Is DNA Damage Response Ready for Action Anywhere?


Abstract
:
1. Introduction
2. Micronuclei
3. Response to Double Strand Breaks in Micronuclear DNA
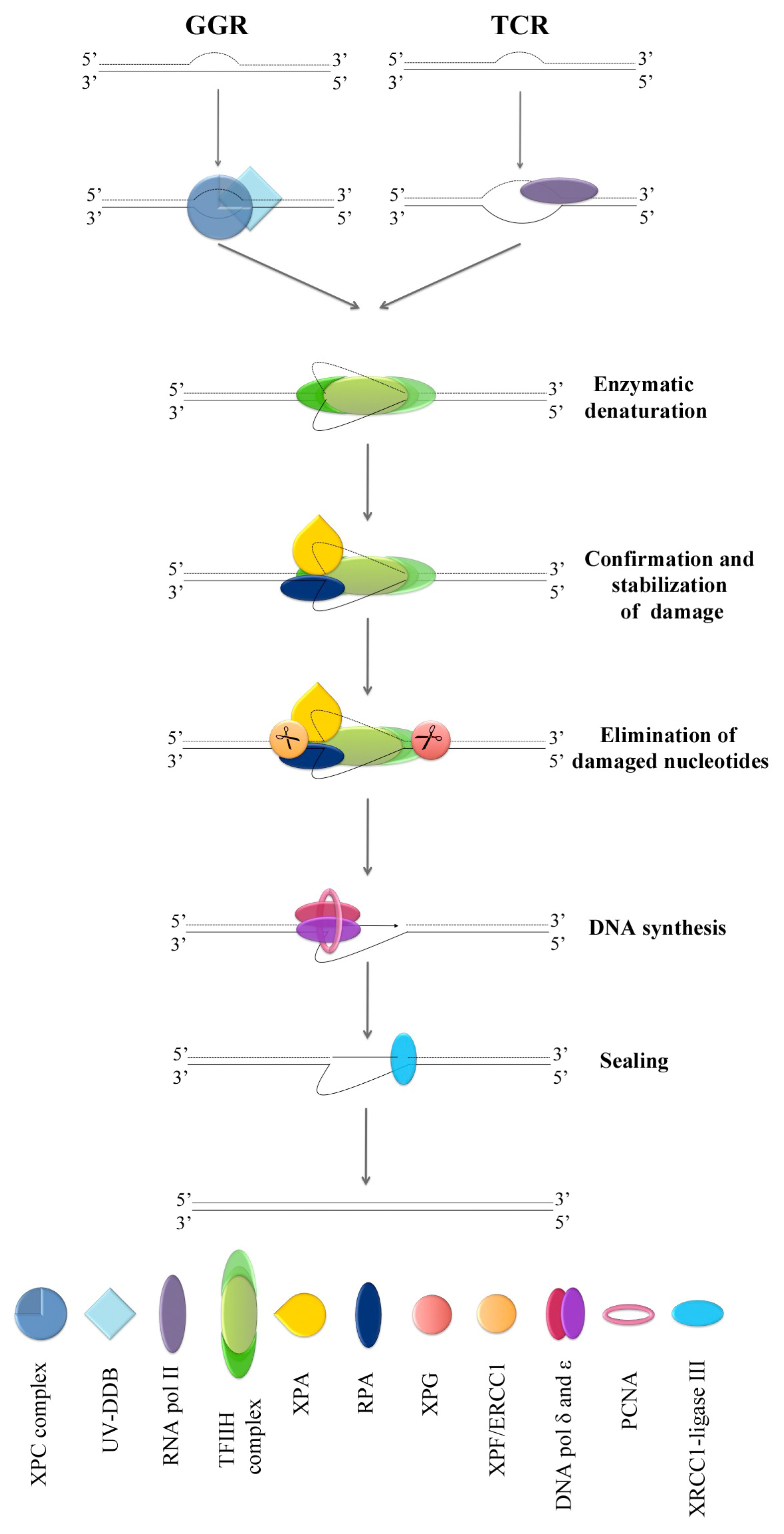
4. NER Factors in Micronuclei
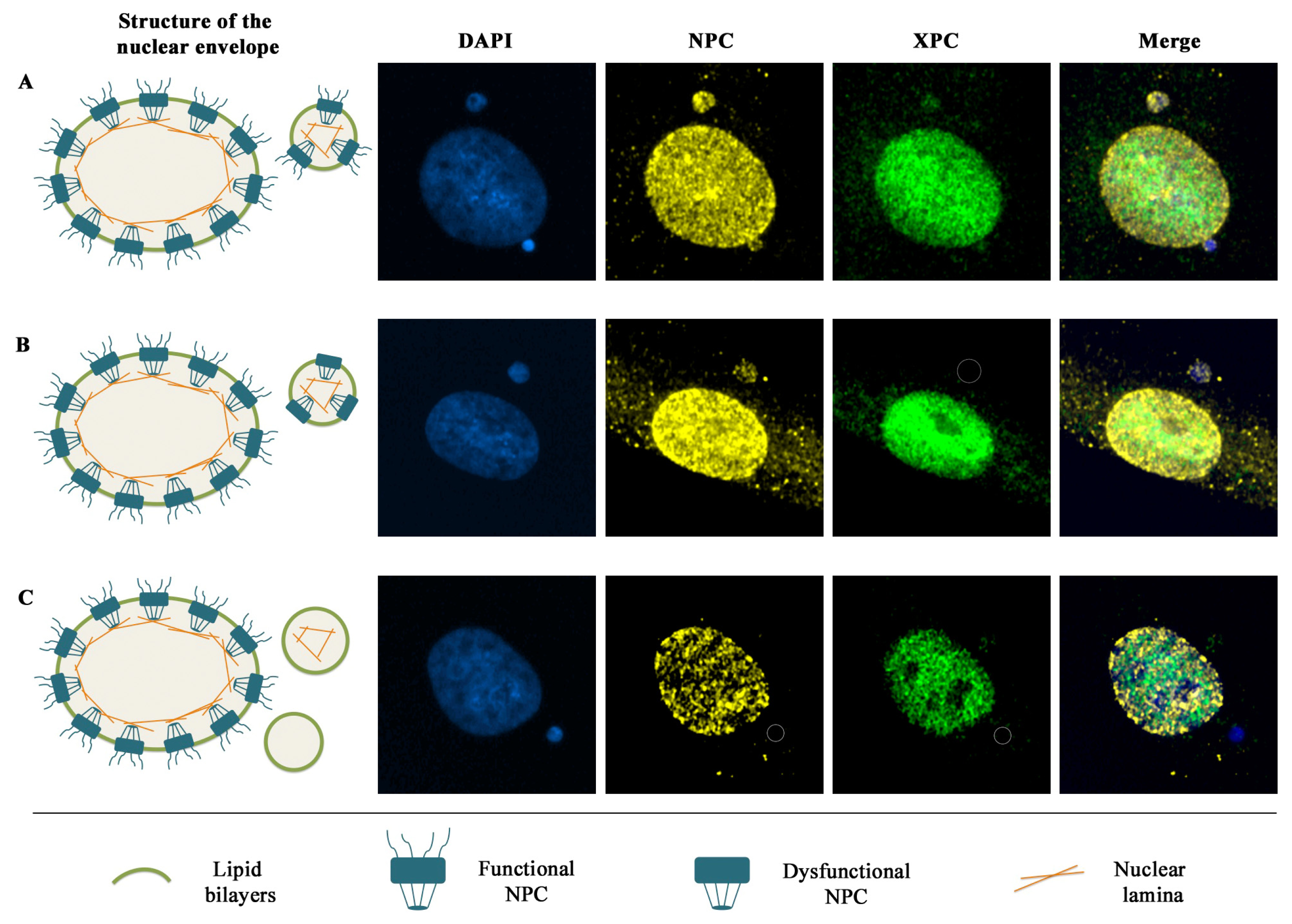
5. Nuclear Envelope Defects Hamper the Recruitment of DDR Factors to Micronuclear DNA Lesions
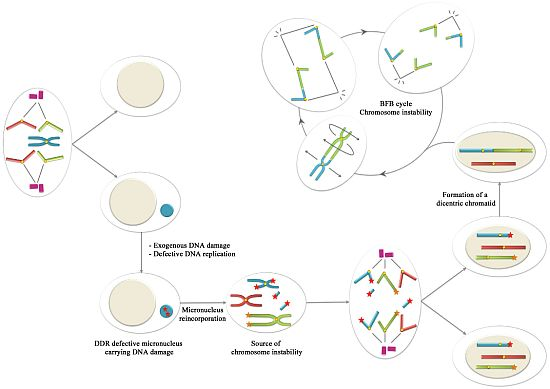
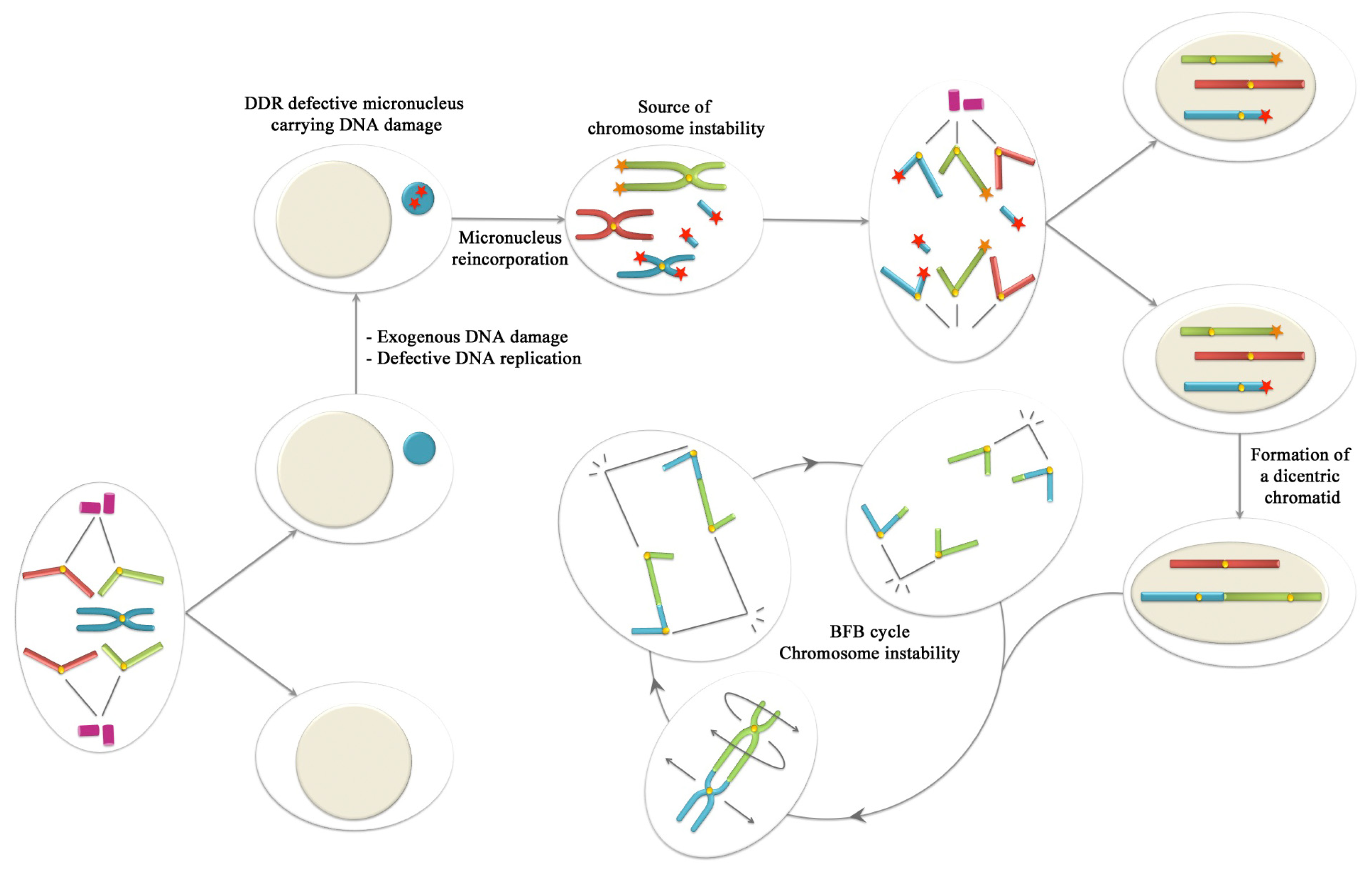
6. Conclusions: Could Be Micronuclei a Source of Chromosome Instability?
Acknowledgments
References
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular Mechanisms of Micronucleus, Nucleoplasmic Bridge and Nuclear Bud Formation in Mammalian and Human Cells. Mutagenesis 2011, 26, 125–132. [Google Scholar]
- Shay, J.W.; Wright, W.E. Senescence and Immortalization: Role of Telomeres and Telomerase. Carcinogenesis 2005, 26, 867–874. [Google Scholar]
- Hande, M.P.; Samper, E.; Lansdorp, P.; Blasco, M.A. Telomere Length Dynamics and Chromosomal Instability in Cells Derived from Telomerase Null Mice. J. Cell Biol 1999, 144, 589–601. [Google Scholar]
- Latre, L.; Tusell, L.; Martin, M.; Miro, R.; Egozcue, J.; Blasco, M.A.; Genesca, A. Shortened Telomeres Join to DNA Breaks Interfering with their Correct Repair. Exp. Cell Res 2003, 287, 282–288. [Google Scholar]
- Hoffelder, D.R.; Luo, L.; Burke, N.A.; Watkins, S.C.; Gollin, S.M.; Saunders, W.S. Resolution of Anaphase Bridges in Cancer Cells. Chromosoma 2004, 112, 389–397. [Google Scholar]
- Pampalona, J.; Soler, D.; Genesca, A.; Tusell, L. Whole Chromosome Loss is Promoted by Telomere Dysfunction in Primary Cells. Genes Chromosomes Cancer 2010, 49, 368–378. [Google Scholar]
- Pampalona, J.; Soler, D.; Genesca, A.; Tusell, L. Telomere Dysfunction and Chromosome Structure Modulate the Contribution of Individual Chromosomes in Abnormal Nuclear Morphologies. Mutat. Res 2010, 683, 16–22. [Google Scholar]
