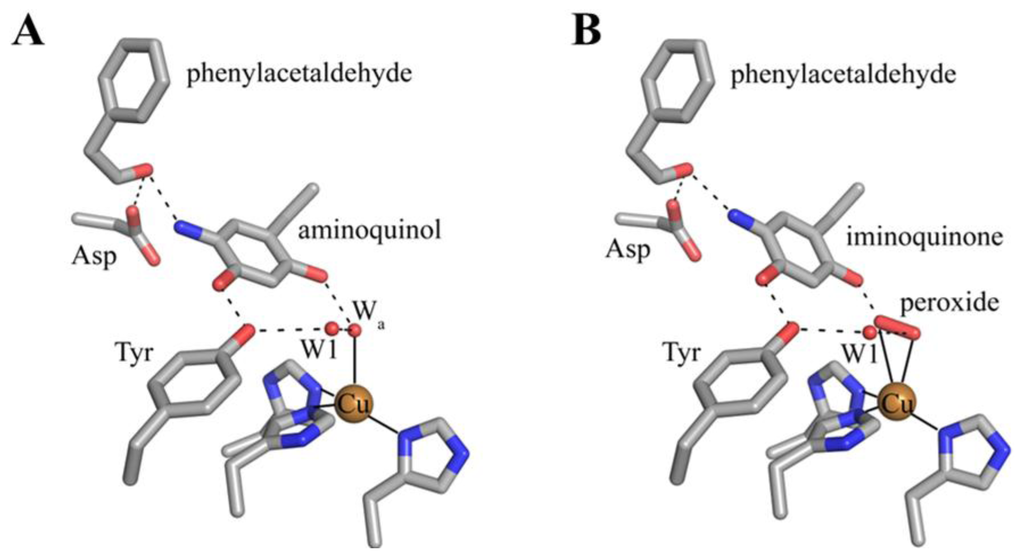
Figure 10.
Structurally characterized intermediates formed during the CAO oxidative half-reaction. (A) Substrate reduced ECAO containing the aminoquinol (PDB code 1d6u) [88]. (B) ECAO containing the iminoquinone (PDB code 1d6z) [88]. Residues are drawn in stick and colored by atom type (carbon, grey). Copper ions are drawn as gold spheres, and water molecules as small red spheres. Hydrogen bonding interactions are indicated by dashed lines, and metal-ligand interactions are indicated by solid lines.
A structure of the iminoquinone intermediate formed during catalysis was solved after the prolonged aerobic exposure of an ECAO crystal to 2-phenylethylamine (Figure 10B) [88]. This steady-state structure contains not only the iminoquinone, but also an oxygen species which has displaced the axial water molecule in the structure containing aminoquinol (Figure 10A). Phenylacetaldehyde product remains bound in the back of the active site in the same position it occupies in the aminoquinol-containing structure (Figure 10A). The side-on geometry of the oxygen species in the presence of iminoquinone and product aldehyde suggested that this was most likely the product hydrogen peroxide. The presence of an oxygen species bound at the copper coincident with product aldehyde demonstrates that in the crystal, the ping-pong kinetics seen in solution have been disrupted.
Finally, it is well documented that CAOs function as obligate dimers, and evidence suggests that some, but not all, CAOs exhibit cooperativity through long-range conformational changes propagated through the CAO dimer from one active site to the other [86,114–118]. BSAO and ANAO exhibit half-site reactivity with regard to hydrazine inhibitors [114,115,118]. In addition, kinetic work using HPAO-1 heterodimers which contain zinc bound in one active site of the HPAO-1 dimer and copper in the second, could not efficiently carry out the oxidative half-reaction at either active site, suggesting that communication between the two metal binding sites influences oxidative chemistry [119]. It has been suggested that metal binding in one CAO active site induces conformational changes through a network of interactions that crosses the dimer interface [119].
8. CAO Channels and Access to the Active Site
The substrates consumed during CAO catalysis (a primary amine and O2) take different paths to the same deeply-buried active site. A distinct substrate amine channel leads from the enzyme surface to the C5 atom of TPQ, the site of nucleophilic attack during catalysis (a distance of ~18 Å in HPAO-1) (Figure 11B). Despite the well-conserved structural homology of the overall CAO fold, the dimensions and shape of the amine substrate channel vary significantly depending on enzyme source. Amongst CAO homologs, only four amino acids within this channel display any sequence homology (besides the invariant aspartate residue which acts as the catalytic base and the consensus sequence containing the precursor tyrosine residue which is converted to TPQ). Position 323 (HPAO-1 numbering) corresponds to an aromatic residue, position 155 corresponds to either a proline or serine residue, position 305 corresponds to a tyrosine residue important in stabilizing TPQ species and off-copper conformations, and position 156 corresponds to a bulky hydrophobic residue. This channel not only allows substrate amine access to the site of catalytic turnover, but also functions as an egress for product aldehyde exiting the active site.
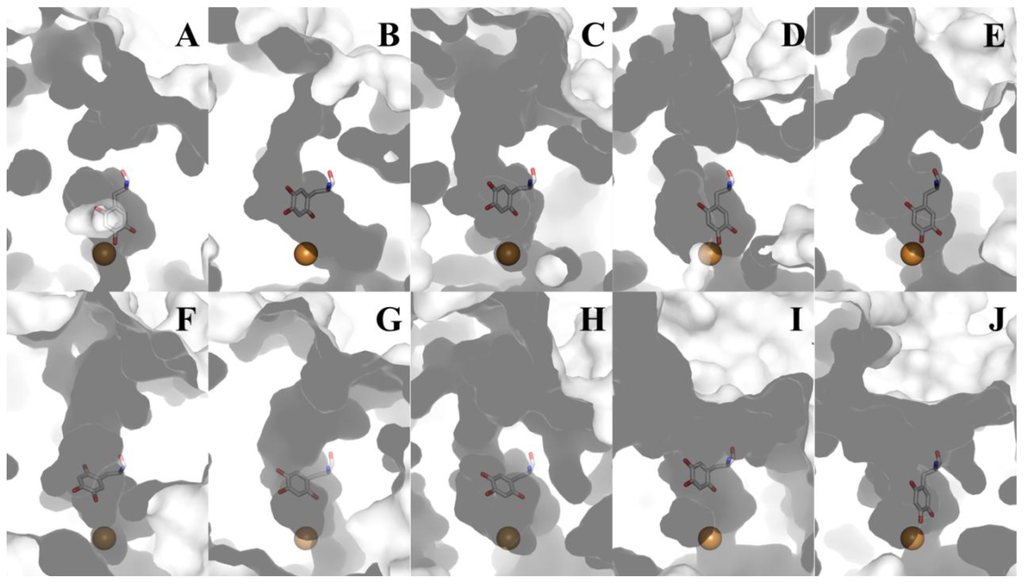
Figure 11.
Cross-section through the amine substrate channels of different CAOs. Each enzyme is drawn as a molecular surface. TPQ is drawn in stick and colored by atom type (carbon, white). Copper ions are drawn as gold spheres. (A) ECAO; (B) HPAO-1; (C) HPAO-2; (D) VAP-1; (E) DAO; (F) PSAO; (G) AGAO; (H) BSAO; (I) ANAO; (J) PPLO (Table 1).
Substrate preference in different CAOs is also determined by the chemical properties of the residues lining the amine substrate/aldehyde product channel, as well as the general shape and size of the channel. These vary significantly depending on enzyme source, with channels ranging from nearly obstructed (ECAO, Figure 11A) to channels so broad that entire peptides can be accommodated and serve as substrates ((I) ANAO and (J) PPLO, Figure 11).
An additional proposed role for the residues lining the amine substrate channel involves the stereospecificity of the proton abstraction step during the CAO reductive half-reaction. The stereospecificity varies depending on enzyme source and the identity of the amine substrate used [86,120–122]. For example, PSAO selectively abstracts the pro-S proton from dopamine, tyramine, and benzylamine during the reductive half-reaction, while BSAO is non-stereoselective when dopamine or tyramine are used as a substrate, but abstracts the pro-S proton from Schiff base complexes formed with benzylamine, p-hydroxybenzylamine, and 3-methylbutylamine [120,121]. In contrast, AGAO is known to selectively abstract the pro-S proton from Schiff base complexes formed with all substrates tested thus far [122]. In all CAO homologs, the pro-S proton from the Schiff base species formed with benzylamine is abstracted in favor of the pro-R proton. Given the strong structural homology between CAO active site residues, it has been suggested that the conformation of the substrate Schiff base complex itself is more important than the positions of active site residues relative to the amine substrate in determining the stereospecificity of the proton abstraction step [123]. The residues lining the amine substrate channel are important for the accommodation of Schiff base intermediates and display relatively low sequence homology amongst CAOs compared to other regions of the protein. The interactions between these residues and the Schiff base complexes consequently influence the stereospecificity of the proton abstraction step during catalysis in a species- and/or substrate-dependent manner.
The second substrate consumed during catalysis, molecular oxygen, had previously been thought to travel to the CAO active site via the “inland lake” where the two monomers in the CAO dimer meet. While it is possible that the inland lake may act as a reservoir for molecular oxygen (based on potential of mean force (PMF) maps which illustrate regions of low free energy for the placement of O2 inside the protein matrix) it has been proposed that the narrow polar channel connecting this region to the active site is more appropriate for exiting H2O2 [64]. This is consistent with both the short length and polar nature of H2O2.
In order to visualize molecular oxygen movement through the protein matrix, crystal structures of the complex between CAO and xenon have been solved from several sources, including PSAO [124], PPLO [124], ECAO [125], AGAO [124], and HPAO-1 [64]. Xenon has the same volume as molecular oxygen and mimics oxygen binding in hydrophobic pockets. Several recurrent O2 binding sites have been identified from these xenon complexes, including in the β-sheet sandwich fold of the catalytic D4 domain, the amine substrate channel, and a channel from the inland lake.
Taken together, crystallographic and PMF data from these studies suggest several species-dependent O2 points of entry into the protein interior (two are shown as black arrows in Figure 12). After its initial entry into the protein matrix, O2 is transiently held within the hydrophobic interior of domain D4, followed by migration closer to the active site where it is activated (Figure 12). Though the interior of domain D4 is largely composed of hydrophobic residues in all CAO homologs, source-specific differences in the primary amino acid sequence of D4 could account for the different points of entry utilized by O2 in different CAO homologs.
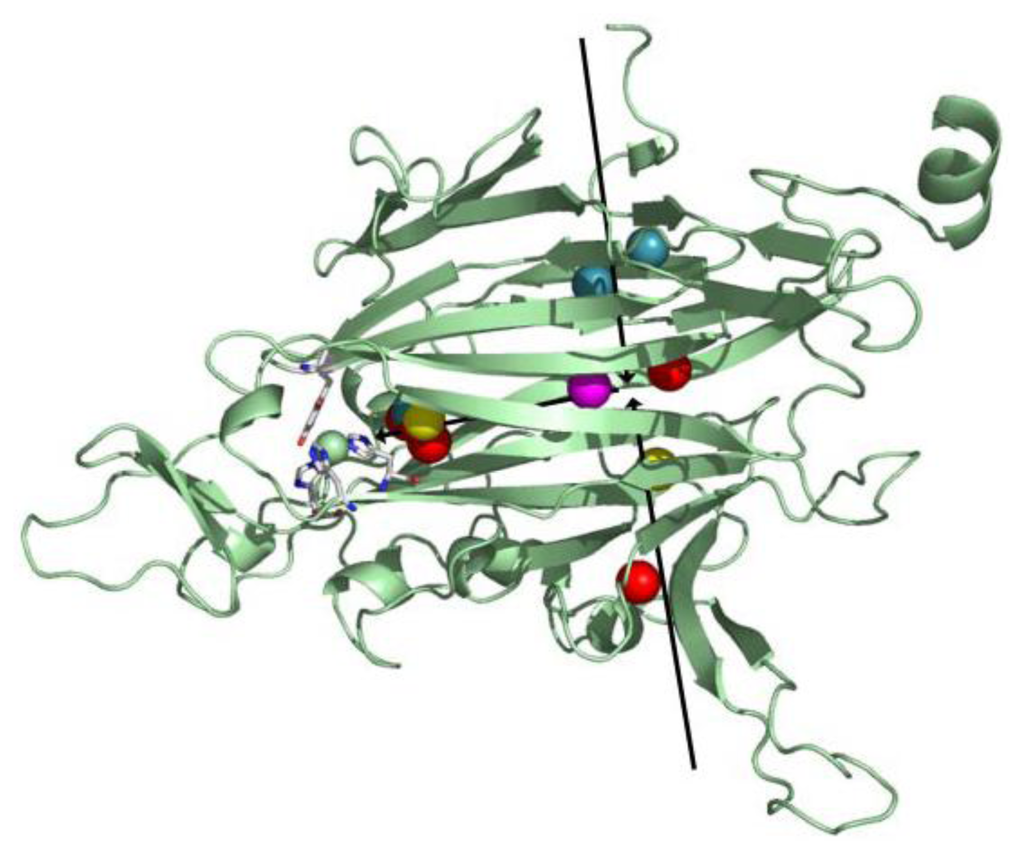
Figure 12.
Overlay of xenon binding sites in xenon/CAO complexes. Domain D4 from one HPAO-1 monomer is drawn as a green cartoon, and active site residues are depicted in stick and colored by atom type (carbon, white). Xenon atoms are drawn as spheres and colored by enzyme source (PSAO, red; PPLO, yellow; AGAO, blue; HPAO-1, magenta). Arrows indicate the direction of O2 movement toward the active site. Figure from [64].
