Nucleotide Excision Repair in Cellular Chromatin: Studies with Yeast from Nucleotide to Gene to Genome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
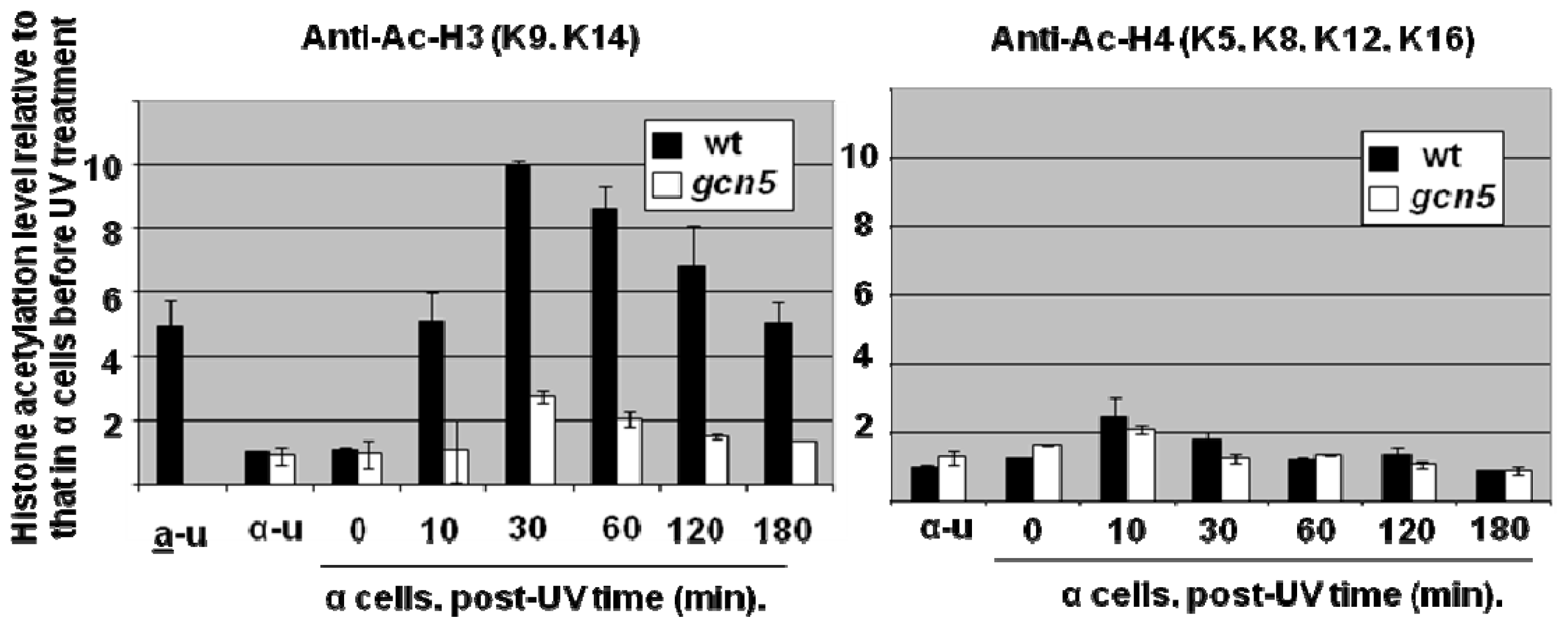{kind=link}
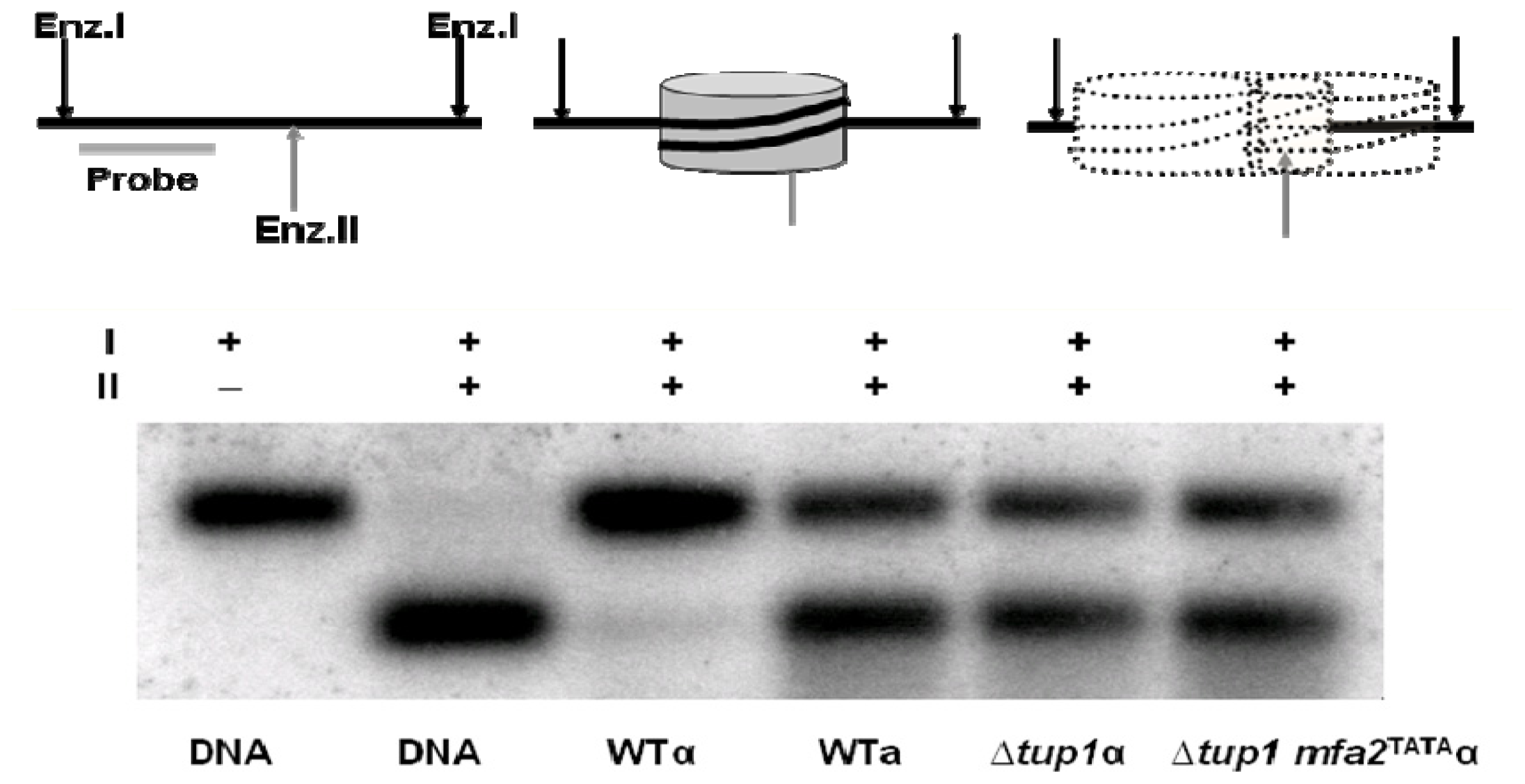{kind=link}
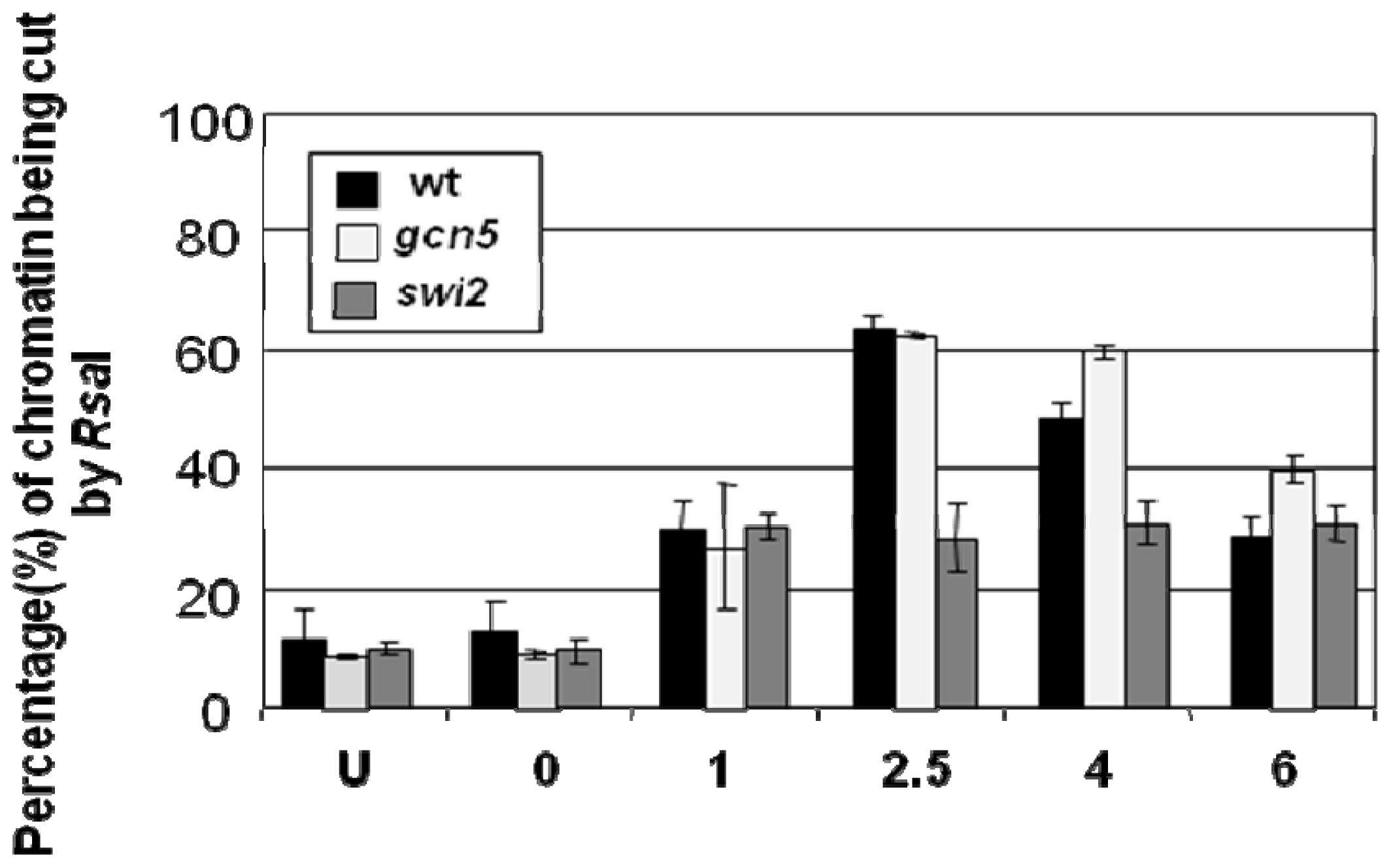{kind=link}
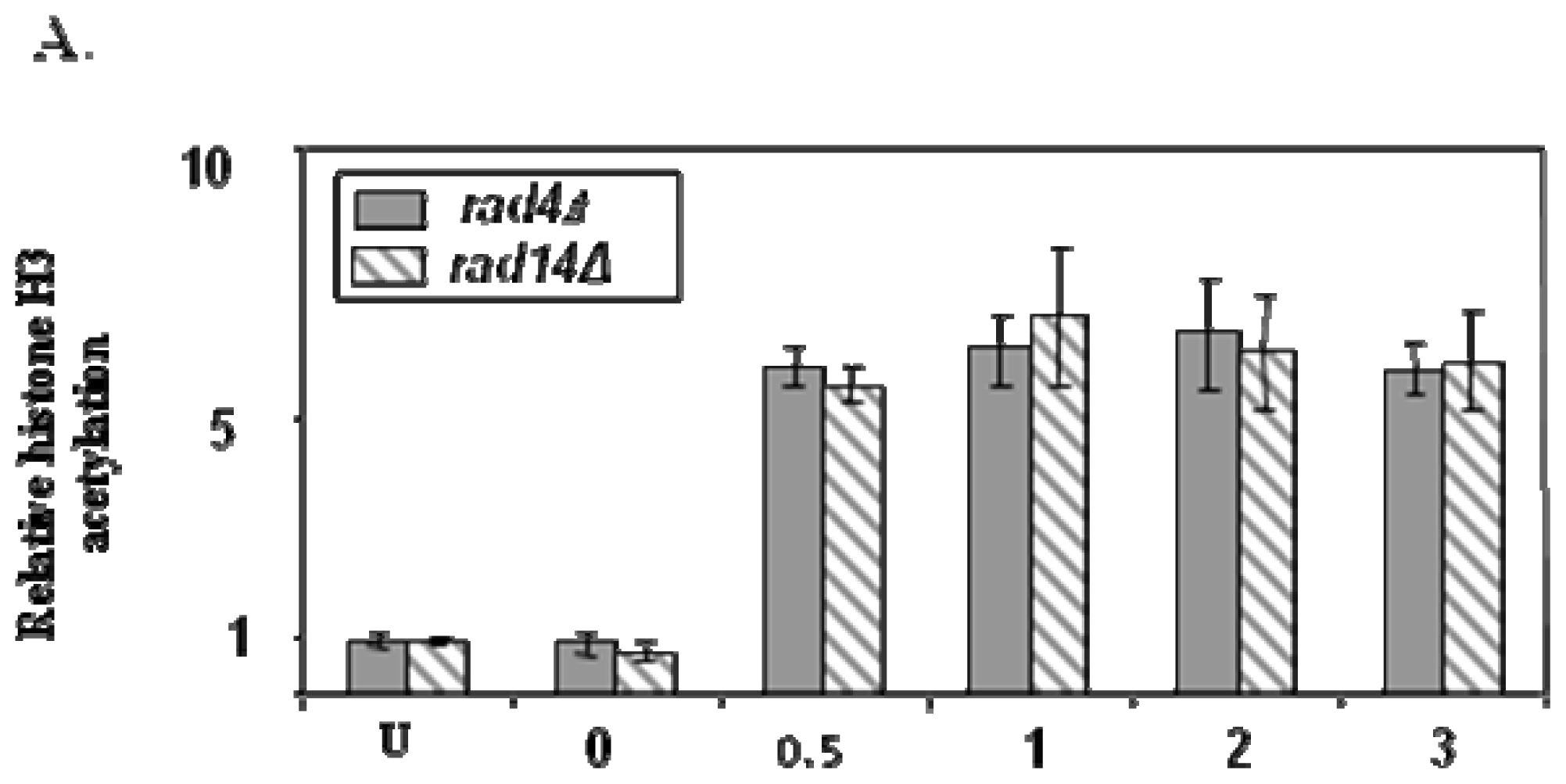{kind=link}
{kind=link}
Abstract
:1. Introduction
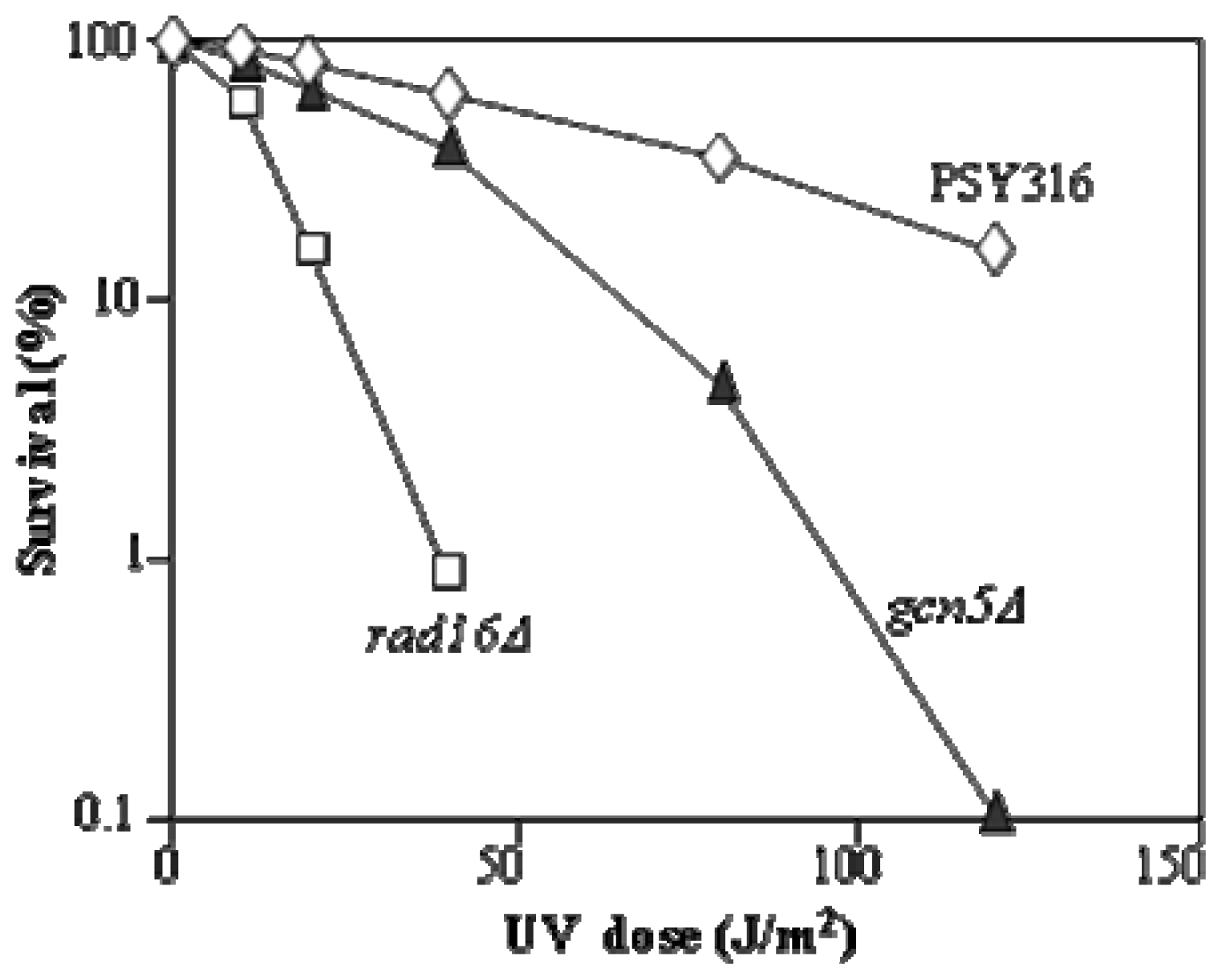
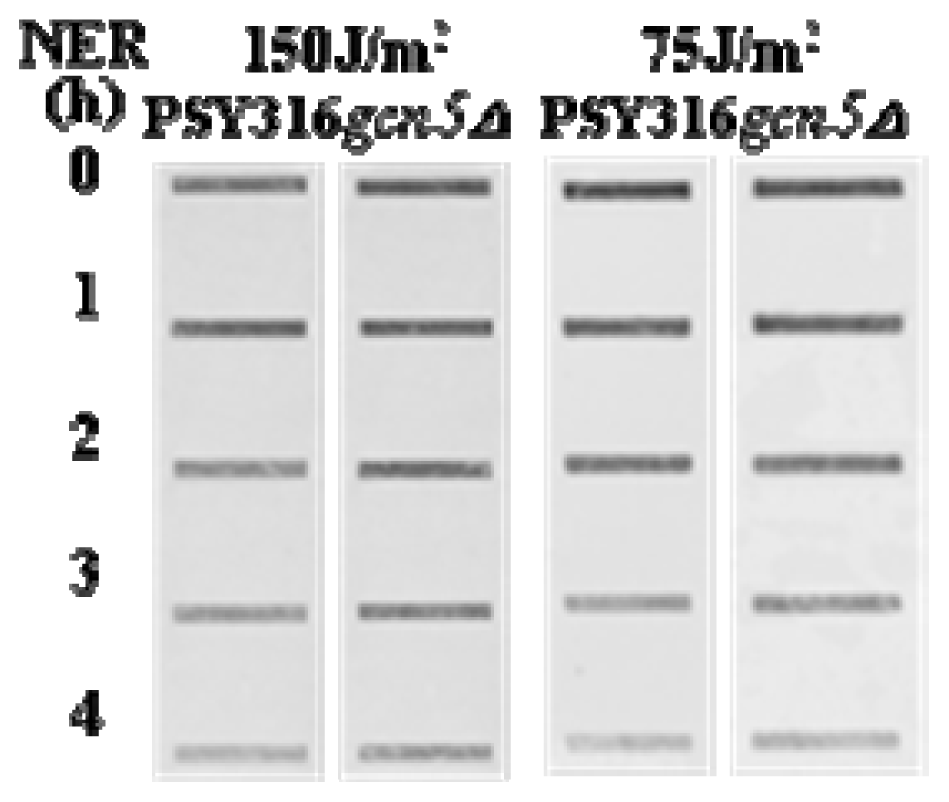
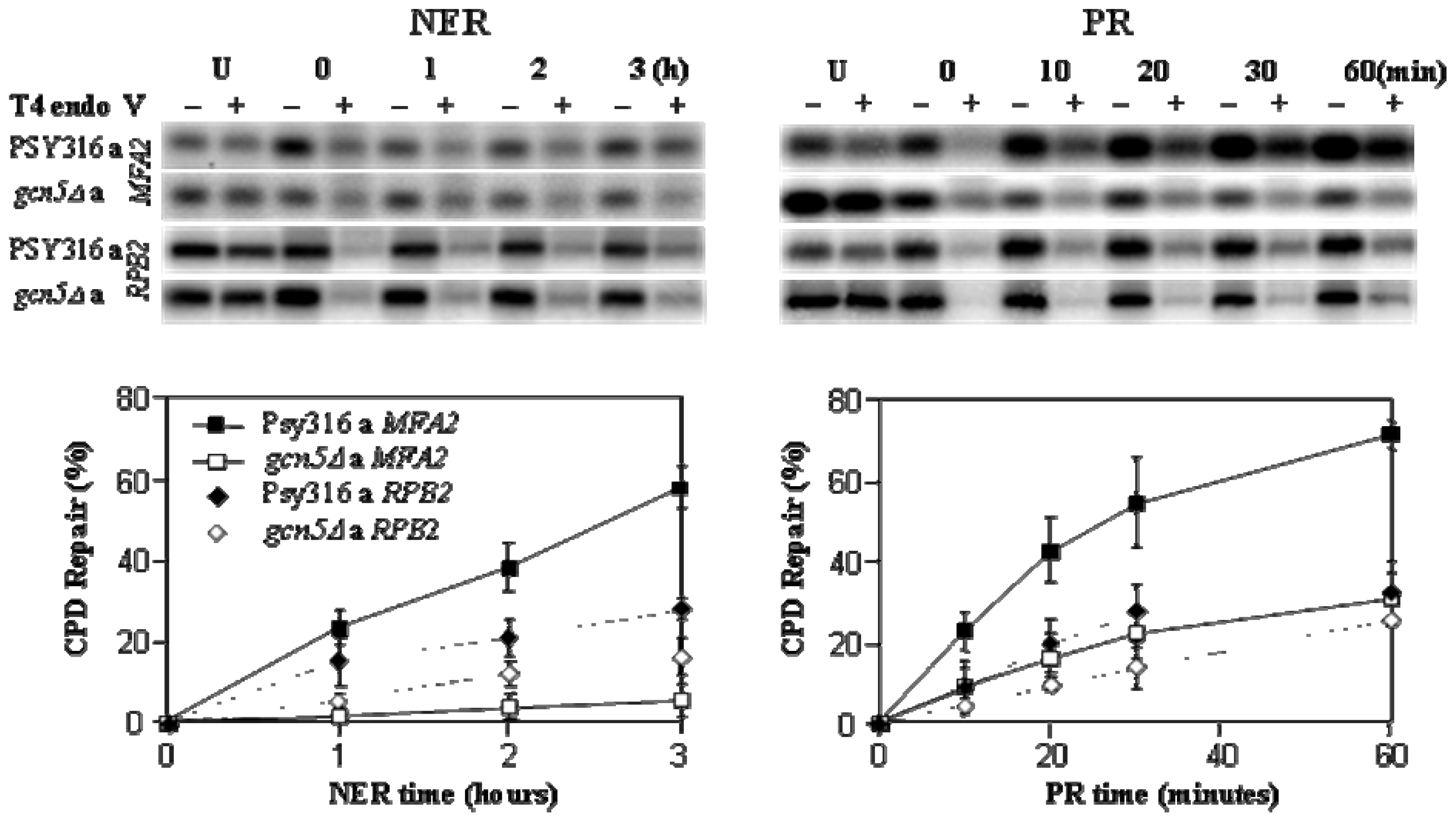
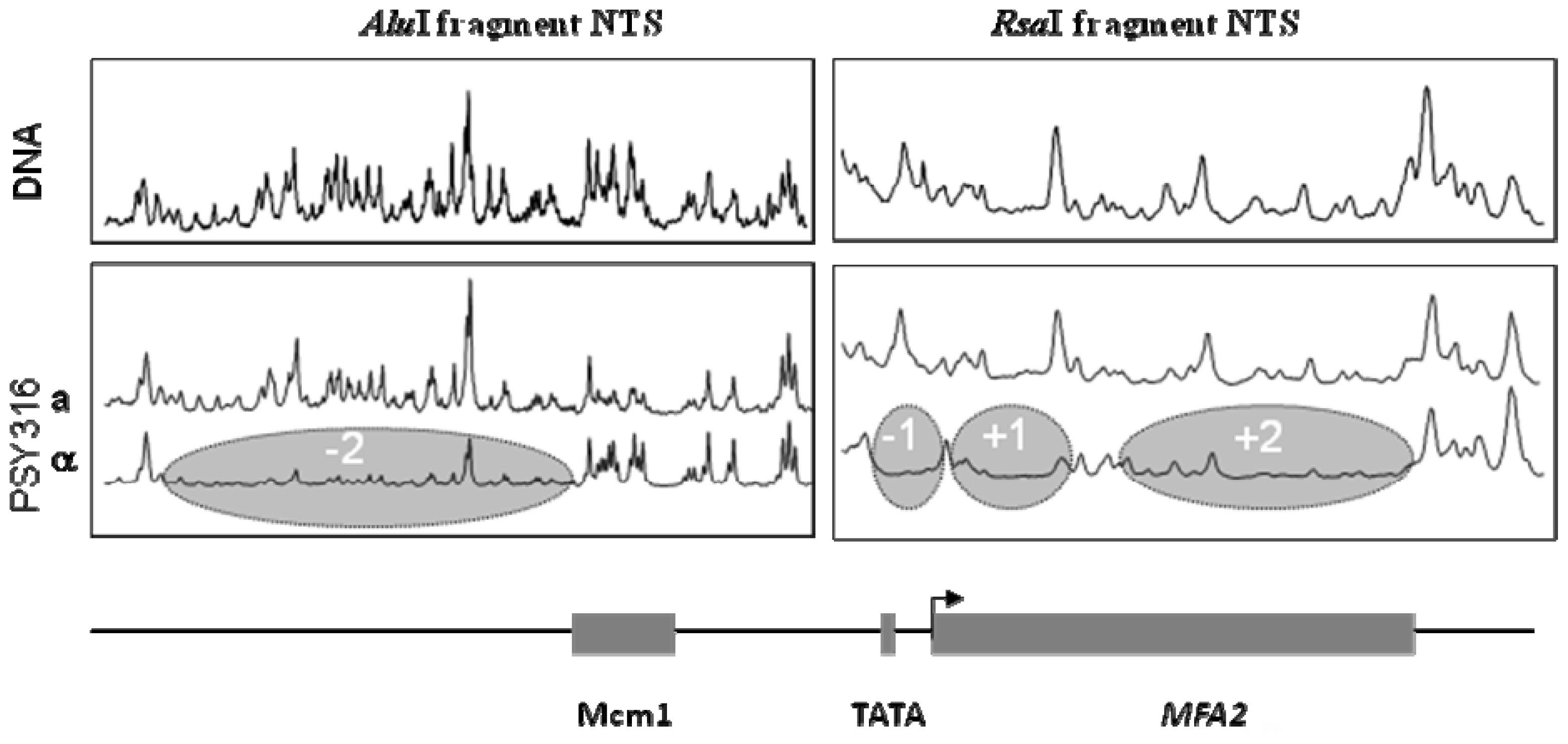
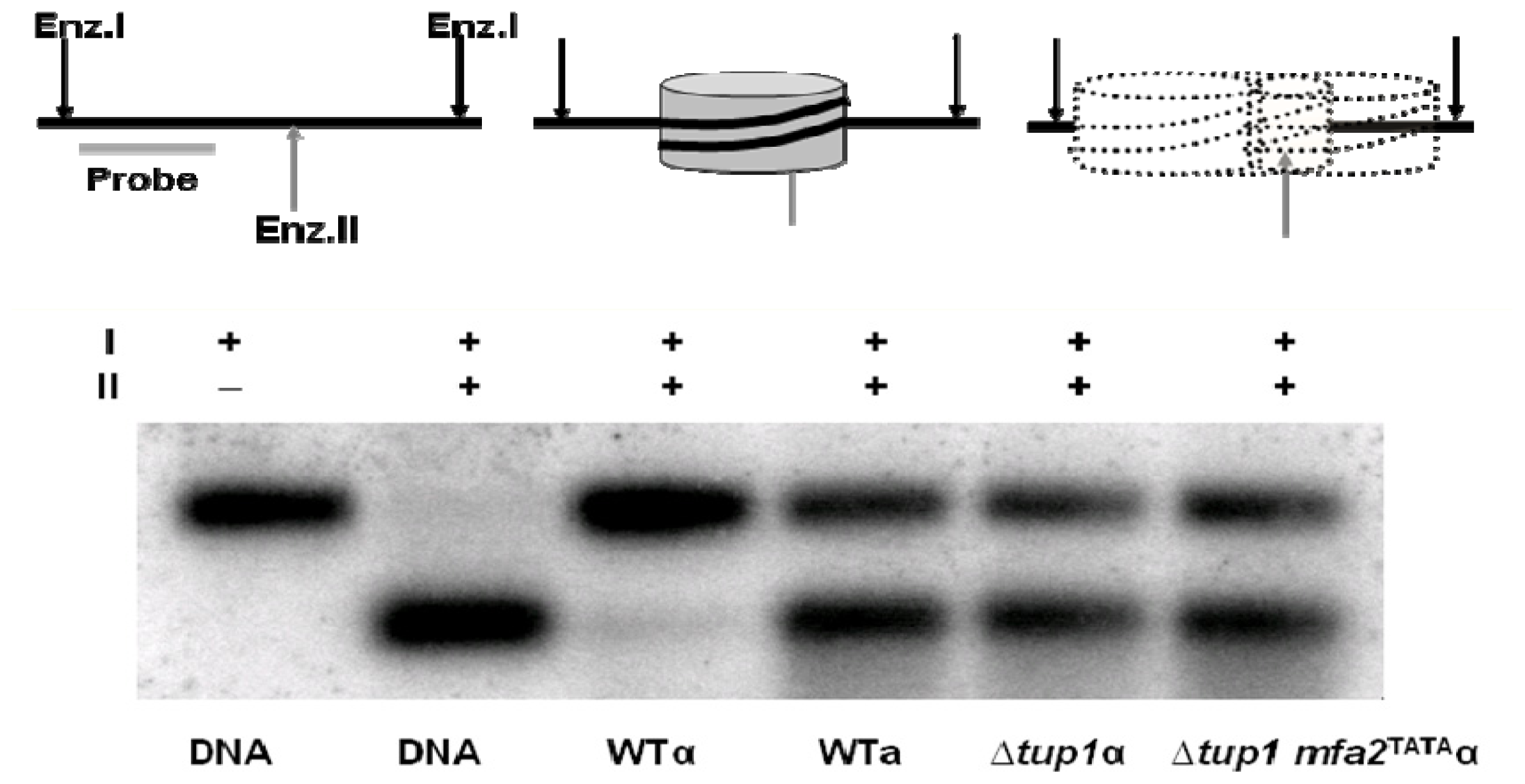
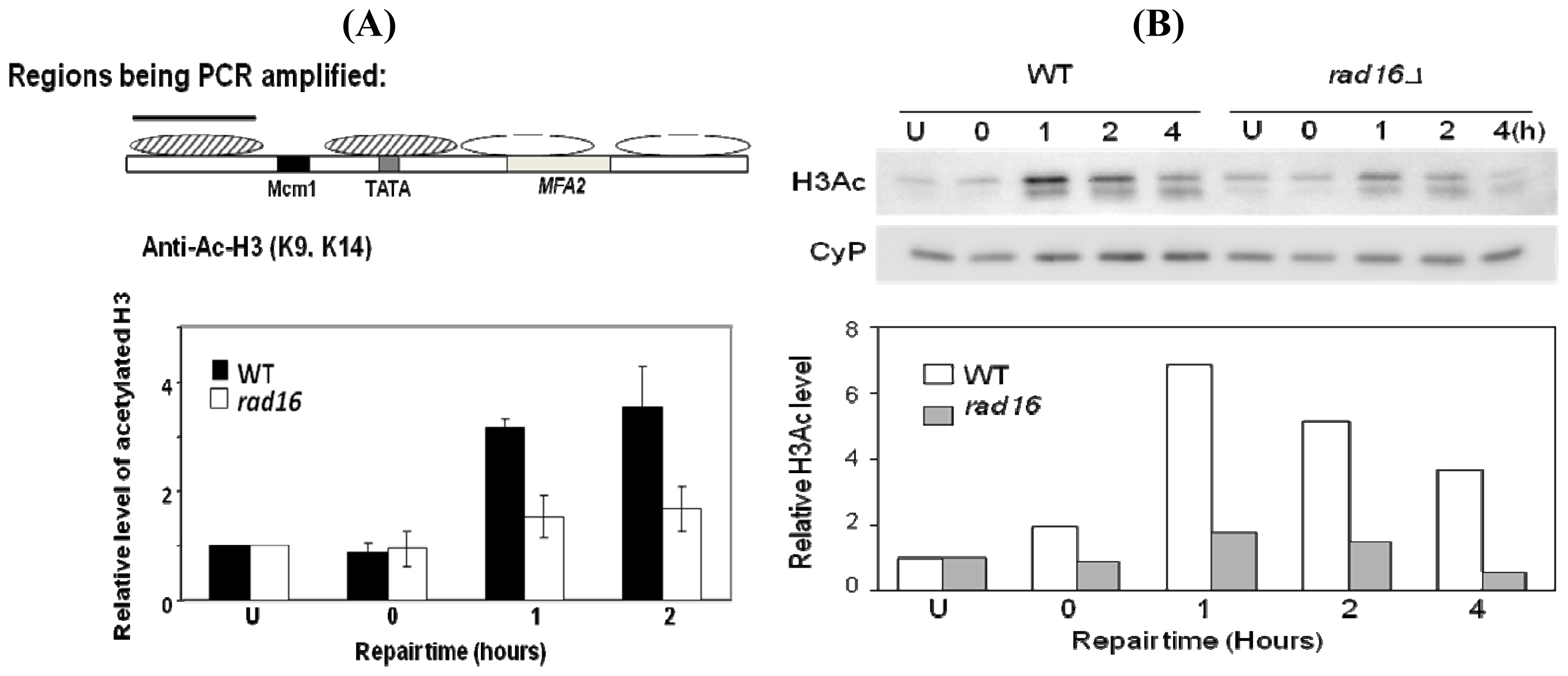
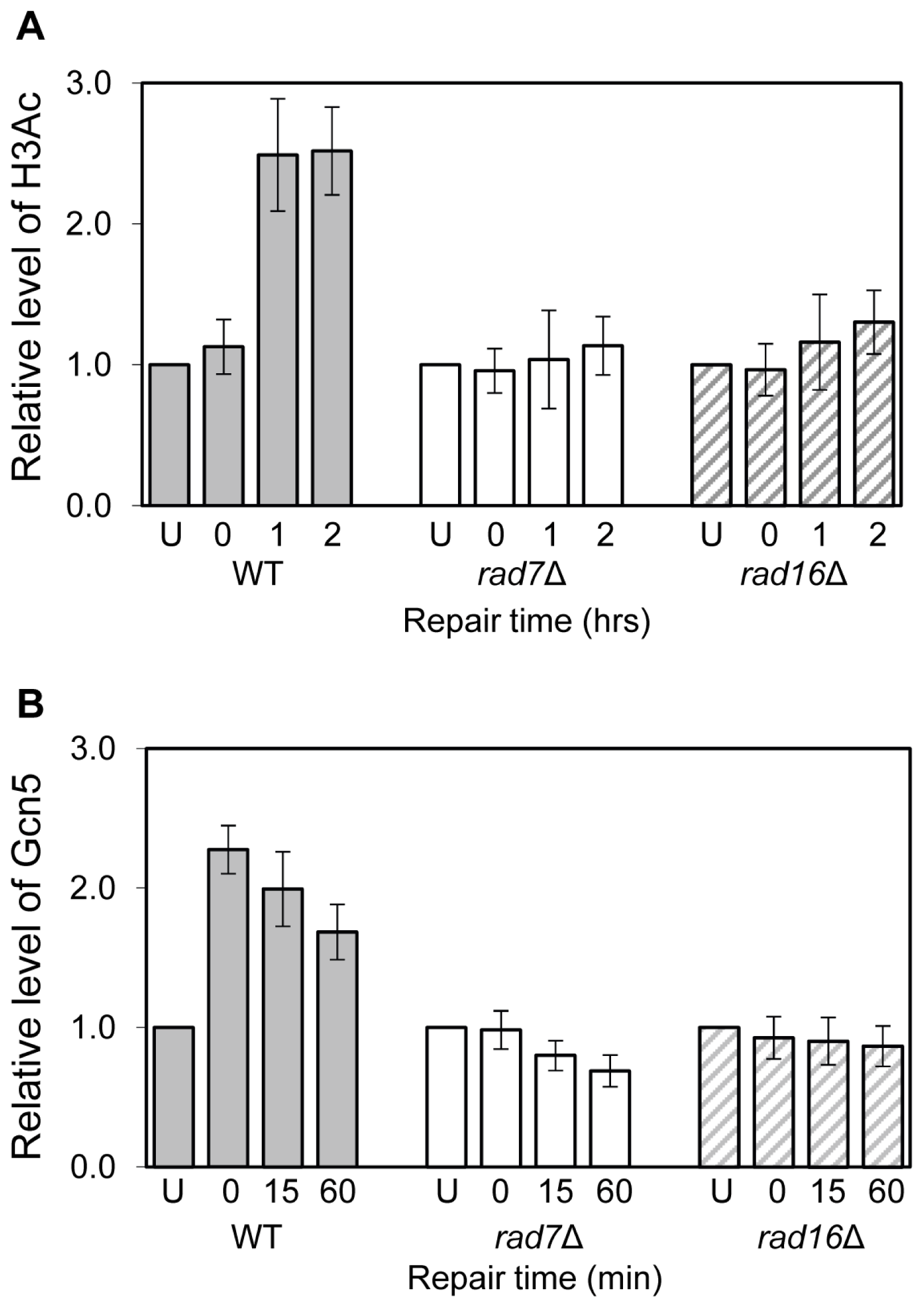
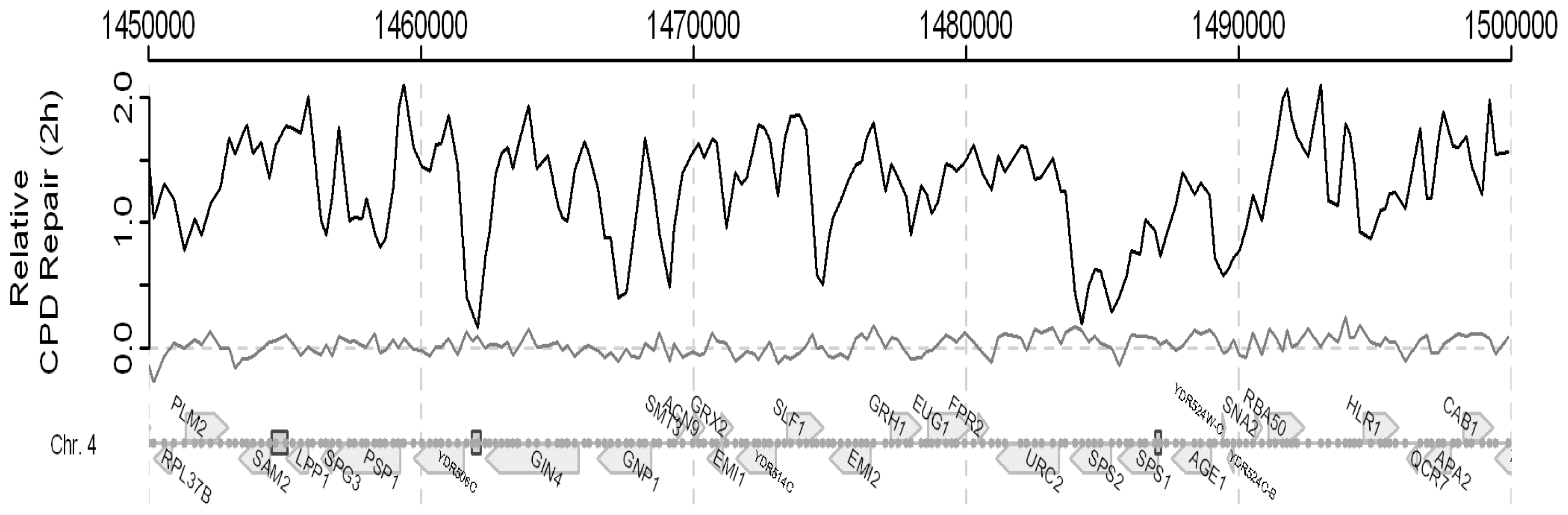