- McClintock, B. The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics 1941, 26, 234–282. [Google Scholar]
- Cimini, D.; Fioravanti, D.; Salmon, E.D.; Degrassi, F. Merotelic Kinetochore Orientation versus Chromosome Mono-Orientation in the Origin of Lagging Chromosomes in Human Primary Cells. J. Cell. Sci 2002, 115, 507–515. [Google Scholar]
- Shimizu, N. Molecular Mechanisms of the Origin of Micronuclei from Extrachromosomal Elements. Mutagenesis 2011, 26, 119–123. [Google Scholar]
- Kanda, T.; Wahl, G.M. The Dynamics of Acentric Chromosomes in Cancer Cells Revealed by GFP-Based Chromosome Labeling Strategies. J. Cell. Biochem 2000, (Suppl 35), 107–114. [Google Scholar]
- Shimizu, N.; Itoh, N.; Utiyama, H.; Wahl, G.M. Selective Entrapment of Extrachromosomally Amplified DNA by Nuclear Budding and Micronucleation during S Phase. J. Cell Biol 1998, 140, 1307–1320. [Google Scholar]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic Instabilities in Human Cancers. Nature 1998, 396, 643–649. [Google Scholar]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The Cancer Genome. Nature 2009, 458, 719–724. [Google Scholar]
- Duffaud, F.; Orsiere, T.; Digue, L.; Villani, P.; Volot, F.; Favre, R.; Botta, A. Micronucleated Lymphocyte Rates from Head-and-Neck Cancer Patients. Mutat. Res 1999, 439, 259–266. [Google Scholar]
- Bonassi, S.; Znaor, A.; Ceppi, M.; Lando, C.; Chang, W.P.; Holland, N.; Kirsch-Volders, M.; Zeiger, E.; Ban, S.; Barale, R.; et al. An Increased Micronucleus Frequency in Peripheral Blood Lymphocytes Predicts the Risk of Cancer in Humans. Carcinogenesis 2007, 28, 625–631. [Google Scholar]
- Bonassi, S.; El-Zein, R.; Bolognesi, C.; Fenech, M. Micronuclei Frequency in Peripheral Blood Lymphocytes and Cancer Risk: Evidence from Human Studies. Mutagenesis 2011, 26, 93–100. [Google Scholar]