9. Summary
The studies on CAOs demonstrate the power and complementarity of X-ray crystallography in understanding catalytic processes when combined with kinetic analysis, mutagenesis and spectroscopy, both in solution and in the crystal. The crystalline environment can dramatically slow down the rates of catalytic steps that depend on enzyme dynamics, and even alter the catalytic mechanism, as seen in the loss of ping-pong kinetics in ECAO catalysis in crystallo. However, when coupled to complementary single crystal spectroscopy any structural changes can effectively be equated to catalytic intermediates observed spectroscopically in solution. X-ray crystallography has been, and will remain, an important tool in the quest for understanding both CAO biogenesis and catalysis at the molecular level.
Acknowledgments
This work was supported by NIH grant GM66569 to C.M.W.
Abbreviations
| AGAO | Arthrobacter globiformis amine oxidase |
| ANAO | Aspergillus nidulans amine oxidase |
| apoCAO | precursor form of copper amine oxidase |
| BSAO | bovine serum amine oxidase |
| CAO | copper amine oxidase |
| CTQ | cysteine tryptophylquinone |
| DAO | diamine oxidase |
| DPQ | dopaquinone |
| ECAO | Escherichia coli amine oxidase |
| HPAO-1 | Hansenula polymorpha amine oxidase that prefers small aliphatic primary amines |
| HPAO-2 | Hansenula polymorpha amine oxidase that prefers aromatic primary amines |
| LMCT | ligand-metal charge transfer |
| LOX | lysyl oxidase |
| LOXL | lysyl oxidase-like proteins |
| LSAO | lentil seedling amine oxidase |
| LTQ | lysyl tyrosylquinone |
| MAO | monoamine oxidase |
| PDB | Protein Data Bank |
| PLP | pyridoxal phosphate |
| PMF | potential of mean force |
| PPLO | Pichia pastoris lysyl oxidase |
| PQQ | pyrroloquinoline quinone |
| PSAO | Pisum sativum amine oxidase |
| rmsd | root-mean-square deviation |
| SSAO | semicarbazide-sensitive amine oxidase |
| topaquinone or TPQ | 2,4,5-trihydroxyphenylalanine quinone |
| aminoquinol or TPQamq | 2,4-dihydroxy-5-aminophenylalanine |
| TPQred | 2,4,5-trihydroxyphenylalanine |
| TTQ | tryptophan tryptophylquinone |
| VAP-1 | vascular adhesion protein-1 |
References
- Shih, J.C.; Chen, K.; Ridd, M.J. Monoamine oxidase: From genes to behavior. Annu. Rev. Neurosci 1999, 22, 197–217. [Google Scholar]
- Seiler, N. Polyamine metabolism. Digestion 1990, 46, 319–330. [Google Scholar]
- Lucero, H.A.; Kagan, H.M. Lysyl oxidase: An oxidative enzyme and effector of cell function. Cell. Mol. Life Sci 2006, 63, 2304–2316. [Google Scholar]
- Okeley, N.M.; van der Donk, W.A. Novel cofactors via post-translational modifications of enzyme active sites. Chem. Biol 2000, 7, 159–171. [Google Scholar]
- Best, C.H. Disappearance of histamine from autolysing lung tissue. J. Physiol 1929, 67, 256. [Google Scholar]
- Parrott, S.; Jones, S.; Cooper, R.A. 2-phenylethylamine catabolism by Escherichia coli K12. J. Gen. Microbiol 1987, 133, 347–351. [Google Scholar]
- Van Dijken, J.P.; Bos, P. Utilization of amines in yeast. Arch. Microbiol 1981, 128, 320–324. [Google Scholar]
- Wimalasekera, R.; Villar, C.; Begum, T.; Scherer, G.F. Copper amine oxidase1 (CuAO1) of Arabidopsis thaliana contributes to abscisic acid- and polyamine-induced nitric oxide biosynthesis and abscisic acid signal transduction. Mol. Plant 2011, 4, 663–678. [Google Scholar]
- Cona, A.; Rea, G.; Angelini, R.; Federico, R.; Tavladoraki, P. Functions of amine oxidases in plant development and defence. Trends Plant Sci 2006, 11, 80–88. [Google Scholar]
- Angelini, R.; Cona, A.; Federico, R.; Fincato, P.; Tavladoraki, P.; Tisi, A. Plant amine oxidases “on the move”: An update. Plant Physiol. Biochem 2010, 48, 560–564. [Google Scholar]
- Campestre, M.P.; Bordenave, C.D.; Origone, A.C.; Menendez, A.B.; Ruiz, O.A.; Rodriguez, A.A.; Maiale, S.J. Polyamine catabolism is involved in response to salt stress in soybean hypocotyls. J. Plant Physiol 2011, 168, 1234–1240. [Google Scholar]
- Schwelberger, H.G. The origin of mammalian plasma amine oxidases. J. Neural Transm 2007, 114, 757–762. [Google Scholar]
- Novotny, W.F.; Chassande, O.; Baker, M.; Lazdunski, M.; Barbry, P. Diamine oxidase is the amiloride-binding protein and is inhibited by amiloride analogues. J. Biol. Chem 1994, 269, 9921–9925. [Google Scholar]
- Kaitaniemi, S.; Elovaara, H.; Grön, K.; Kidron, H.; Liukkonen, J.; Salminen, T.; Salmi, M.; Jalkanen, S.; Elima, K. The unique substrate specificity of human Aoc2, a semi-carbazide-sensitive amine oxidase. Cell. Mol. Life Sci 2009, 66, 2743–2757. [Google Scholar]