2. Results and Discussion
3. Experimental Section
3.1. Yeast Strains
3.2. UV Irradiation of Yeast Cells
3.3. UV Survival Assays
3.4. Preparation of Yeast DNA
3.5. Preparation of Yeast Chromatin
- UV irradiation of yeast cells took place as described above. However after re-suspension of the cells into YPD 3 mL of 37.5% Formaldehyde was added per 100 mL of culture to form protein-DNA and protein-protein cross-links. The culture underwent incubation on a shaking platform at room temperature for between 10 min and 40 min dependent on the protein of interest. Following this incubation 5.5 mL of 2.5 M glycine was added and incubated at room temperature on a shaking platform for 5 min. This step stopped the crosslinking. All samples were collected by centrifugation and re-suspended in approximately 40 mL PBS, where-after the cells were kept on ice and in the dark.
- Cells in 40 mL PBS were pelleted, before washing and transferring to a 2 mL microcentrifuge tube using 1 mL of FA/SDS. The cells were pelleted at 4 °C, the supernatant was removed and the pellet was re-suspended in 500 μL FA/SDS( + PMSF).
- Mechanical lysing of the cell wall occurred by adding 0.5 mL of glass beads to each sample, before vortexing for 10 min at 4 °C. The chromatin was purified from the beads by puncturing a hole in the 2 mL microcentrifuge tube with a hot needle prior to the placement of the microcentrifuge tube in a 15 mL Falcon tube.