- Abraham, R.T.; Tibbetts, R.S. Cell Biology. Guiding ATM to Broken DNA. Science 2005, 308, 510–511. [Google Scholar]
- Stracker, T.H.; Morales, M.; Couto, S.S.; Hussein, H.; Petrini, J.H. The Carboxy Terminus of NBS1 is Required for Induction of Apoptosis by the MRE11 Complex. Nature 2007, 447, 218–221. [Google Scholar]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA Double-Stranded Breaks Induce Histone H2AX Phosphorylation on Serine 139. J. Biol. Chem 1998, 273, 5858–5868. [Google Scholar]
- Paull, T.T.; Rogakou, E.P.; Yamazaki, V.; Kirchgessner, C.U.; Gellert, M.; Bonner, W.M. A Critical Role for Histone H2AX in Recruitment of Repair Factors to Nuclear Foci after DNA Damage. Curr. Biol 2000, 10, 886–895. [Google Scholar]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase Chromatin Domains Involved in DNA Double-Strand Breaks in Vivo. J. Cell Biol 1999, 146, 905–916. [Google Scholar]
- Bekker-Jensen, S.; Mailand, N. Assembly and Function of DNA Double-Strand Break Repair Foci in Mammalian Cells. DNA Repair (Amst) 2010, 9, 1219–1228. [Google Scholar]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 Ubiquitylates Histones at DNA Double-Strand Breaks and Promotes Assembly of Repair Proteins. Cell 2007, 131, 887–900. [Google Scholar]
- Helleday, T.; Lo, J.; van Gent, D.C.; Engelward, B.P. DNA Double-Strand Break Repair: From Mechanistic Understanding to Cancer Treatment. DNA Repair (Amst) 2007, 6, 923–935. [Google Scholar]
- Branzei, D.; Foiani, M. Regulation of DNA Repair Throughout the Cell Cycle. Nat. Rev. Mol. Cell Biol 2008, 9, 297–308. [Google Scholar]
- Shrivastav, M.; de Haro, L.P.; Nickoloff, J.A. Regulation of DNA Double-Strand Break Repair Pathway Choice. Cell Res 2008, 18, 134–147. [Google Scholar]