- Salmi, M.; Jalkanen, S. VAP-1: An adhesin and an enzyme. Trends Immunol 2001, 22, 211–216. [Google Scholar]
- Yraola, F.; Zorzano, A.; Albericio, F.; Royo, M. Structure-activity relationships of SSAO/VAP-1 arylalkylamine-based substrates. ChemMedChem 2009, 4, 495–503. [Google Scholar]
- Lyles, G.A. Substrate-specificity of mammalian tissue-bound semicarbazide-sensitive amine oxidase. Prog. Brain Res 1995, 106, 293–303. [Google Scholar]
- Gong, B.; Boor, P.J. The role of amine oxidases in xenobiotic metabolism. Expert Opin. Drug Metab. Toxicol 2006, 2, 559–571. [Google Scholar]
- Maintz, L.; Schwarzer, V.; Bieber, T.; van der ven, K.; Novak, N. Effects of histamine and diamine oxidase activities on pregnancy: A critical review. Hum. Reprod. Update 2008, 14, 485–495. [Google Scholar]
- McGrath, A.P.; Hilmer, K.M.; Collyer, C.A.; Shepard, E.M.; Elmore, B.O.; Brown, D.E.; Dooley, D.M.; Guss, J.M. Structure and inhibition of human diamine oxidase. Biochemistry 2009, 48, 9810–9822. [Google Scholar]
- Toninello, A.; Pietrangeli, P.; de Marchi, U.; Salvi, M.; Mondovi, B. Amine oxidases in apoptosis and cancer. Biochim. Biophys. Acta Rev. Cancer 2006, 1765, 1–13. [Google Scholar]
- Taksande, B.G.; Kotagale, N.R.; Patel, M.R.; Shelkar, G.P.; Ugale, R.R.; Chopde, C.T. Agmatine, an endogenous imidazoline receptor ligand modulates ethanol anxiolysis and withdrawal anxiety in rats. Eur. J. Pharmacol 2010, 637, 89–101. [Google Scholar]
- Dunkel, P.; Gelain, A.; Barlocco, D.; Haider, N.; Gyires, K.; Sperlagh, B.; Magyar, K.; Maccioni, E.; Fadda, A.; Matyus, P. Semicarbazide-sensitive amine oxidase/vascular adhesion protein 1: Recent developments concerning substrates and inhibitors of a promising therapeutic target. Curr. Med. Chem 2008, 15, 1827–1839. [Google Scholar]
- McDonald, A.; Tipton, K.; O'Sullivan, J.; Olivieri, A.; Davey, G.; Coonan, A.M.; Fu, W. Modelling the roles of MAO and SSAO in glucose transport. J. Neural Transm 2007, 114, 783–786. [Google Scholar]
- Kivi, E.; Elima, K.; Aalto, K.; Nymalm, Y.; Auvinen, K.; Koivunen, E.; Otto, D.M.; Crocker, P.R.; Salminen, T.A.; Salmi, M.; et al. Human Siglec-10 can bind to vascular adhesion protein-1 and serves as its substrate. Blood 2009, 114, 5385–5392. [Google Scholar]
- Stolen, C.M.; Marttila-Ichihara, F.; Koskinen, K.; Yegutkin, G.G.; Turja, R.; Bono, P.; Skurnik, M.; Hanninen, A.; Jalkanen, S.; Salmi, M. Absence of the endothelial oxidase Aoc3 leads to abnormal leukocyte traffic in vivo. Immunity 2005, 22, 105–115. [Google Scholar]
- Mathys, K.C.; Ponnampalam, S.N.; Padival, S.; Nagaraj, R.H. Semicarbazide-sensitive amine oxidase in aortic smooth muscle cells mediates synthesis of a methylglyoxal-AGE: Implications for vascular complications in diabetes. Biochem. Biophys. Res. Commun 2002, 297, 863–869. [Google Scholar]
- Jiang, Z.J.; Richardson, J.S.; Yu, P.H. The contribution of cerebral vascular semicarbazide-sensitive amine oxidase to cerebral amyloid angiopathy in Alzheimer’s disease. Neuropathol. Appl. Neurobiol 2008, 34, 194–204. [Google Scholar]
- Janes, S.M.; Mu, D.; Wemmer, D.; Smith, A.J.; Kaur, S.; Maltby, D.; Burlingame, A.L.; Klinman, J.P. A new redox cofactor in eukaryotic enzymes: 6-hydroxydopa at the active site of bovine serum amine oxidase. Science 1990, 248, 981–987. [Google Scholar]
- Klabunde, T.; Eicken, C.; Sacchettini, J.C.; Krebs, B. Crystal structure of a plant catechol oxidase containing a dicopper center. Nat. Struct. Biol 1998, 5, 1084–1090. [Google Scholar]
- Lerch, K. Primary structure of tyrosinase from Neurospora crassa: Purification and amino acid sequence and chemical structure of a tripeptide containing an unusual thioether. J. Biol. Chem 1982, 257, 6414–6419. [Google Scholar]
- Bravo, J.; Fita, I.; Ferrer, J.C.; Ens, W.; Hillar, A.; Switala, J.; Loewen, P.C. Identification of a novel bond between a histidine and the essential tyrosine in catalase HPII of Escherichia coli. Protein Sci 1997, 6, 1016–1023. [Google Scholar]
- Yoshikawa, S.; Shinzawa-Itoh, K.; Nakashima, R.; Yaono, R.; Yamashita, E.; Inoue, N.; Yao, M.; Fei, M.J.; Libeu, C.P.; Mizushima, T.; et al. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science 1998, 280, 1723–1729. [Google Scholar]