- The lysate was collected in the 15 mL Falcon tube by centrifugation, before the glass beads were washed with 500 μL of FA/SAS buffer (+PMSF) and subjected to a further centrifugation.
- The cell lysate should have exited the microcentrifuge tube through the hole generated and have collected in the 15 mL Falcon tube. In order to remove any soluble proteins not crosslinked to the DNA, the cell lysate was transferred to a 2 mL microcentrifuge tube and centrifuged at 12,000 rpm for 20 min at 4 °C in a microfuge (Beckman Coulter, 22R centrifuge). The supernatant was removed by aspiration and the remaining pellet re-suspended and transferred to a 15 mL Corning tube with 900 μL FA/SDS (+PMSF).
- Sonication was carried out using a Bioruptor, set to High and at 4 °C, for 20 s ON followed by 40 s OFF. Metal probes were cleaned with 100% ethanol and then with H2O before being screwed into the 15 mL Corning tubes and placed in the Bioruptor water bath. The settings on the Bioruptor were checked, and the programme started for 6 cycles.
- Following sonication, the 15 mL Corning tubes were centrifuged at 4000 rpm for 10 min. The supernatant was transferred to a 1.5 mL microcentrifuge tube and centrifuged at 12,000 rpm in a microfuge for 20 min.
- Finally the supernatant was transferred to a fresh 1.5 mL microcentrifuge tube. The 1 mL of chromatin was snap frozen with liquid nitrogen and stored at −80 °C DNA gel electrophoresis.
3.6. Chromatin Immunoprecipitation (ChIP)
- For each sample to be IP’d, 50 μL of Dynabeads (anti-Rabbit Ig G, Invitrogen) was taken and washed 3 times with 500 μL PBS-BSA (0.1%). After the washes, the Dynabeads were re-suspended in 100 μL PBS-BSA (0.1%) per sample before the addition of a specific antibody. An antibody titration experiment was performed beforehand to determine the amount of antibody to add per sample for optimal immunoprecipitation. For this protocol, usually 2 μg of antibody per sample was required.
- The mixture of Dynabeads and antibody was incubated at 30 °C, for 30 min at 1300 rpm in an Eppendorf thermomixer. At this stage the antibody should attach to the Dynabeads.