- Terradas, M.; Martin, M.; Tusell, L.; Genesca, A. DNA Lesions Sequestered in Micronuclei Induce a Local Defective-Damage Response. DNA Repair (Amst) 2009, 8, 1225–1234. [Google Scholar]
- Nelson, G.; Buhmann, M.; von Zglinicki, T. DNA Damage Foci in Mitosis are Devoid of 53BP1. Cell. Cycle 2009, 8, 3379–3383. [Google Scholar]
- Giunta, S.; Jackson, S.P. Give Me a Break, but Not in Mitosis: The Mitotic DNA Damage Response Marks DNA Double-Strand Breaks with Early Signaling Events. Cell. Cycle 2011, 10, 1215–1221. [Google Scholar]
- Yoshikawa, T.; Kashino, G.; Ono, K.; Watanabe, M. Phosphorylated H2AX Foci in Tumor Cells have no Correlation with their Radiation Sensitivities. J. Radiat. Res (Tokyo) 2009, 50, 151–160. [Google Scholar]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA Breaks and Chromosome Pulverization from Errors in Mitosis. Nature 2012, 482, 53–58. [Google Scholar]
- Medvedeva, N.G.; Panyutin, I.V.; Panyutin, I.G.; Neumann, R.D. Phosphorylation of Histone H2AX in Radiation-Induced Micronuclei. Radiat. Res 2007, 168, 493–498. [Google Scholar]
- MacPhail, S.H.; Banath, J.P.; Yu, Y.; Chu, E.; Olive, P.L. Cell Cycle-Dependent Expression of Phosphorylated Histone H2AX: Reduced Expression in Unirradiated but Not X-Irradiated G1-Phase Cells. Radiat. Res 2003, 159, 759–767. [Google Scholar]
- Kim, J.A.; Kruhlak, M.; Dotiwala, F.; Nussenzweig, A.; Haber, J.E. Heterochromatin is Refractory to Gamma-H2AX Modification in Yeast and Mammals. J. Cell Biol 2007, 178, 209–218. [Google Scholar]
- Karagiannis, T.C.; Harikrishnan, K.N.; El-Osta, A. Disparity of Histone Deacetylase Inhibition on Repair of Radiation-Induced DNA Damage on Euchromatin and Constitutive Heterochromatin Compartments. Oncogene 2007, 26, 3963–3971. [Google Scholar]
- Cowell, I.G.; Sunter, N.J.; Singh, P.B.; Austin, C.A.; Durkacz, B.W.; Tilby, M.J. γH2AX Foci Form Preferentially in Euchromatin after Ionising-Radiation. PLoS One 2007, 2, e1057. [Google Scholar]