- Ostermeier, C.; Harrenga, A.; Ermler, U.; Michel, H. Structure at 2.7 Angstrom resolution of the Paracoccus denitrificans two-subunit cytochrome c oxidase complexed with an antibody F-V fragment. Proc. Natl. Acad. Sci. USA 1997, 94, 10547–10553. [Google Scholar]
- Ito, N.; Phillips, S.E.V.; Stevens, C.; Ogel, Z.B.; McPherson, M.J.; Keen, J.N.; Yadav, K.D.S.; Knowles, P.F. Novel thioether bond revealed by a 1.7 Å crystal structure of galactose oxidase. Nature 1991, 350, 87–90. [Google Scholar]
- Gielens, C.; DeGeest, N.; Xin, X.Q.; Devreese, B.; van Beeumen, J.; Preaux, G. Evidence for a cysteine-histidine thioether bridge in functional units of molluscan haemocyanins and location of the disulfide bridges in functional units d and g of the beta(c)-haemocyanin of Helix pomatia. Eur. J. Biochem 1997, 248, 879–888. [Google Scholar]
- Taylor, T.C.; Andersson, I. The structure of the complex between rubisco and its natural substrate ribulose 1,5-bisphosphate. J. Mol. Biol 1997, 265, 432–444. [Google Scholar]
- Jabri, E.; Carr, M.B.; Hausinger, R.P.; Karplus, P.A. The crystal structure of urease from Klebsiella aerogenes. Science 1995, 268, 998–1004. [Google Scholar]
- Benning, M.M.; Kuo, J.M.; Raushel, F.M.; Holden, H.M. 3-dimensional structure of the binuclear metal center of phosphotriesterase. Biochemistry 1995, 34, 7973–7978. [Google Scholar]
- Yeh, J.I.; Claiborne, A.; Hol, W.G. Structure of the native cysteine-sulfenic acid redox center of enterococcal NADH peroxidase refined at 2.8 Å resolution. Biochemistry 1996, 35, 9951–9957. [Google Scholar]
- Schmidt, B.; Selmer, T.; Ingendoh, A.; von Figura, K. A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell 1995, 82, 271–278. [Google Scholar]
- Nagashima, S.; Nakasako, M.; Dohmae, N.; Tsujimura, M.; Takio, K.; Odaka, M.; Yohda, M.; Kamiya, N.; Endo, I. Novel non-heme iron center of nitrile hydratase with a claw setting of oxygen atoms. Nat. Struct. Biol 1998, 5, 347–351. [Google Scholar]
- Claiborne, A.; Yeh, J.I.; Mallett, T.C.; Luba, J.; Crane, E.J.; Charrier, V.; Parsonage, D. Protein-sulfenic acids: Diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry 1999, 38, 15407–15416. [Google Scholar]
- Wang, S.X.; Mure, M.; Medzihradszky, K.F.; Burlingame, A.L.; Brown, D.E.; Dooley, D.M.; Smith, A.J.; Kagan, H.M.; Klinman, J.P. A crosslinked cofactor in lysyl oxidase: Redox function for amino acid side chains. Science 1996, 273, 1078–1084. [Google Scholar]
- Mcintire, W.S.; Wemmer, D.E.; Chistoserdov, A.; Lidstrom, M.E. A new cofactor in a prokaryotic enzyme: Tryptophan tryptophylquinone as the redox prosthetic group in methylamine dehydrogenase. Science 1991, 252, 817–824. [Google Scholar]
- Vandenberghe, I.; Kim, J.K.; Devreese, B.; Hacisalihoglu, A.; Iwabuki, H.; Okajima, T.; Kuroda, S.; Adachi, O.; Jongejan, J.A.; Duine, J.A.; et al. The covalent structure of the small subunit from Pseudomonas putida amine dehydrogenase reveals the presence of three novel types of internal cross-linkages, all involving cysteine in a thioether bond. J. Biol. Chem 2001, 276, 42923–42931. [Google Scholar]
- Yasunobu, K.T.; Ishizaki, H.; Minamiura, N. The molecular mechanistic and immunological properties of amine oxidases. Mol. Cell. Biochem 1976, 13, 3–29. [Google Scholar]
- Ameyama, M.; Hayashi, M.; Matsushita, K.; Shinagawa, E.; Adachi, O. Microbial production of pyrroloquinoline quinone. Agric. Biol. Chem 1984, 48, 561–565. [Google Scholar]
- Lobenstein-Verbeek, C.L.; Jongejan, J.A.; Frank, J.; Duine, J.A. Bovine serum amine oxidase—A mammalian enzyme having covalently bound PQQ as prosthetic group. FEBS Lett 1984, 170, 305–309. [Google Scholar]
- Moog, R.S.; McGuirl, M.A.; Cote, C.E.; Dooley, D.M. Evidence for methoxatin (pyrroloquinolinequinone) as the cofactor in bovine plasma amine oxidase from resonance Raman spectroscopy. Proc. Natl. Acad. Sci. USA 1986, 83, 8435–8439. [Google Scholar]
- Knowles, P.F.; Pandeya, K.B.; Rius, F.X.; Spencer, C.M.; Moog, R.S.; McGuirl, M.A.; Dooley, D.M. The organic cofactor in plasma amine oxidase—Evidence for pyrroloquinoline quinone and against pyridoxal phosphate. Biochem. J 1987, 241, 603–608. [Google Scholar]
- Airenne, T.T.; Nymalm, Y.; Kidron, H.; Smith, D.J.; Pihlavisto, M.; Salmi, M.; Jalkanen, S.; Johnson, M.S.; Salminen, T.A. Crystal structure of the human vascular adhesion protein-1: Unique structural features with functional implications. Protein Sci 2005, 14, 1964–1974. [Google Scholar]