- The Dynabeads were collected using a magnet held against the tube, and washed 3 times with 500 μL PBS-BSA (0.1%). Finally the Dynabeads were re-suspended in 50 μL of PBS-BSA (0.1%) per sample. From this master mix, 50 μL of beads was added to 100 μL of sonicated chromatin. In addition 30 μL 10× PBS-BSA (10 mg/mL) was added and the final volume adjusted to 300 μL in total with PBS. This was incubated at 21 °C at 1300 rpm for 3 h in an Eppendorf Thermomixer.
- After the incubation, the Dynabeads (plus antibody plus DNA) were collected using a magnet and the supernatant removed. The beads were washed with 500 μL FA/SDS buffer. This was followed by a series of washes: 3 washes with 1 mL FA/SDS + NaCl buffer; 1 wash with 500 μL LiCl buffer and finally with 500 μL cold TE.
- After the final wash, the DNA was eluted off the Dynabeads with 125 μL of Pronase buffer at 65 °C, at 900 rpm for 20 min in an Eppendorf Thermomixer. Following this the supernatant was transferred to a fresh 1.5 mL microcentrifuge tube and 6.25 μL of Pronase was added before incubation overnight at 65 °C in a water bath.
- For the input samples, 50 μL of sonicated chromatin was adjusted to 100 μL with TE buffer, before the addition of 25 μL 5× Pronase Buffer. Like the IP samples, the input samples all had 6.25 μL of Pronase added and were incubated overnight at 65 °C in a water bath.
- The following day, the IP and input samples were all treated with 2 μL of RNase and incubated for 1 h at 37 °C. After the incubation, the DNA was purified using Qiagen PCR purification kit (followed as per manufacturer’s instructions) into 50 μL elution buffer. From this stage the IP and input DNA could either be used in a qRT-PCR or used as the starting point for genome-wide experiments.
3.7. Quantitative Real-Time PCR (qRT-PCR)
3.8. Preparation of Yeast Whole Cell Extract (WCE)
4. Conclusions
Acknowledgments
References
- Ramanathan, B.; Smerdon, M.J. Changes in nuclear protein acetylation in UV-damaged human cells. Carcinogenesis 1986, 7, 1087–1094. [Google Scholar]
- Ramanathan, B.; Smerdon, M.J. Enhanced DNA repair synthesis in hyperacetylated nucleosomes. J. Biol. Chem 1989, 264, 11026–11094. [Google Scholar]
- Smerdon, M.J.; Thoma, F. Site-specific DNA repair at the nucleosome level in a yeast minichromosome. Cell 1990, 61, 675–684. [Google Scholar]
- Wellinger, R.E.; Thoma, F. Nucleosome structure and positioning modulate nucleotide excision repair in the non-transcribed strand of an active gene. EMBO J 1997, 16, 5046–5056. [Google Scholar]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis; ASM Press: Washington, DC, USA, 2005. [Google Scholar]
- Li, S.; Waters, R. Nucleotide level detection of cyclobutane pyrimidine dimers using oligonucleotides and magnetic beads to facilitate labeling of DNA fragments incised at dimers and chemical sequencing reference ladders. Carcinogenesis 1996, 17, 1549–1552. [Google Scholar]
- Teng, Y.; Li, S.; Waters, R.; Reed, S.H. Excision repair at the level of the nucleotide in the Saccharomyces cerevisiae MFA2 gene: The mapping of where enhanced repair in the transcribed strand begins or ends and the identification of only a partial Rad16 requisite for repairing upstream control sequences. J. Mol. Biol 1997, 267, 324–337. [Google Scholar]
- Teng, Y.; Longhese, M.P.; McDonough, G.; Waters, R. Mutants with changes in different domains of yeast replication protein A exhibit differences in repairing the control region, the transcribed strand and the non-transcribed strand of the Saccharomyces cerevisiae MFA2 gene. J. Mol. Biol 1998, 280, 355–363. [Google Scholar]
- Teng, Y.; Waters, R. Excision repair at the level of the nucleotide in the upstream control region, the coding sequence and the region where transcription terminates of the S. cerevisiae MFA2 gene and the role of the RAD26. Nucleic Acids Res 2000, 28, 1114–1119. [Google Scholar]
- Yu, S.; Teng, Y.; Lowndes, N.F.; Waters, R. RAD9, RAD24, RAD16 and RAD26 are required for the inducible nucleotide excision repair of UV-induced cyclobutane pyrimidine dimers from the transcribed and non transcribed regions of the Saccharomyces cerevisiae MFA2 gene. Mutat. Res 2001, 485, 229–236. [Google Scholar]
- Teng, Y.; Yu, Y.; Waters, R. The Saccharomyces cerevisiae histone acetyltransferase Gcn5 has a role in the photoreactivation and nucleotide excision repair of UV induced cyclobutane pyrimidine dimers in the MFA2 gene. J. Mol. Biol 2002, 316, 489–499. [Google Scholar]
- Ferreiro, J.A.; Powell, N.G.; Karabetsou, N.; Kent, N.A.; Mellor, J.; Waters, R. Cbf1p modulates chromatin structure, transcription and repair at the S. cerevisiae MET16 locus. Nucleic Acids Res 2004, 32, 1617–1626. [Google Scholar]
- Tremblay, M.; Teng, Y.; Paquette, M.; Waters, R.; Conconi, A. Complementary roles of yeast Rad4p and Rad34p in nucleotide excision repair of active and inactive rDNA chromatin. Mol. Cell Biol 2008, 28, 7504–7513. [Google Scholar]
- Taschner, M.; Harreman, M.; Teng, Y.; Gill, H.; Anindya, R.; Maslen, S.L.; Skehel, J.M.; Waters, R.; Svejstrup, J.Q. A role for checkpoint kinase-dependent Rad26 phosphorylation in transcription-coupled DNA repair in Saccharomyces cerevisiae. Mol. Cell Biol 2010, 30, 436–446. [Google Scholar]
- Teng, Y.; Yu, S.; Waters, R. The mapping of nucleosomes and regulatory protein binding sites on the Saccharomyces cerevisiae MFA2 gene: A high resolution approach. Nucleic Acids Res 2001, 29, e64. [Google Scholar]
- Yu, Y.; Teng, Y.; Reed, S.H.; Waters, R. UV irradiation stimulates histone acetylation and chromatin remodeling at a repressed yeast locus. Proc. Natl. Acad. Sci. USA 2005, 102, 8650–8655. [Google Scholar]
- Teng, Y.; Yu, Y.; Ferreiro, J.A.; Waters, R. Histone acetylation, chromatin remodeling, transcription and nucleotide excision repair in S. cerevisiae: studies with two model genes. DNA Repair 2005, 4, 870–883. [Google Scholar]
- Ferreiro, J.A.; Powell, N.G.; Karabetsou, N.; Mellor, J.; Waters, R. Roles for Gcn5p and Ada2p in transcription and nucleotide excision repair at the Saccharomyces cerevisiae MET16 gene. Nucleic Acids Res 2006, 34, 976–985. [Google Scholar]
- Teng, Y.; Liu, H.; Gill, H.W.; Yu, Y.; Waters, R.; Reed, S.H. Saccharomyces cerevisiae Rad16 mediates ultraviolet-dependent histone H3 acetylation required for efficient global genome nucleotide-excision repair. EMBO Rep 2008, 9, 97–102. [Google Scholar]
- Yu, S.; Teng, Y.; Waters, R.; Reed, S.H. How chromatin is remodeled during DNA repair of UV-induced DNA damage in Saccharomyces cerevisiae. PLoS Genet 2011, 7, e1002124. [Google Scholar]