- Falk, M.; Lukasova, E.; Kozubek, S. Chromatin Structure Influences the Sensitivity of DNA to Gamma-Radiation. Biochim. Biophys. Acta 2008, 1783, 2398–2414. [Google Scholar]
- Puerto, S.; Ramirez, M.J.; Marcos, R.; Creus, A.; Surralles, J. Radiation-Induced Chromosome Aberrations in Human Euchromatic (17cen-p53) and Heterochromatic (1cen-1q12) Regions. Mutagenesis 2001, 16, 291–296. [Google Scholar]
- Goodarzi, A.A.; Noon, A.T.; Deckbar, D.; Ziv, Y.; Shiloh, Y.; Lobrich, M.; Jeggo, P.A. ATM Signaling Facilitates Repair of DNA Double-Strand Breaks Associated with Heterochromatin. Mol. Cell 2008, 31, 167–177. [Google Scholar]
- Goodarzi, A.A.; Jeggo, P.; Lobrich, M. The Influence of Heterochromatin on DNA Double Strand Break Repair: Getting the Strong, Silent Type to Relax. DNA Repair (Amst) 2010, 9, 1273–1282. [Google Scholar]
- Cann, K.L.; Dellaire, G. Heterochromatin and the DNA Damage Response: The Need to Relax. Biochem. Cell Biol 2011, 89, 45–60. [Google Scholar]
- Gillet, L.C.; Scharer, O.D. Molecular Mechanisms of Mammalian Global Genome Nucleotide Excision Repair. Chem. Rev 2006, 106, 253–276. [Google Scholar]
- Naegeli, H.; Sugasawa, K. The Xeroderma Pigmentosum Pathway: Decision Tree Analysis of DNA Quality. DNA Repair (Amst) 2011, 10, 673–683. [Google Scholar]
- Hoeijmakers, J.H. Genome Maintenance Mechanisms for Preventing Cancer. Nature 2001, 411, 366–374. [Google Scholar]
- Sugasawa, K.; Akagi, J.; Nishi, R.; Iwai, S.; Hanaoka, F. Two-Step Recognition of DNA Damage for Mammalian Nucleotide Excision Repair: Directional Binding of the XPC Complex and DNA Strand Scanning. Mol. Cell 2009, 36, 642–653. [Google Scholar]
- Coin, F.; Oksenych, V.; Egly, J.M. Distinct Roles for the XPB/p52 and XPD/p44 Subcomplexes of TFIIH in Damaged DNA Opening during Nucleotide Excision Repair. Mol. Cell 2007, 26, 245–256. [Google Scholar]