- Jakobsson, E.; Nilsson, J.; Ogg, D.; Kleywegt, G.J. Structure of human semicarbazide-sensitive amine oxidase/vascular adhesion protein-1. Acta Crystallogr. Sect. D Biol. Crytallogr 2005, 61, 1550–1562. [Google Scholar]
- Lunelli, M.; di Paolo, M.L.; Biadene, M.; Calderone, V.; Battistutta, R.; Scarpa, M.; Rigo, A.; Zanotti, G. Crystal structure of amine oxidase from bovine serum. J. Mol. Biol 2005, 346, 991–1004. [Google Scholar]
- Li, R.; Klinman, J.P.; Mathews, F.S. Copper amine oxidase from Hansenula polymorpha: The crystal structure determined at 2.4 Å resolution reveals the active conformation. Structure 1998, 6, 293–307. [Google Scholar]
- Chang, C.M.; Klema, V.J.; Johnson, B.J.; Mure, M.; Klinman, J.P.; Wilmot, C.M. Kinetic and structural analysis of substrate specificity in two copper amine oxidases from Hansenula polymorpha. Biochemistry 2010, 49, 2540–2550. [Google Scholar]
- Duff, A.P.; Cohen, A.E.; Ellis, P.J.; Kuchar, J.A.; Langley, D.B.; Shepard, E.M.; Dooley, D.M.; Freeman, H.C.; Guss, J.M. The crystal structure of Pichia pastoris lysyl oxidase. Biochemistry 2003, 42, 15148–15157. [Google Scholar]
- Wilce, M.C.; Dooley, D.M.; Freeman, H.C.; Guss, J.M.; Matsunami, H.; McIntire, W.S.; Ruggiero, C.E.; Tanizawa, K.; Yamaguchi, H. Crystal structures of the copper-containing amine oxidase from Arthrobacter globiformis in the holo and apo forms: Implications for the biogenesis of topaquinone. Biochemistry 1997, 36, 16116–1633. [Google Scholar]
- Parsons, M.R.; Convery, M.A.; Wilmot, C.M.; Yadav, K.D.; Blakeley, V.; Corner, A.S.; Phillips, S.E.; McPherson, M.J.; Knowles, P.F. Crystal structure of a quinoenzyme: copper amine oxidase of Escherichia coli at 2 Å resolution. Structure 1995, 3, 1171–1184. [Google Scholar]
- Kumar, V.; Dooley, D.M.; Freeman, H.C.; Guss, J.M.; Harvey, I.; McGuirl, M.A.; Wilce, M.C.; Zubak, V.M. Crystal structure of a eukaryotic (pea seedling) copper-containing amine oxidase at 2.2 Å resolution. Structure 1996, 4, 943–955. [Google Scholar]
- McGrath, A.P.; Mithieux, S.M.; Collyer, C.A.; Bakhuis, J.G.; van den Berg, M.; Sein, A.; Heinz, A.; Schmelzer, C.; Weiss, A.S.; Guss, J.M. Structure and activity of Aspergillus nidulans copper amine oxidase. Biochemistry 2011, 50, 5718–5730. [Google Scholar]
- Smith, M.A.; Pirrat, P.; Pearson, A.R.; Kurtis, C.R.; Trinh, C.H.; Gaule, T.G.; Knowles, P.F.; Phillips, S.E.; McPherson, M.J. Exploring the roles of the metal ions in Escherichia coli copper amine oxidase. Biochemistry 2010, 49, 1268–1280. [Google Scholar]
- Sebela, M.; Luhova, L.; Frebort, I.; Hirota, S.; Faulhammer, H.G.; Stuzka, V.; Pec, P. Confirmation of the presence of a Cu(II) topa quinone active site in the amine oxidase from fenugreek seedlings. J. Exp. Bot 1997, 48, 1897–1907. [Google Scholar]
- Johnson, B.J.; Cohen, J.; Welford, R.W.; Pearson, A.R.; Schulten, K.; Klinman, J.P.; Wilmot, C.M. Exploring molecular oxygen pathways in Hansenula polymorpha copper-containing amine oxidase. J. Biol. Chem 2007, 282, 17767–17776. [Google Scholar]
- Cai, D.; Klinman, J.P. Evidence of a self-catalytic mechanism of 2,4,5-trihydroxyphenylalanine quinone biogenesis in yeast copper amine oxidase. J. Biol. Chem 1994, 269, 32039–32042. [Google Scholar]
- Matsuzaki, R.; Fukui, T.; Sato, H.; Ozaki, Y.; Tanizawa, K. Generation of the topa quinone cofactor in bacterial monoamine oxidase by cupric ion-dependent autooxidation of a specific tyrosyl residue. FEBS Lett 1994, 351, 360–364. [Google Scholar]
- Ruggiero, C.E.; Dooley, D.M. Stoichiometry of the topa quinone biogenesis reaction in copper amine oxidases. Biochemistry 1999, 38, 9556–9556. [Google Scholar]
- Mu, D.; Janes, S.M.; Smith, A.J.; Brown, D.E.; Dooley, D.M.; Klinman, J.P. Tyrosine codon corresponds to topa quinone at the active site of copper amine oxidases. J. Biol. Chem 1992, 267, 7979–7982. [Google Scholar]
- Kim, M.; Okajima, T.; Kishishita, S.; Yoshimura, M.; Kawamori, A.; Tanizawa, K.; Yamaguchi, H. X-ray snapshots of quinone cofactor biogenesis in bacterial copper amine oxidase. Nat. Struct. Biol 2002, 9, 591–596. [Google Scholar]
- Schwartz, B.; Dove, J.E.; Klinman, J.P. Kinetic analysis of oxygen utilization during cofactor biogenesis in a copper-containing amine oxidase from yeast. Biochemistry 2000, 39, 3699–3707. [Google Scholar]
- Klema, V.J.; Johnson, B.J.; Klinman, J.P.; Wilmot, C.M. The precursor form of Hansenula polymorpha copper amine oxidase-1 in complex with Cu(I) and Co(II). Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun 2012, 68, 501–510. [Google Scholar]