- Flaus, A.; Owen-Hughes, T. Mechanisms for ATP-dependent chromatin remodeling: The means to the end. FEBS J 2011, 278, 3579–3595. [Google Scholar]
- Ferreira, H.; Flaus, A.; Owen-Hughes, T. Histone modifications influence the action of Snf2 family remodeling enzymes by different mechanisms. J. Mol. Biol 2007, 374, 563–579. [Google Scholar]
- Svejstrup, J.Q. Mechanisms of transcription-coupled DNA repair. Nat. Rev. Mol. Cell. Biol 2002, 3, 21–29. [Google Scholar]
- Li, S.; Smerdon, M.J. Rpb4 and Rpb9 mediate subpathways of transcription-coupled DNA repair in Saccharomyces cerevisiae. EMBO J 2002, 21, 5921–5929. [Google Scholar]
- Verhage, R.; Zeeman, A.M.; de Groot, N.; Gleig, F.; Bang, D.D.; van de Putte, P.; Brouwer, J. The RAD7 and RAD16 genes, which are essential for pyrimidine dimmer removal from the silent mating type loci, are also required for repair of the nontranscribed strand of an active gene in Saccharomyces cerevisiae. Mol. Cell. Biol 1994, 14, 6135–6142. [Google Scholar]
- Reed, S.H.; Akiyama, M.; Stillman, B.; Friedberg, E.C. Yeast autonomously replicating sequence binding factor is involved in nucleotide excision repair. Genes Dev 1999, 13, 3052–3058. [Google Scholar]
- Yu, S.; Smirnova, J.B.; Friedberg, E.C.; Stillman, B.; Akiyama, M.; Owen-Hughes, T.; Waters, R.; Reed, S.H. ABF1 binding sites promote efficient global genome nucleotide excision repair. J. Biol.Chem 2009, 284, 966–973. [Google Scholar]
- Bang, D.D.; Verhage, R.; Goosen, N.; Brouwer, J.; van de Putte, P. Molecular cloning of RAD16, a gene involved in differential repair in Saccharomyces cerevisiae. Nucleic Acids Res 1992, 20, 3925–3931. [Google Scholar]
- Eisen, J.A.; Sweder, K.S.; Hanawalt, P.C. Evolution of the SNF2 family of proteins: Subfamilies with distinct sequences and functions. Nucleic Acids Res 1995, 23, 2715–2723. [Google Scholar]
- Whitehouse, I.; Flaus, A.; Cairns, B.R.; White, M.F.; Workman, J.L.; Owen-Hughes, T. Nucleosome mobilization catalysed by the yeast SWI/SNF complex. Nature 1999, 400, 784–787. [Google Scholar]
- Havas, K.; Flaus, A.; Phelan, M.; Kingston, R.; Wade, P.A.; Lilley, D.M.; Owen-Hughes, T. Generation of superhelical torsion by ATP-dependent chromatin remodeling activities. Cell 2000, 103, 1133–1142. [Google Scholar]
- Van Komen, S.; Petukhova, G.; Sigurdson, S.; Stratton, S.; Sung, P. Superhelicity-driven homologous DNA pairing by yeast recombination factors Rad51 and Rad54. Mol. Cell 2000, 6, 563–572. [Google Scholar]
- Yu, S.; Owen-Hughes, T.; Friedberg, E.C.; Waters, R.; Reed, S.H. The yeast Rad7/Rad16/Abf1 complex generates superhelical torsion in DNA that is required for nucleotide excision repair. DNA Repair 2004, 3, 277–287. [Google Scholar]
- Wang, Z.; Wei, S.; Reed, S.H.; Wu, X.; Svejstrup, J.Q.; Feaver, W.J.; Kornberg, R.D.; Friedberg, E.C. The RAD7, RAD16 and RAD23 genes of Saccharomyces cerevisiae: Requirement for transcription-independent nucleotide excision repair in vitro and interactions between the gene products. Mol. Cell Biol 1997, 17, 635–643. [Google Scholar]
- Guzder, S.N.; Sung, P.; Prakash, L.; Prakash, S. Yeast Rad7-Rad16 complex, specific for the nucleotide excision repair of the nontranscribed DNA strand, is an ATP-dependent DNA damage sensor. J. Biol. Chem 1997, 272, 21665–21668. [Google Scholar]
- Gillette, T.G.; Yu, S.; Zhou, Z.; Waters, R.; Johnston, S.A.; Reed, S.H. Distinct functions of the ubiquitin-proteasome pathway influence nucleotide excision repair. EMBO J 2006, 25, 2529–2539. [Google Scholar]
- Diffley, J. Early events in eukaryotic DNA replication. Trends Cell Biol 1992, 2, 298–303. [Google Scholar]
- Yarragudi, A.; Parfrey, L.W.; Morse, R.H. Genome-wide analysis of transcriptional dependence and probable target sites for Abf1 and Rap1 in Saccharomyces cerevisiae. Nucleic Acids Res 2007, 35, 193–202. [Google Scholar]
- Bone, J.R.; Roth, S.Y. Recruitment of the yeast Tup1p-Ssn6p repressor is associated with localized decreases in histone acetylation. J. Biol. Chem 2001, 276, 1808–1818. [Google Scholar]
- Malavé, T.M.; Dent, S.Y. Transcriptional repression by Tup1-Ssn6. Biochem. Cell Biol 2006, 84, 437–443. [Google Scholar]
- Irizar, A.; Yu, Y.; Reed, S.H.; Louis, E.J.; Waters, R. Silenced yeast chromatin is maintained by Sir2 in preference to permitting histone acetylations for efficient NER. Nucleic Acids Res 2010, 38, 4675–4686. [Google Scholar]
- Loney, E.R.; Inglis, P.W.; Sharp, S.; Pryde, F.E.; Kent, N.A.; Mellor, J.; Louis, E.J. Repressive and non-repressive chromatin at native telomeres in Saccharomyces cerevisiae. Epigenet. Chromatin 2009, 2, 18. [Google Scholar]
- Mockler, T.C.; Chan, S.; Sundaresan, A.; Chen, H.; Jacobsen, S.E.; Ecker, J.R. Applications of DNA tiling arrays for whole-genome analysis. Genomics 2005, 85, 1–15. [Google Scholar]
- Pokholok, D.K.; Harbison, C.T.; Levine, S.; Cole, M.; Hannett, N.M.; Lee, T.I.; Bell, G.W.; Walker, K.; Rolfe, P.A.; Herbolsheimer, E.; et al. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell 2005, 122, 517–527. [Google Scholar]
- Teng, Y.; Bennett, M.; Evans, K.E.; Zhuang-Jackson, H.; Higgs, A.; Reed, S.H.; Waters, R. A novel method for the genome-wide high resolution analysis of DNA damage. Nucleic Acids Res 2011, 39, e10. [Google Scholar]














© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Waters, R.; Evans, K.; Bennett, M.; Yu, S.; Reed, S. Nucleotide Excision Repair in Cellular Chromatin: Studies with Yeast from Nucleotide to Gene to Genome. Int. J. Mol. Sci. 2012, 13, 11141-11164. https://doi.org/10.3390/ijms130911141
Waters R, Evans K, Bennett M, Yu S, Reed S. Nucleotide Excision Repair in Cellular Chromatin: Studies with Yeast from Nucleotide to Gene to Genome. International Journal of Molecular Sciences. 2012; 13(9):11141-11164. https://doi.org/10.3390/ijms130911141
Chicago/Turabian StyleWaters, Raymond, Katie Evans, Mark Bennett, Shirong Yu, and Simon Reed. 2012. "Nucleotide Excision Repair in Cellular Chromatin: Studies with Yeast from Nucleotide to Gene to Genome" International Journal of Molecular Sciences 13, no. 9: 11141-11164. https://doi.org/10.3390/ijms130911141
APA StyleWaters, R., Evans, K., Bennett, M., Yu, S., & Reed, S. (2012). Nucleotide Excision Repair in Cellular Chromatin: Studies with Yeast from Nucleotide to Gene to Genome. International Journal of Molecular Sciences, 13(9), 11141-11164. https://doi.org/10.3390/ijms130911141