- Jones, C.J.; Wood, R.D. Preferential Binding of the Xeroderma Pigmentosum Group A Complementing Protein to Damaged DNA. Biochemistry 1993, 32, 12096–12104. [Google Scholar]
- Mu, D.; Hsu, D.S.; Sancar, A. Reaction Mechanism of Human DNA Repair Excision Nuclease. J. Biol. Chem 1996, 271, 8285–8294. [Google Scholar]
- Moser, J.; Kool, H.; Giakzidis, I.; Caldecott, K.; Mullenders, L.H.; Fousteri, M.I. Sealing of Chromosomal DNA Nicks during Nucleotide Excision Repair Requires XRCC1 and DNA Ligase III Alpha in a Cell-Cycle-Specific Manner. Mol. Cell 2007, 27, 311–323. [Google Scholar]
- Pfeifer, G.P.; You, Y.H.; Besaratinia, A. Mutations Induced by Ultraviolet Light. Mutat. Res 2005, 571, 19–31. [Google Scholar]
- Rademakers, S.; Volker, M.; Hoogstraten, D.; Nigg, A.L.; Mone, M.J.; van Zeeland, A.A.; Hoeijmakers, J.H.; Houtsmuller, A.B.; Vermeulen, W. Xeroderma Pigmentosum Group A Protein Loads as a Separate Factor Onto DNA Lesions. Mol. Cell. Biol 2003, 23, 5755–5767. [Google Scholar]
- Hoogstraten, D.; Bergink, S.; Ng, J.M.; Verbiest, V.H.; Luijsterburg, M.S.; Geverts, B.; Raams, A.; Dinant, C.; Hoeijmakers, J.H.; Vermeulen, W.; et al. Versatile DNA Damage Detection by the Global Genome Nucleotide Excision Repair Protein XPC. J. Cell. Sci 2008, 121, 2850–2859. [Google Scholar]
- Terradas, M.; Martin, M.; Hernandez, L.; Tusell, L.; Genesca, A. Nuclear Envelope Defects Impede a Proper Response to Micronuclear DNA Lesions. Mutat. Res 2012, 729, 35–40. [Google Scholar]
- Rasala, B.A.; Ramos, C.; Harel, A.; Forbes, D.J. Capture of AT-Rich Chromatin by ELYS Recruits POM121 and NDC1 to Initiate Nuclear Pore Assembly. Mol. Biol. Cell 2008, 19, 3982–3996. [Google Scholar]