- Dove, J.E.; Schwartz, B.; Williams, N.K.; Klinman, J.P. Investigation of spectroscopic intermediates during copper-binding and TPQ formation in wild-type and active-site mutants of a copper-containing amine oxidase from yeast. Biochemistry 2000, 39, 3690–3698. [Google Scholar]
- Goto, Y.; Klinman, J.P. Binding of dioxygen to non-metal sites in proteins: exploration of the importance of binding site size versus hydrophobicity in the copper amine oxidase from Hansenula polymorpha. Biochemistry 2002, 41, 13637–13643. [Google Scholar]
- Dubois, J.; Klinman, J.P. The nature of O2 reactivity leading to topa quinone in the copper amine oxidase from Hansenula polymoropha and its relationship to catalytic turnover. Biochemistry 2005, 44, 11381–11388. [Google Scholar]
- Chen, Z.; Schwartz, B.; Williams, N.K.; Li, R.; Klinman, J.P.; Mathews, F.S. Crystal structure at 2.5 Å resolution of zinc-substituted copper amine oxidase of Hansenula polymorpha expressed in Escherichia coli. Biochemistry 2000, 39, 9709–9717. [Google Scholar]
- McGrath, A.P.; Caradoc-Davies, T.; Collyer, C.A.; Guss, J.M. Correlation of active site metal content in human diamine oxidase with trihydroxyphenylalanine quinone cofactor biogenesis. Biochemistry 2010, 49, 8316–8324. [Google Scholar]
- Okajima, T.; Kishishita, S.; Chiu, Y.C.; Murakawa, T.; Kim, M.; Yamaguchi, H.; Hirota, S.; Kuroda, S.; Tanizawa, K. Reinvestigation of metal ion specificity for quinone cofactor biogenesis in bacterial copper amine oxidase. Biochemistry 2005, 44, 12041–12048. [Google Scholar]
- Samuels, N.M.; Klinman, J.P. 2,4,5-trihydroxyphenylalanine quinone biogenesis in the copper amine oxidase from Hansenula polymorpha with the alternate metal nickel. Biochemistry 2005, 44, 14308–14317. [Google Scholar]
- Samuels, N.M.; Klinman, J.P. Investigation of Cu(I)-dependent 2,4,5-trihydroxyphenylalanine quinone biogenesis in Hansenula polymorpha amine oxidase. J. Biol. Chem 2006, 281, 21114–21118. [Google Scholar]
- Drummond, J.T.; Matthews, R.G. Nitrous oxide degradation by cobalamin-dependent methionine synthase: Characterization of the reactants and products in the inactivation reaction. Biochemistry 1994, 33, 3732–4371. [Google Scholar]
- Gomes, L.; Pereira, E.; de Castro, B. Nickel(II) complexes with N2OS and N2S2 co-ordination spheres: Reduction and spectroscopic study of the corresponding Ni(I) complexes. J. Chem. Soc. Dalton Trans 2000, 8, 1373–1379. [Google Scholar]
- Chen, Z.W.; Datta, S.; Dubois, J.L.; Klinman, J.P.; Mathews, F.S. Mutation at a strictly conserved, active site tyrosine in the copper amine oxidase leads to uncontrolled oxygenase activity. Biochemistry 2010, 49, 7393–7402. [Google Scholar]
- Schwartz, B.; Green, E.L.; Sanders-Loehr, J.; Klinman, J.P. Relationship between conserved consensus site residues and the productive conformation for the TPQ cofactor in a copper-containing amine oxidase from yeast. Biochemistry 1998, 37, 16591–16600. [Google Scholar]
- DuBois, J.L.; Klinman, J.P. Methods for characterizing TPQ-containing proteins. Methods Enzymol 2004, 378, 17–31. [Google Scholar]
- Holm, R.H.; Kennepohl, P.; Solomon, E.I. Structural and functional aspects of metal sites in biology. Chem. Rev 1996, 96, 2239–2314. [Google Scholar]
- Wilmot, C.M.; Murray, J.M.; Alton, G.; Parsons, M.R.; Convery, V.B.; Corner, A.S.; Palcic, M.M.; Knowles, P.F.; McPherson, M.J.; Phillips, S.E. Catalytic mechanism of the quinoenzyme amine oxidase from Escherichia coli: Exploring the reductive half-reaction. Biochemistry 1997, 36, 1608–1620. [Google Scholar]
- Murray, J.M.; Saysell, C.G.; Wilmot, C.M.; Tambyrajah, W.S.; Jaeger, J.; Knowles, P.F.; Phillips, S.E.; McPherson, M.J. The active site base controls cofactor reactivity in Escherichia coli amine oxidase: X-ray crystallographic studies with mutational variants. Biochemistry 1999, 38, 8217–8227. [Google Scholar]
- Wilmot, C.M.; Hajdu, J.; McPherson, M.J.; Phillips, S.E. Visualization of dioxygen bound to copper during enzyme catalysis. Science 1999, 286, 1724–1728. [Google Scholar]
- Pietrangeli, P.; Nocera, S.; Mondovi, B.; Morpurgo, L. Is the catalytic mechanism of bacteria, plant, and mammal copper-TPQ amine oxidases identical? Biochim. Biophys. Acta 2003, 1647, 152–156. [Google Scholar]
- Dooley, D.M.; McGuirl, M.A.; Brown, D.E.; Turowski, P.N.; McIntire, W.S.; Knowles, P.F. A Cu(I)-semiquinone state in substrate-reduced amine oxidases. Nature 1991, 349, 262–264. [Google Scholar]