- Labidi, B.; Gregoire, M.; Frackowiak, S.; Hernandez-Verdun, D.; Bouteille, M. RNA Polymerase Activity in PtK1 Micronuclei Containing Individual Chromosomes. an in vitro and in situ Study. Exp. Cell Res 1987, 169, 233–244. [Google Scholar]
- Geraud, G.; Laquerriere, F.; Masson, C.; Arnoult, J.; Labidi, B.; Hernandez-Verdun, D. Three-Dimensional Organization of Micronuclei Induced by Colchicine in PtK1 Cells. Exp. Cell Res 1989, 181, 27–39. [Google Scholar]
- Huang, Y.; Fenech, M.; Shi, Q. Micronucleus Formation Detected by Live-Cell Imaging. Mutagenesis 2011, 26, 133–138. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DDR factor | Medvedeva et al. 2009 [33] | Yoshikawa et al. 2009 [31] | Terradas et al. 2009 [28] | Crasta et al. 2012 [32] |
|---|---|---|---|---|
| ATM | ✓ | 5%–16% | - | - |
| DNA-PKcs | - | <1%–7% | - | - |
| 53BP1 | ✘ | 5%–6% | 14.1% | ✓ |
| MRE11 | ✘ | - | 27.6% | - |
| MDC1 | ✓ | - | - | - |
| RAD17 | ✘ | - | - | - |
| RAD50 | ✘ | - | - | - |
| ATR | - | - | - | ✓ |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Terradas, M.; Martín, M.; Hernández, L.; Tusell, L.; Genescà, A. Is DNA Damage Response Ready for Action Anywhere? Int. J. Mol. Sci. 2012, 13, 11569-11583. https://doi.org/10.3390/ijms130911569
Terradas M, Martín M, Hernández L, Tusell L, Genescà A. Is DNA Damage Response Ready for Action Anywhere? International Journal of Molecular Sciences. 2012; 13(9):11569-11583. https://doi.org/10.3390/ijms130911569
Chicago/Turabian StyleTerradas, Mariona, Marta Martín, Laia Hernández, Laura Tusell, and Anna Genescà. 2012. "Is DNA Damage Response Ready for Action Anywhere?" International Journal of Molecular Sciences 13, no. 9: 11569-11583. https://doi.org/10.3390/ijms130911569
APA StyleTerradas, M., Martín, M., Hernández, L., Tusell, L., & Genescà, A. (2012). Is DNA Damage Response Ready for Action Anywhere? International Journal of Molecular Sciences, 13(9), 11569-11583. https://doi.org/10.3390/ijms130911569