- Welford, R.W.; Lam, A.; Mirica, L.M.; Klinman, J.P. Partial conversion of Hansenula polymorpha amine oxidase into a “plant” amine oxidase: Implications for copper chemistry and mechanism. Biochemistry 2007, 46, 10817–10827. [Google Scholar]
- Medda, R.; Padiglia, A.; Bellelli, A.; Pedersen, J.Z.; Agro, A.F.; Floris, G. Cu(I)-semiquinone radical species in plant copper-amine oxidases. FEBS Lett 1999, 453, 1–5. [Google Scholar]
- Shepard, E.M.; Dooley, D.M. Intramolecular electron transfer rate between active-site copper and TPQ in Arthrobacter globiformis amine oxidase. J. Biol. Inorg. Chem 2006, 11, 1039–1048. [Google Scholar]
- Mukherjee, A.; Smirnov, V.V.; Lanci, M.P.; Brown, D.E.; Shepard, E.M.; Dooley, D.M.; Roth, J.P. Inner-sphere mechanism for molecular oxygen reduction catalyzed by copper amine oxidases. J. Am. Chem. Soc 2008, 130, 9459–9473. [Google Scholar]
- Turowski, P.N.; McGuirl, M.A.; Dooley, D.M. Intramolecular electron transfer rate between active-site copper and topa quinone in pea seedling amine oxidase. J. Biol. Chem 1993, 268, 17680–17682. [Google Scholar]
- Barker, R.; Boden, N.; Cayley, G.; Charlton, S.C.; Henson, R.; Holmes, M.C.; Kelly, I.D.; Knowles, P.F. Properties of cupric ions in benzylamine oxidase from pig plasma as studied by magnetic-resonance and kinetic methods. Biochem. J 1979, 177, 289–302. [Google Scholar]
- Juda, G.A.; Shepard, E.M.; Elmore, B.O.; Dooley, D.M. A comparative study of the binding and inhibition of four copper-containing amine oxidases by azide: Implications for the role of copper during the oxidative half-reaction. Biochemistry 2006, 45, 8788–8800. [Google Scholar]
- Su, Q.; Klinman, J.P. Probing the mechanism of proton coupled electron transfer to dioxygen: The oxidative half-reaction of bovine serum amine oxidase. Biochemistry 1998, 37, 12513–12525. [Google Scholar]
- Mills, S.A.; Klinman, J.P. Evidence against reduction of Cu2+ to Cu+ during dioxygen activation in a copper amine oxidase from yeast. J. Am. Chem. Soc 2000, 122, 9897–9904. [Google Scholar]
- Mills, S.A.; Goto, Y.; Su, Q.; Plastino, J.; Klinman, J.P. Mechanistic comparison of the cobalt-substituted and wild-type copper amine oxidase from Hansenula polymorpha. Biochemistry 2002, 41, 10577–10584. [Google Scholar]
- Padiglia, A.; Medda, R.; Pedersen, J.Z.; Finazzi, A.A.; Lorrai, A.; Murgia, B.; Floris, G. Effect of metal substitution in copper amine oxidase from lentil seedlings. J. Biol. Inorg. Chem 1999, 4, 608–613. [Google Scholar]
- Schwartz, B.; Olgin, A.K.; Klinman, J.P. The role of copper in topa quinone biogenesis and catalysis, as probed by azide inhibition of a copper amine oxidase from yeast. Biochemistry 2001, 40, 2954–2963. [Google Scholar]
- Dawkes, H.C.; Phillips, S.E. Copper amine oxidase: Cunning cofactor and controversial copper. Curr. Opin. Struct. Biol 2001, 11, 666–673. [Google Scholar]
- Mills, S.A.; Brown, D.E.; Dang, K.; Sommer, D.; Bitsimis, A.; Nguyen, J.; Dooley, D.M. Cobalt substitution supports an inner-sphere electron transfer mechanism for oxygen reduction in pea seedling amine oxidase. J. Biol. Inorg. Chem 2012, 17, 507–515. [Google Scholar]
- Mure, M.; Klinman, J.P. Synthesis and spectroscopic characterization of model compounds for the active site cofactor in copper amine oxidases. J. Am. Chem. Soc 1993, 115, 7117–7127. [Google Scholar]
- Hartmann, C.; Brzovic, P.; Klinman, J.P. Spectroscopic detection of chemical intermediates in the reaction of para-substituted benzylamines with bovine serum amine oxidase. Biochemistry 1993, 32, 2234–2241. [Google Scholar]
- Cai, D.; Dove, J.; Nakamura, N.; Sanders-Loehr, J.; Klinman, J.P. Mechanism-based inactivation of a yeast methylamine oxidase mutant: Implications for the functional role of the consensus sequence surrounding topaquinone. Biochemistry 1997, 36, 11472–11478. [Google Scholar]
- Hevel, J.M.; Mills, S.A.; Klinman, J.P. Mutation of a strictly conserved, active-site residue alters substrate specificity and cofactor biogenesis in a copper amine oxidase. Biochemistry 1999, 38, 3683–3693. [Google Scholar]
- Hirota, S.; Iwamoto, T.; Kishishita, S.; Okajima, T.; Yamauchi, O.; Tanizawa, K. Spectroscopic observation of intermediates formed during the oxidative half-reaction of copper/topa quinone-containing phenylethylamine oxidase. Biochemistry 2001, 40, 15789–15796. [Google Scholar]
- Mure, M.; Klinman, J.P. Model studies of topa quinone: Synthesis and characterization of topa quinone derivatives. Methods Enzymol 1995, 258, 39–52. [Google Scholar]
- Nakamura, N.; Matsuzaki, R.; Choi, Y.H.; Tanizawa, K.; Sanders-Loehr, J. Biosynthesis of topa quinone cofactor in bacterial amine oxidases. Solvent origin of C-2 oxygen determined by Raman spectroscopy. J. Biol. Chem 1996, 271, 4718–4724. [Google Scholar]
- Murakawa, T.; Hayashi, H.; Taki, M.; Yamamoto, Y.; Kawano, Y.; Tanizawa, K.; Okajima, T. Structural insights into the substrate specificity of bacterial copper amine oxidase obtained by using irreversible inhibitors. J. Biochem 2012, 151, 167–178. [Google Scholar]
- Chiu, Y.C.; Okajima, T.; Murakawa, T.; Uchida, M.; Taki, M.; Hirota, S.; Kim, M.; Yamaguchi, H.; Kawano, Y.; Kamiya, N.; et al. Kinetic and structural studies on the catalytic role of the aspartic acid residue conserved in copper amine oxidase. Biochemistry 2006, 45, 4105–4120. [Google Scholar]
- Frebort, I.; Toyama, H.; Matsushita, K.; Adachi, O. Half-site reactivity with p-nitrophenylhydrazine and subunit separation of the dimeric copper-containing amine oxidase from Aspergillus niger Biochem. Mol. Biol. Int 1995, 36, 1207–1216. [Google Scholar]
- Morpurgo, L.; Agostinelli, E.; Mondovi, B.; Avigliano, L.; Silvestri, R.; Stefancich, G.; Artico, M. Bovine serum amine oxidase: Half-site reactivity with phenylhydrazine, semicarbazide, and aromatic hydrazides. Biochemistry 1992, 31, 2615–2621. [Google Scholar]
- Agostinelli, E.; Morpurgo, L.; Wang, C.; Giartosio, A.; Mondovi, B. Properties of cobalt-substituted bovine serum amine oxidase. Eur. J. Biochem 1994, 222, 727–732. [Google Scholar]
- Morpurgo, L.; Agostinelli, E.; Muccigrosso, J.; Martini, F.; Mondovi, B.; Avigliano, L. Benzylhydrazine as a pseudo-substrate of bovine serum amine oxidase. Biochem. J 1989, 260, 19–25. [Google Scholar]
- de Biase, D.; Agostinelli, E.; de Matteis, G.; Mondovi, B.; Morpurgo, L. Half-of-the-sites reactivity of bovine serum amine oxidase: Reactivity and chemical identity of the second site. Eur. J. Biochem 1996, 237, 93–99. [Google Scholar]
- Takahashi, K.; Klinman, J.P. Relationship of stopped flow to steady state parameters in the dimeric copper amine oxidase from Hansenula polymorpha and the role of zinc in inhibiting activity at alternate copper-containing subunits. Biochemistry 2006, 45, 4683–4694. [Google Scholar]
- Coleman, A.A.; Hindsgaul, O.; Palcic, M.M. Stereochemistry of copper amine oxidase reactions. J. Biol. Chem 1989, 264, 19500–19505. [Google Scholar]
- Scaman, C.H.; Palcic, M.M. Stereochemical course of tyramine oxidation by semicarbazide-sensitive amine oxidase. Biochemistry 1992, 31, 6829–6841. [Google Scholar]
- Uchida, M.; Ohtani, A.; Kohyama, N.; Okajima, T.; Tanizawa, K.; Yamamoto, Y. Stereochemistry of 2-phenylethylamine oxidation catalyzed by bacterial copper amine oxidase. Biosci. Biotechnol. Biochem 2003, 67, 2664–2667. [Google Scholar]
- Taki, M.; Murakawa, T.; Nakamoto, T.; Uchida, M.; Hayashi, H.; Tanizawa, K.; Yamamoto, Y.; Okajima, T. Further insight into the mechanism of stereoselective proton abstraction by bacterial copper amine oxidase. Biochemistry 2008, 47, 7726–7733. [Google Scholar]
- Duff, A.P.; Trambaiolo, D.M.; Cohen, A.E.; Ellis, P.J.; Juda, G.A.; Shepard, E.M.; Langley, D.B.; Dooley, D.M.; Freeman, H.C.; Guss, J.M. Using xenon as a probe for dioxygen-binding sites in copper amine oxidases. J. Mol. Biol 2004, 344, 599–607. [Google Scholar]
- Pirrat, P.; Smith, M.A.; Pearson, A.R.; McPherson, M.J.; Phillips, S.E. Structure of a xenon derivative of Escherichia coli copper amine oxidase: Confirmation of the proposed oxygen-entry pathway. Acta. Crystallogr. Sect. F Struct. Biol. Cryst. Commun 2008, 64, 1105–1109. [Google Scholar]
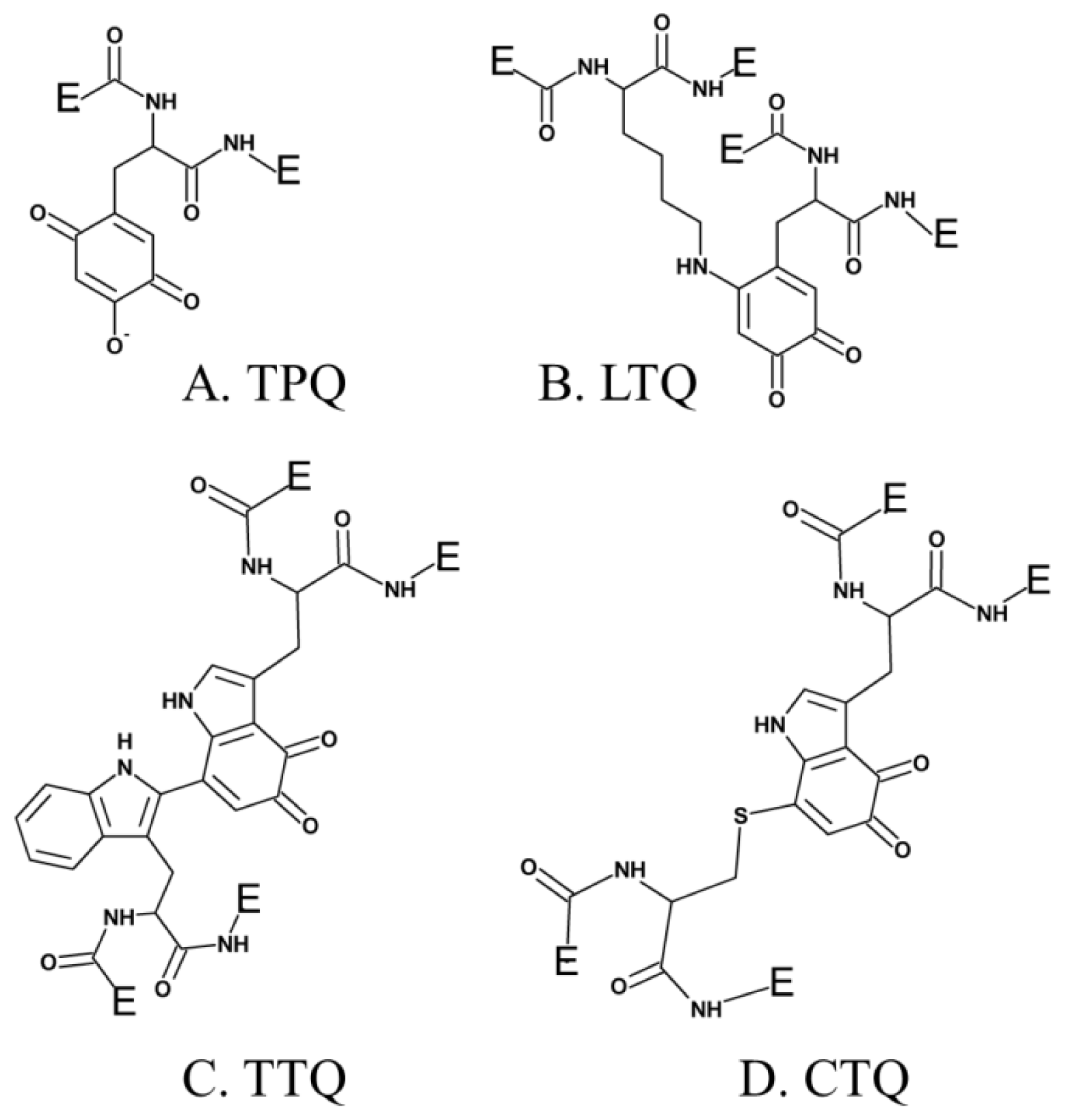
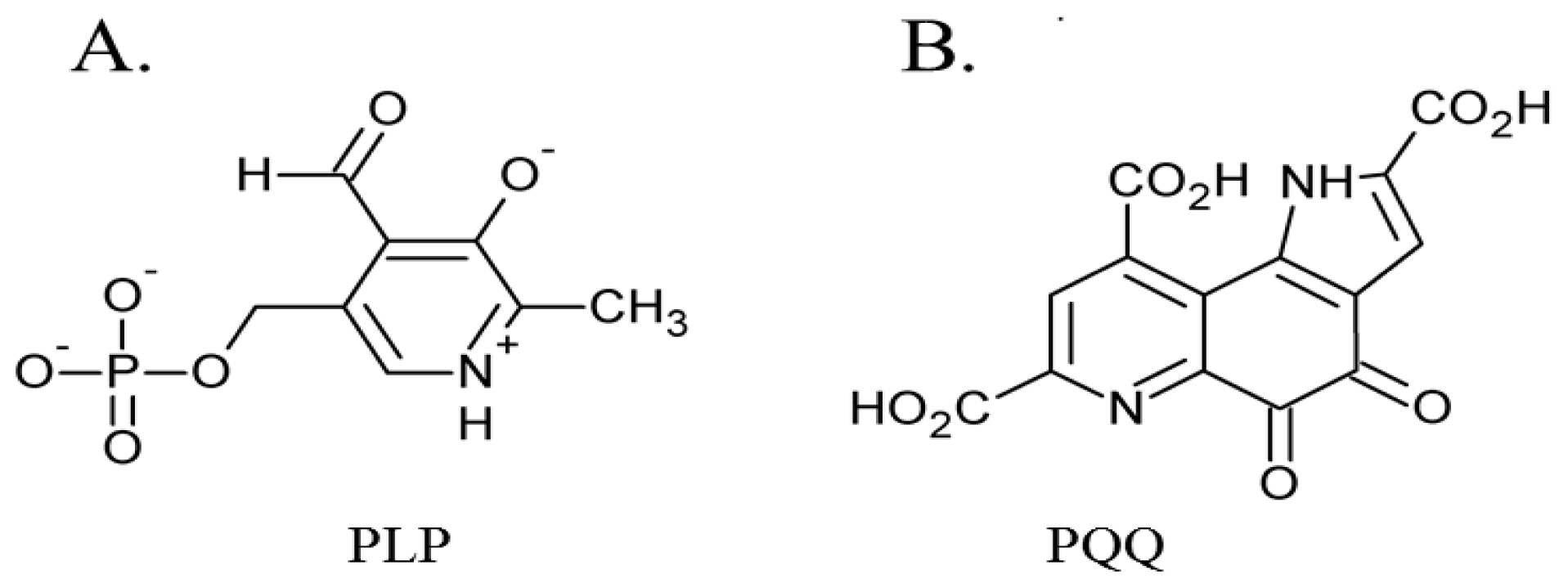
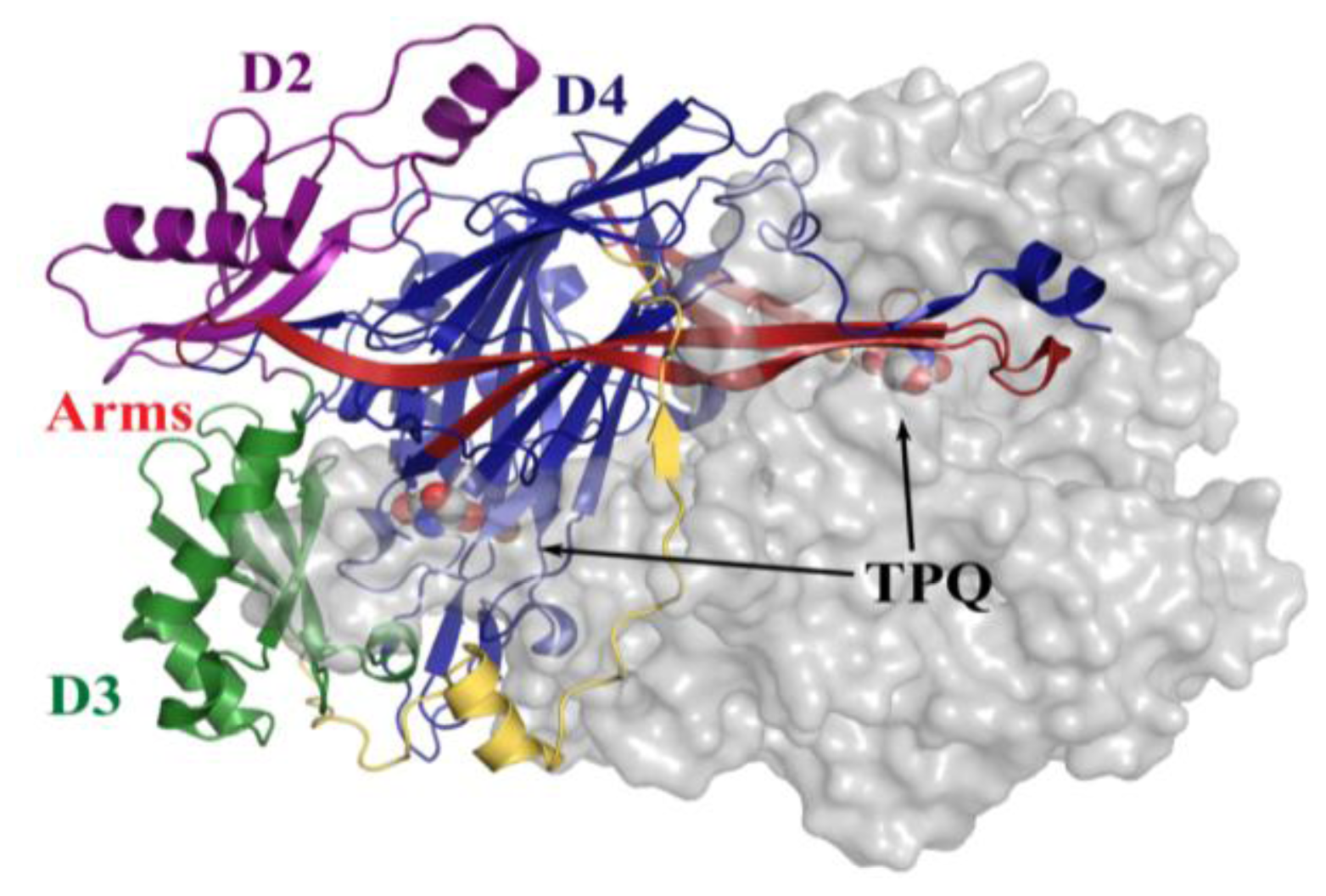
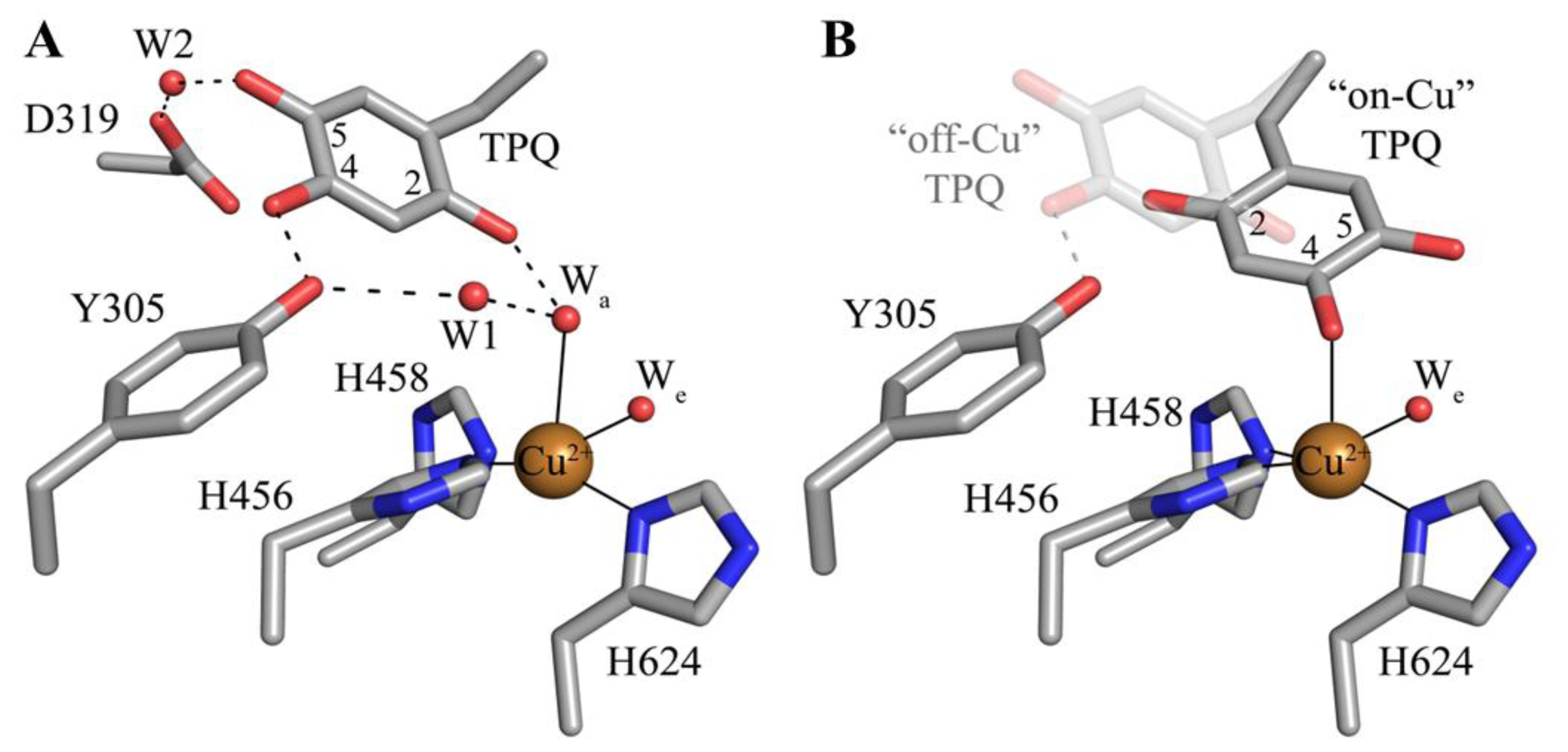
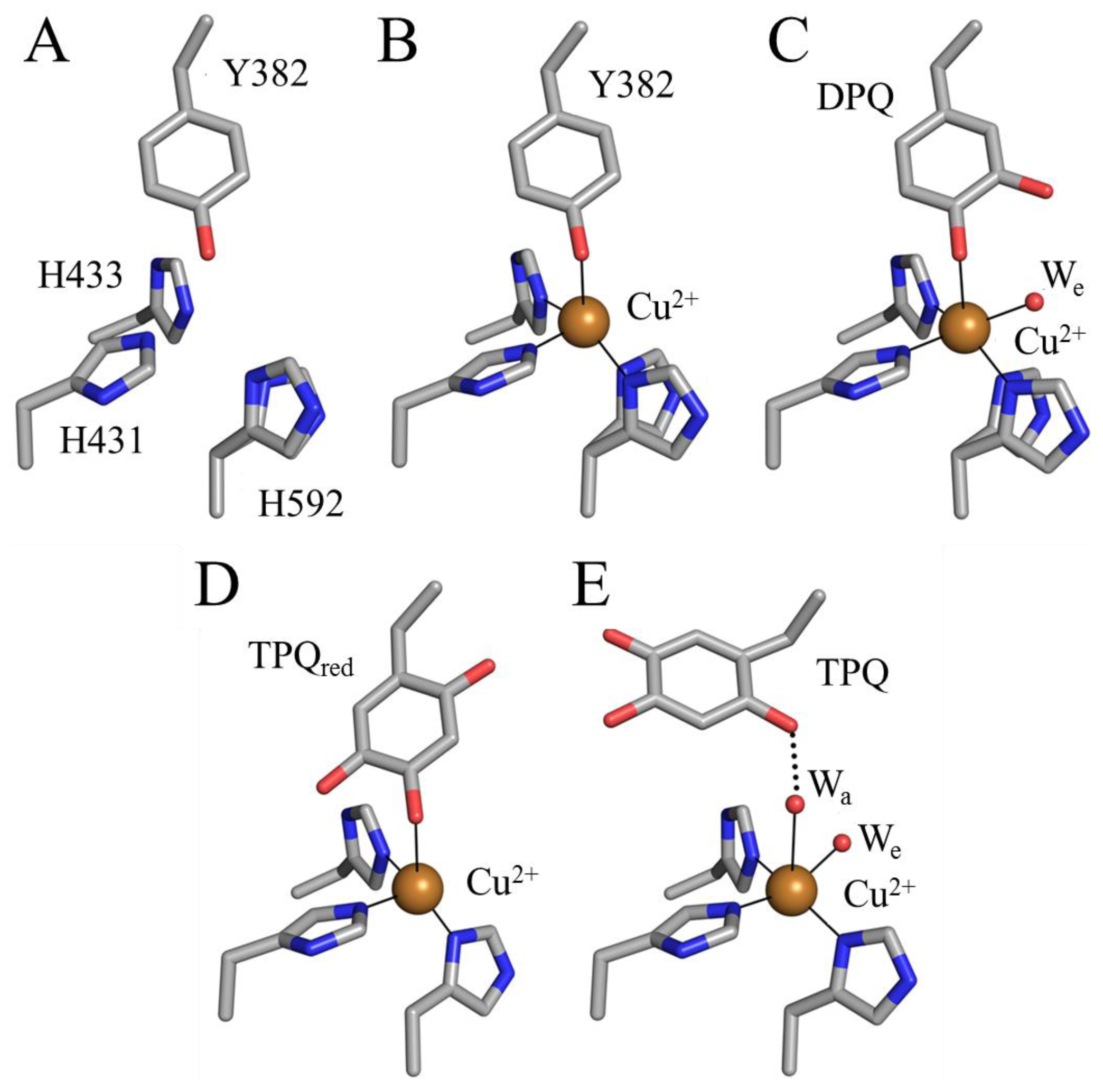
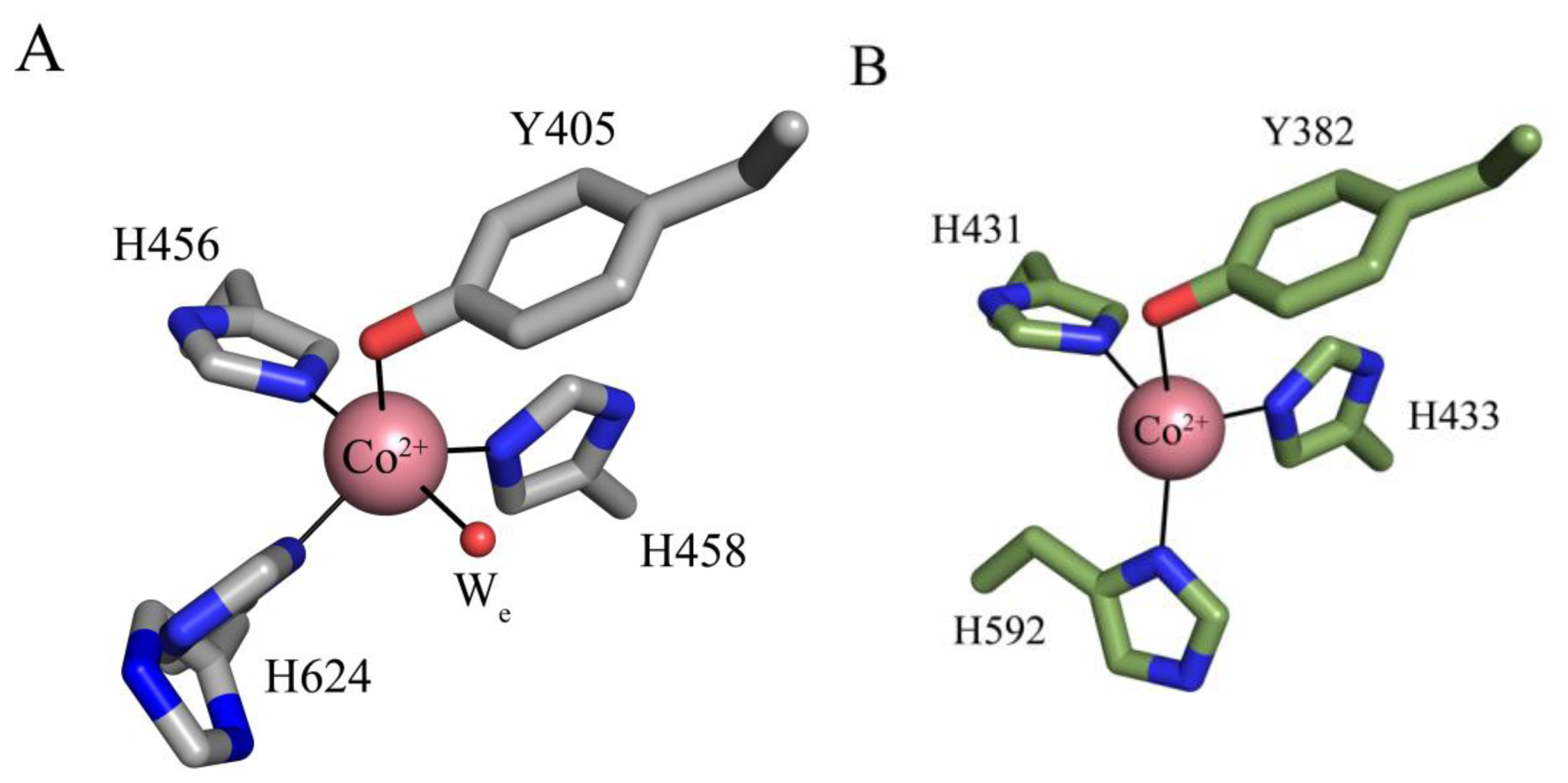
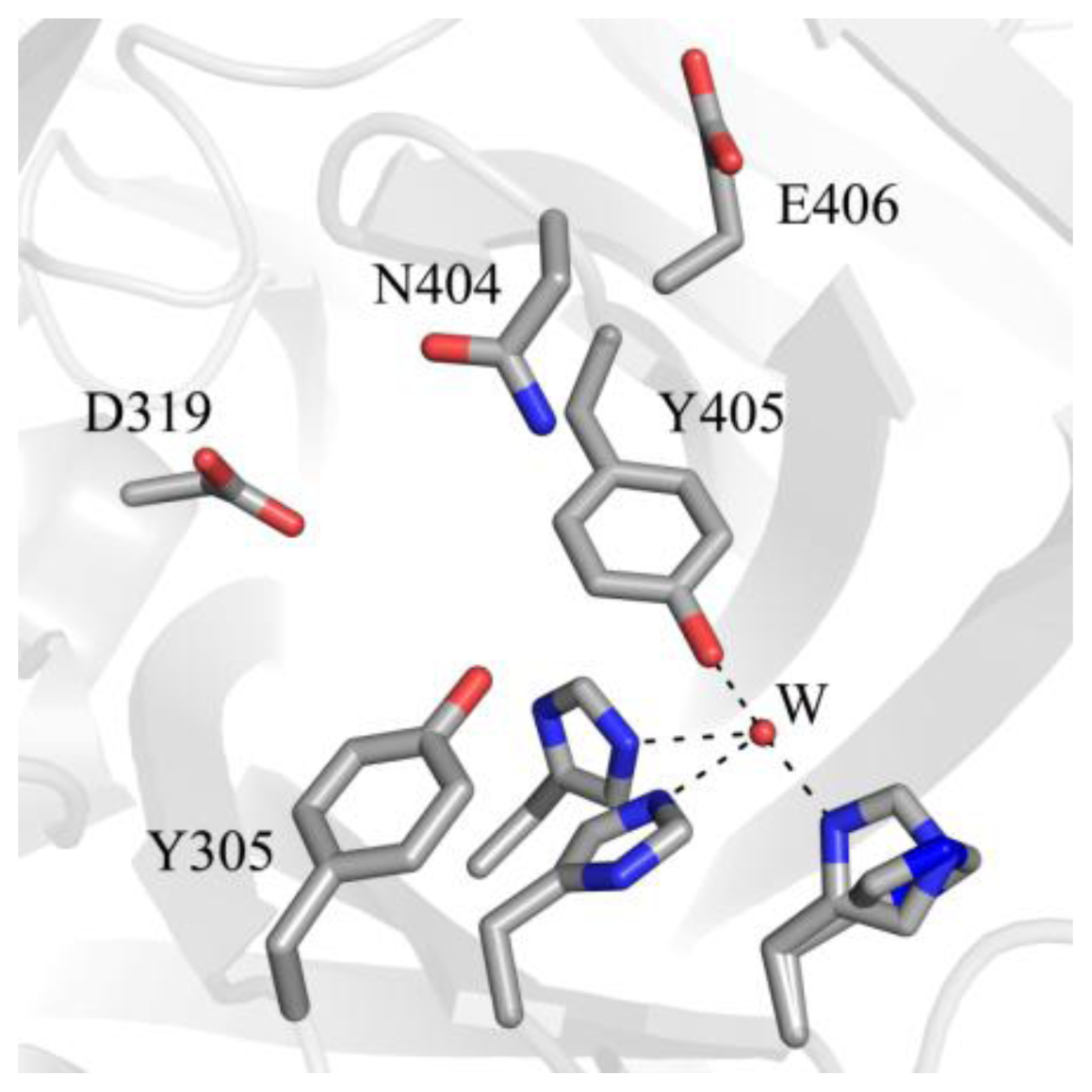
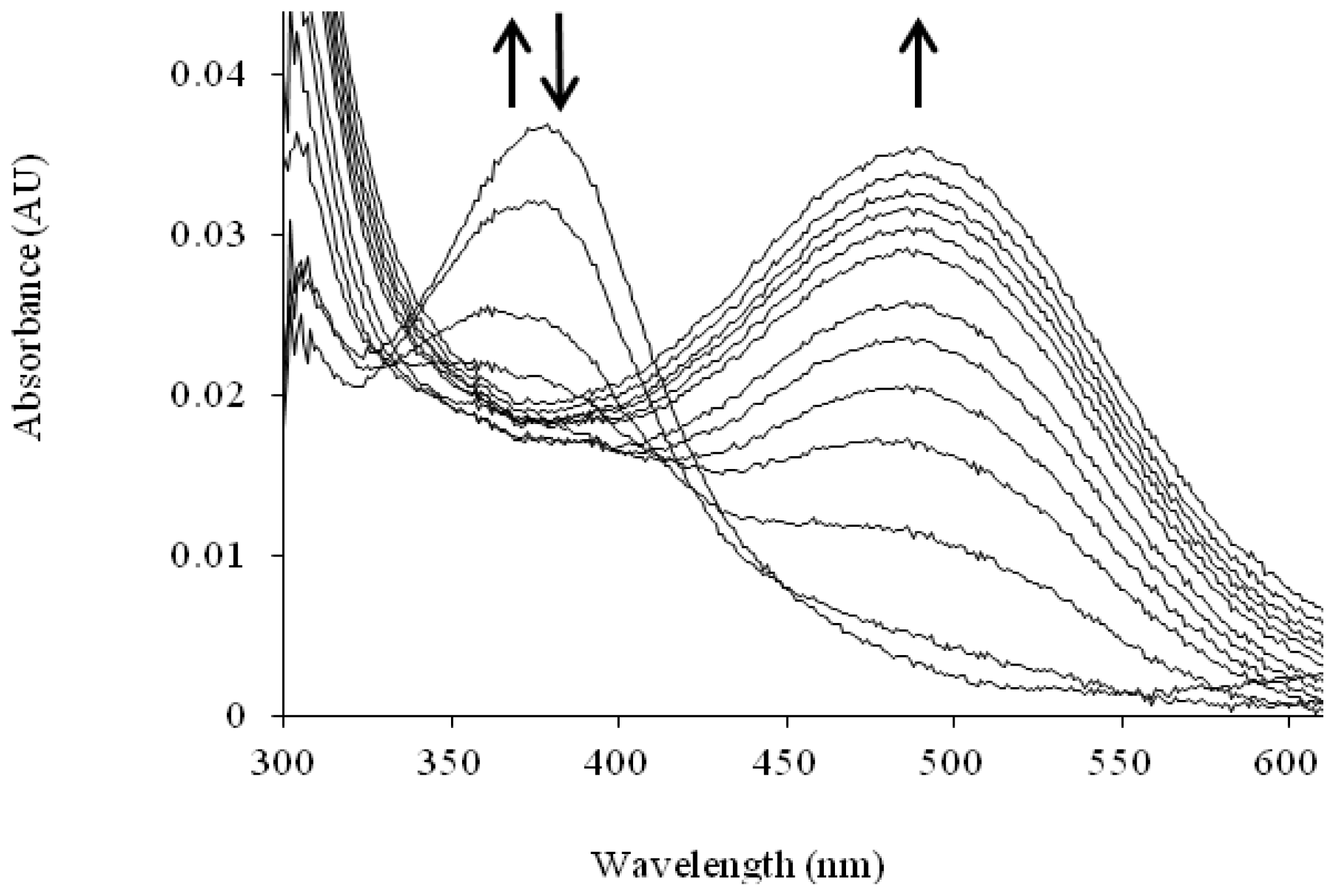
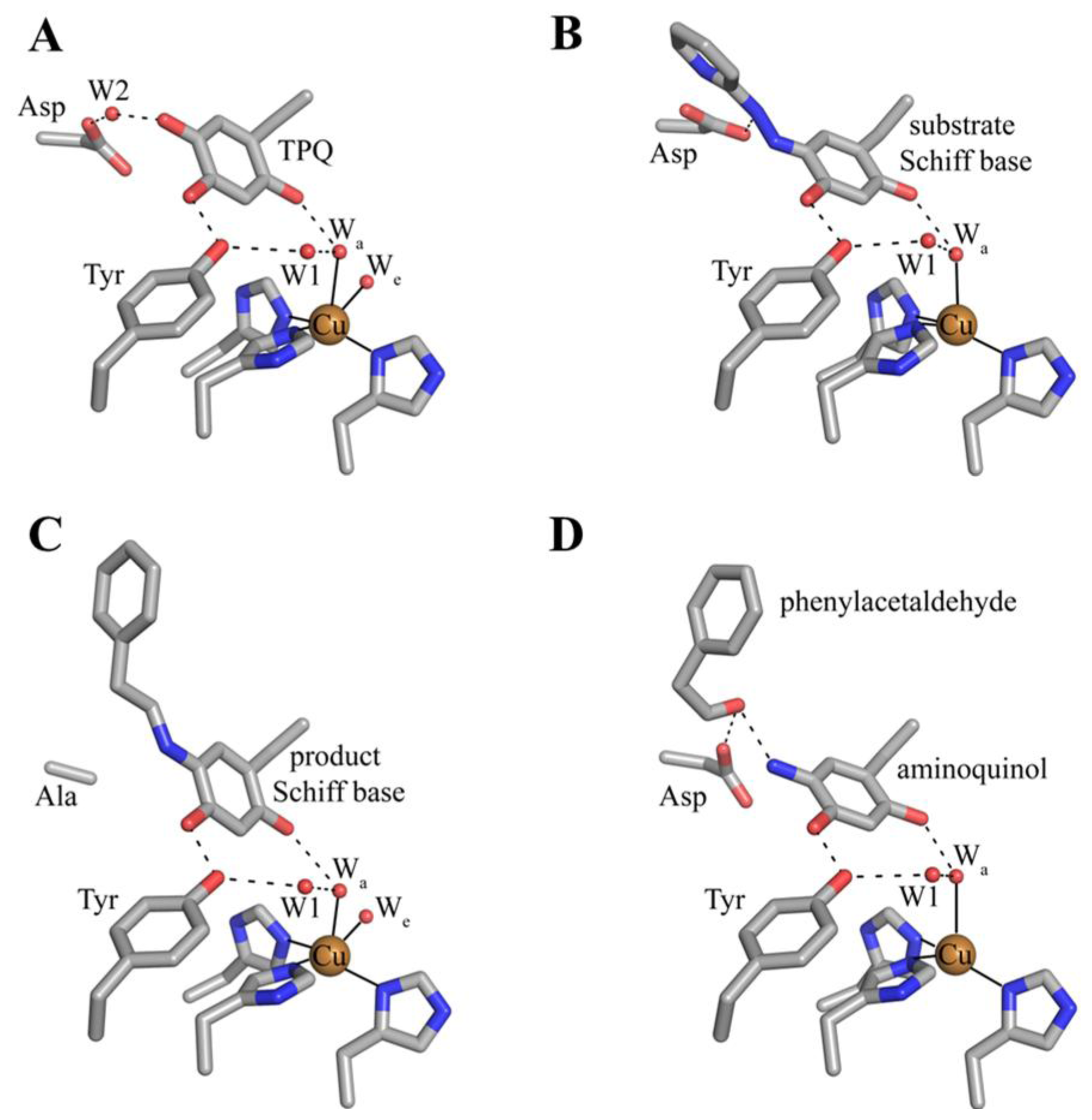
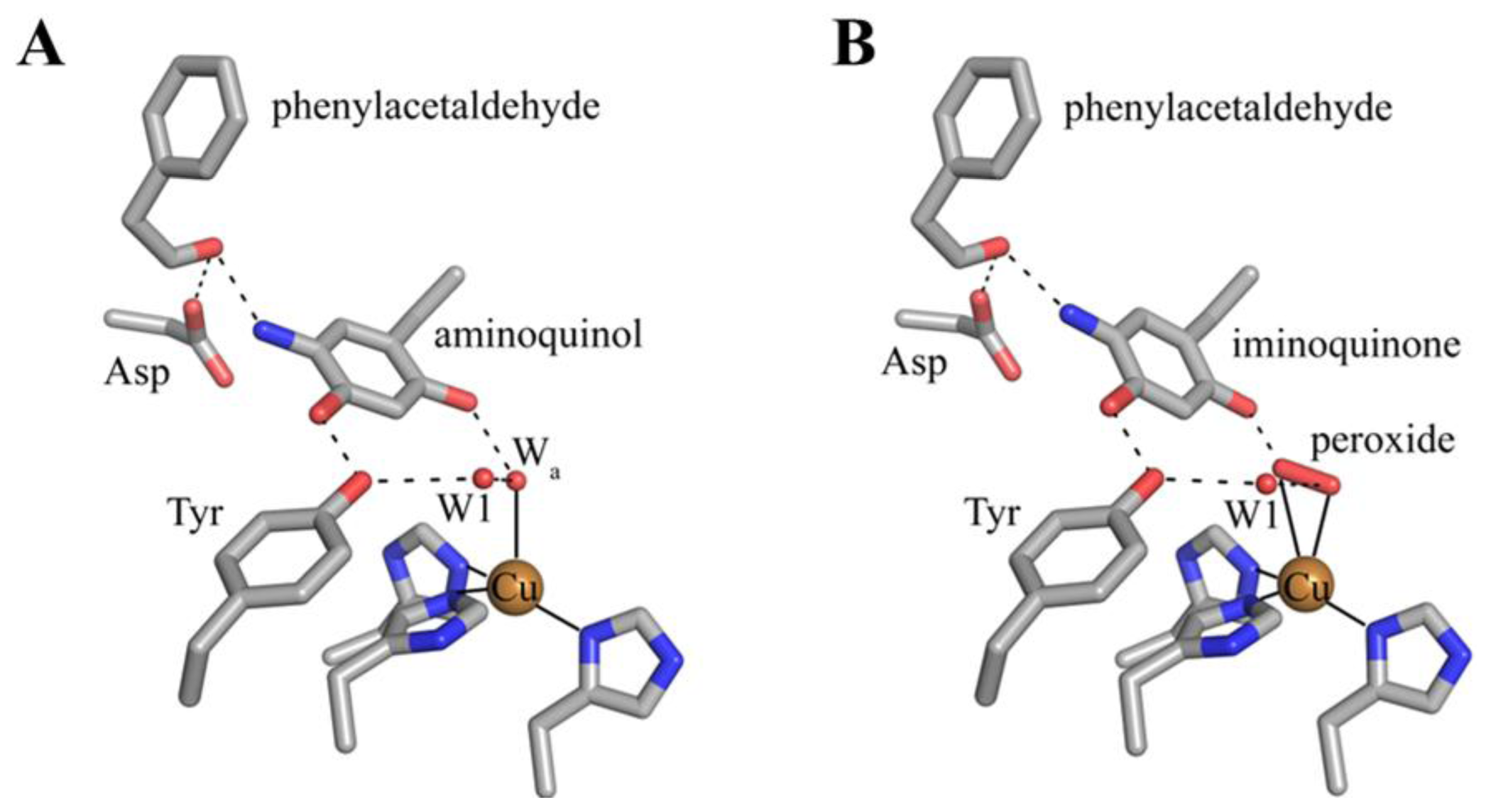
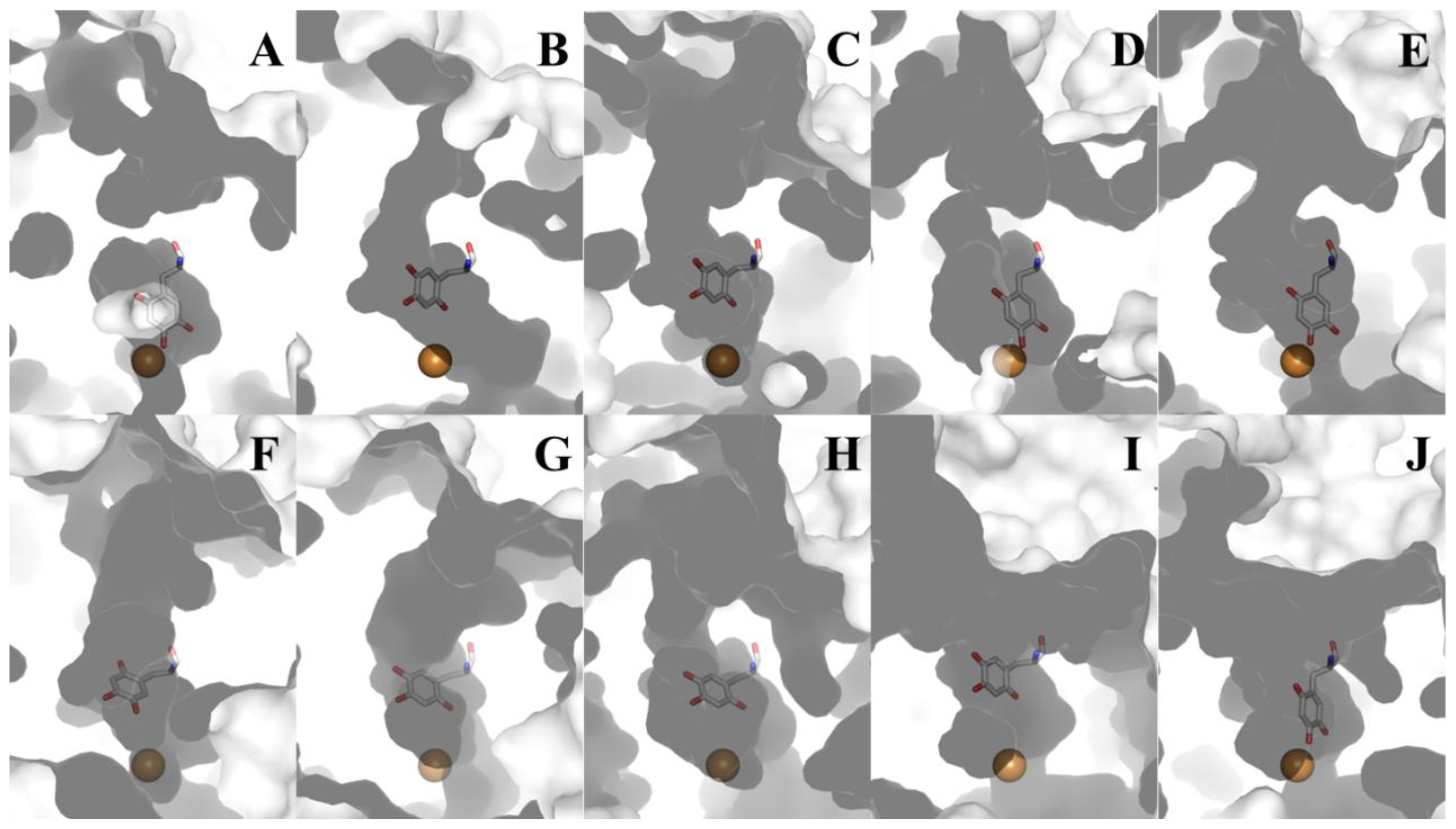
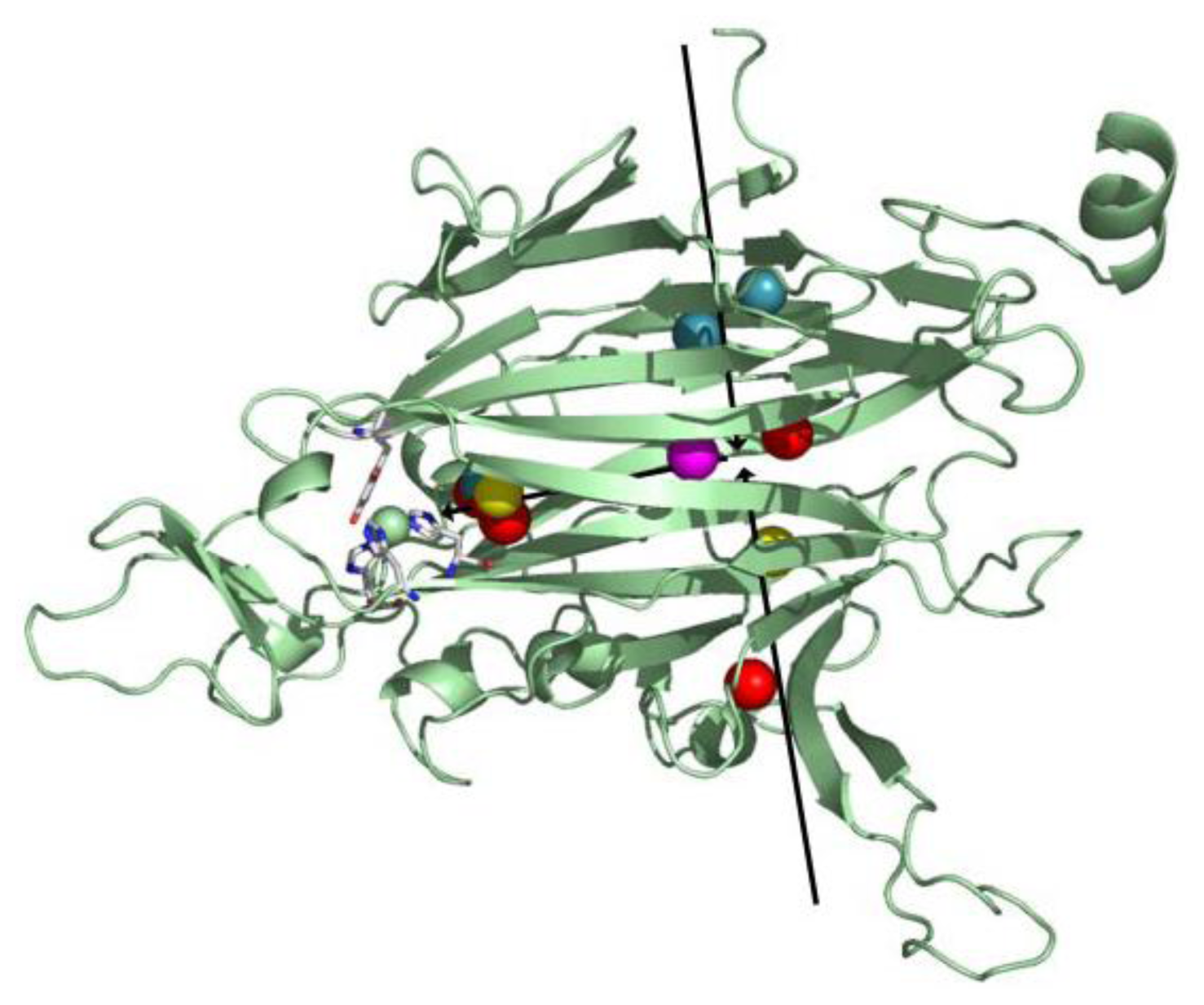
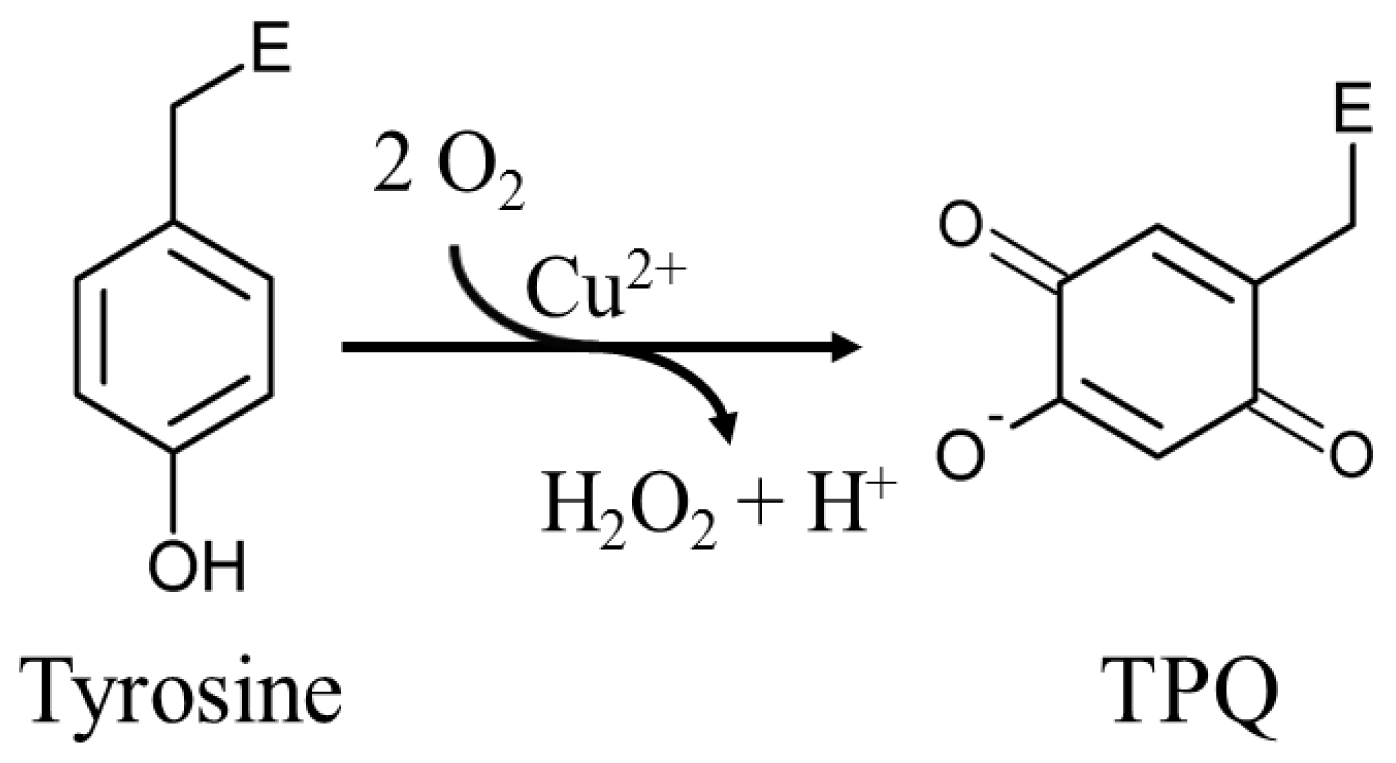
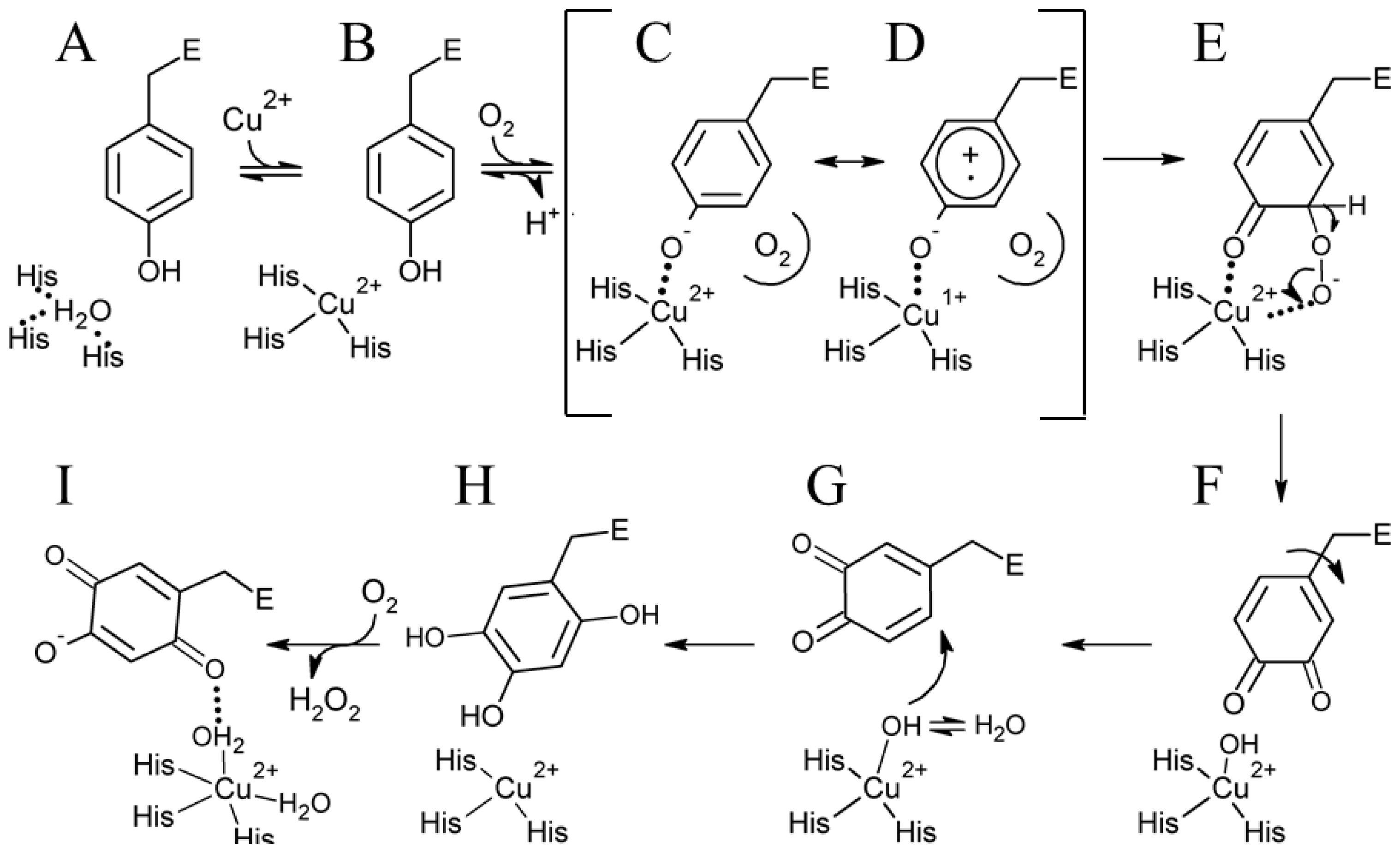
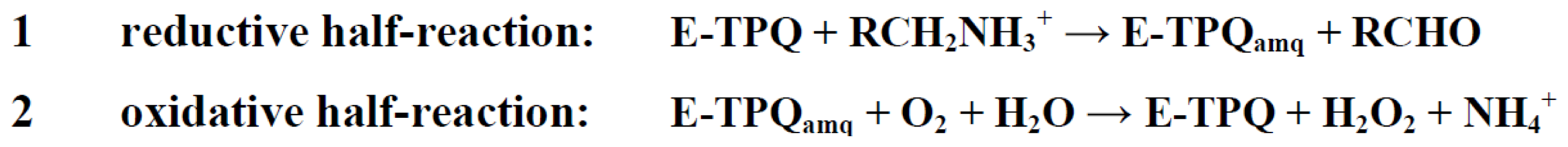
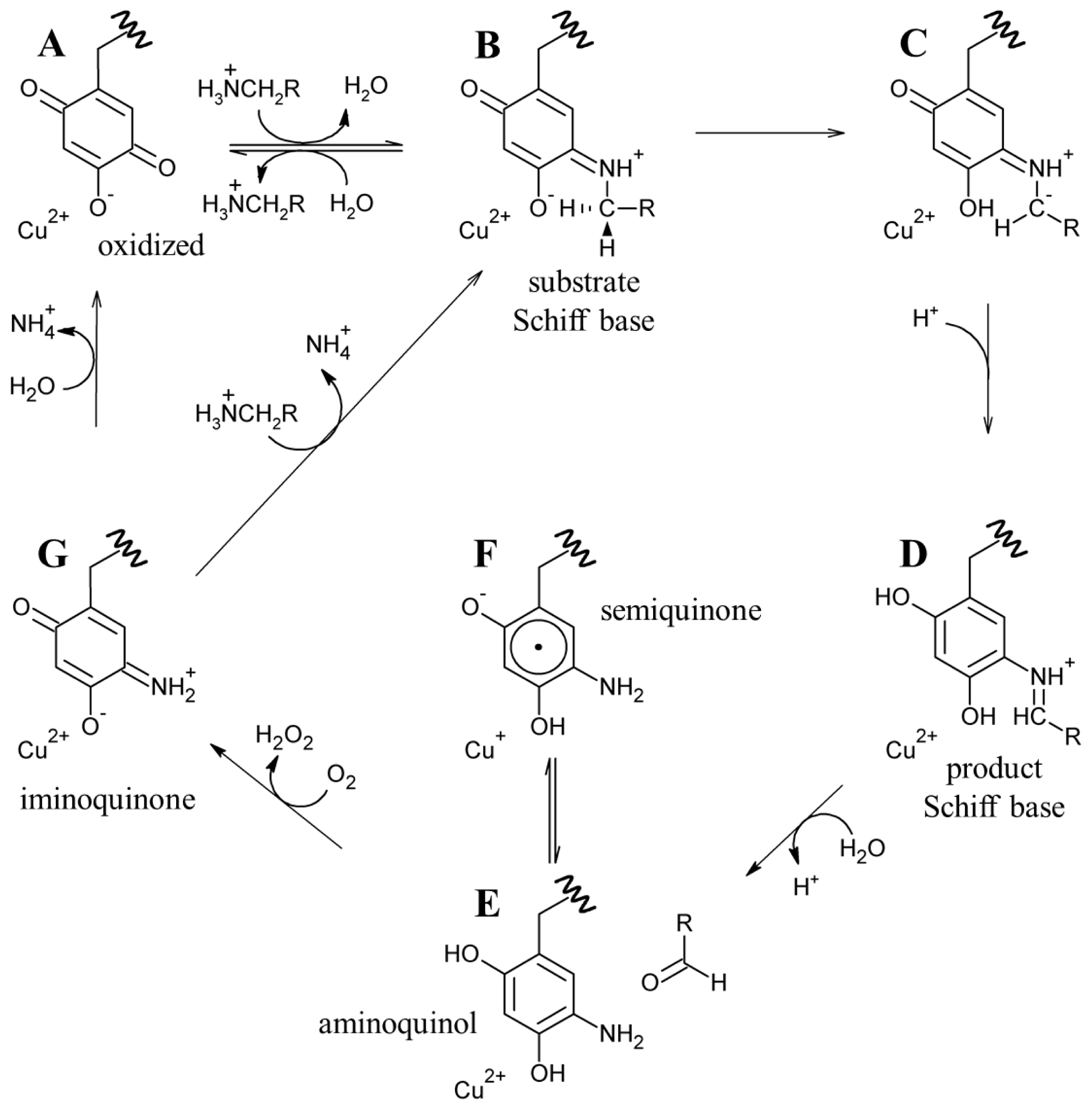
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).