Purification and Properties of an Insecticidal Metalloprotease Produced by Photorhabdus luminescens Strain 0805-P5G, the Entomopathogenic Nematode Symbiont



Abstract
:
1. Introduction
2. Results and Discussion
2.1. Strain Isolation and Identification
2.2. Purification of Protease from P. luminescens
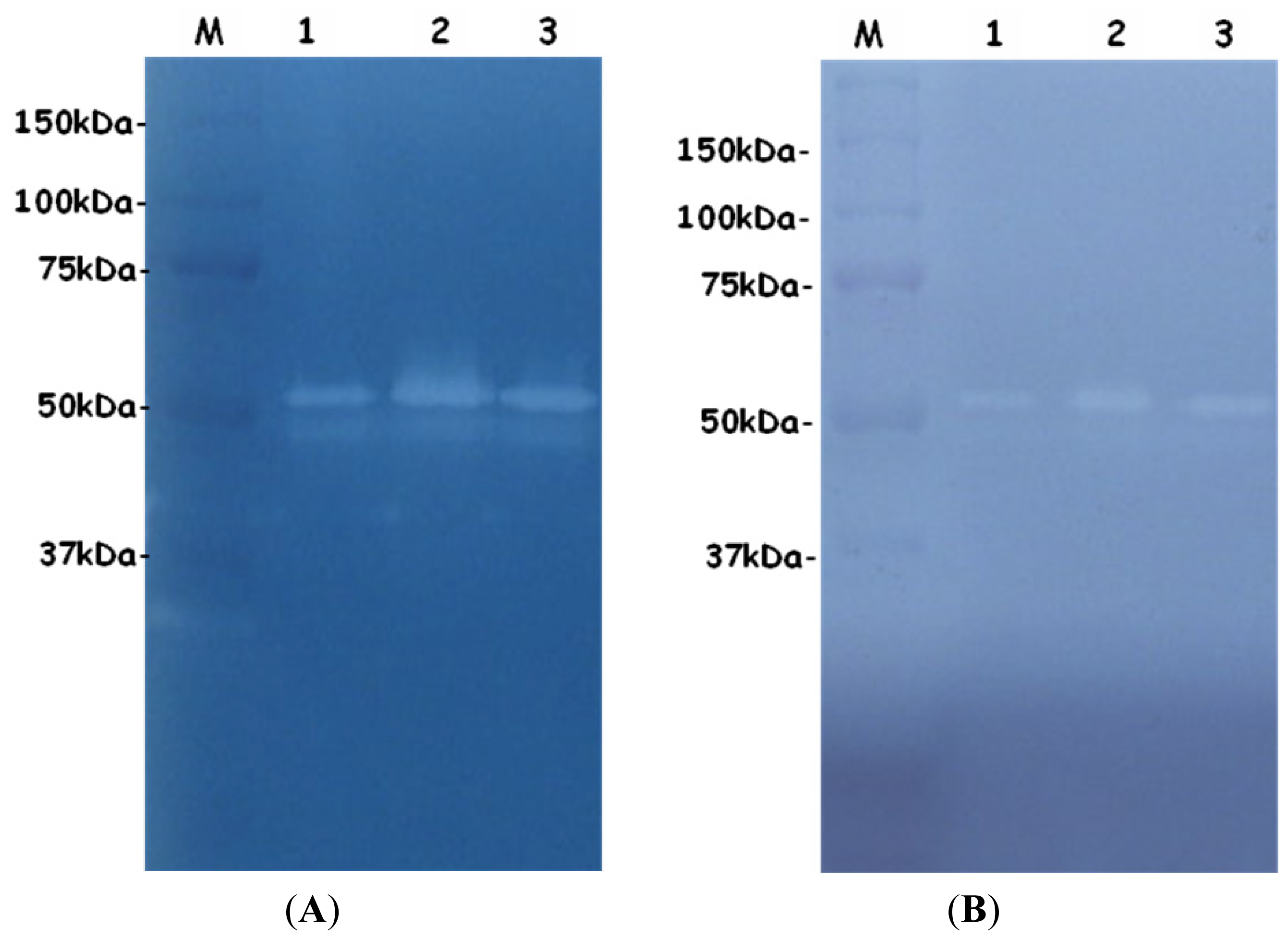
2.3. SDS-PAGE and Zymograms
2.4. N-terminal Amino Acid Sequence and Mass Spectrometry
2.5. Effect of pH on Enzyme Activity and Stability
2.6. Effect of Temperature on Enzyme Activity and Stability
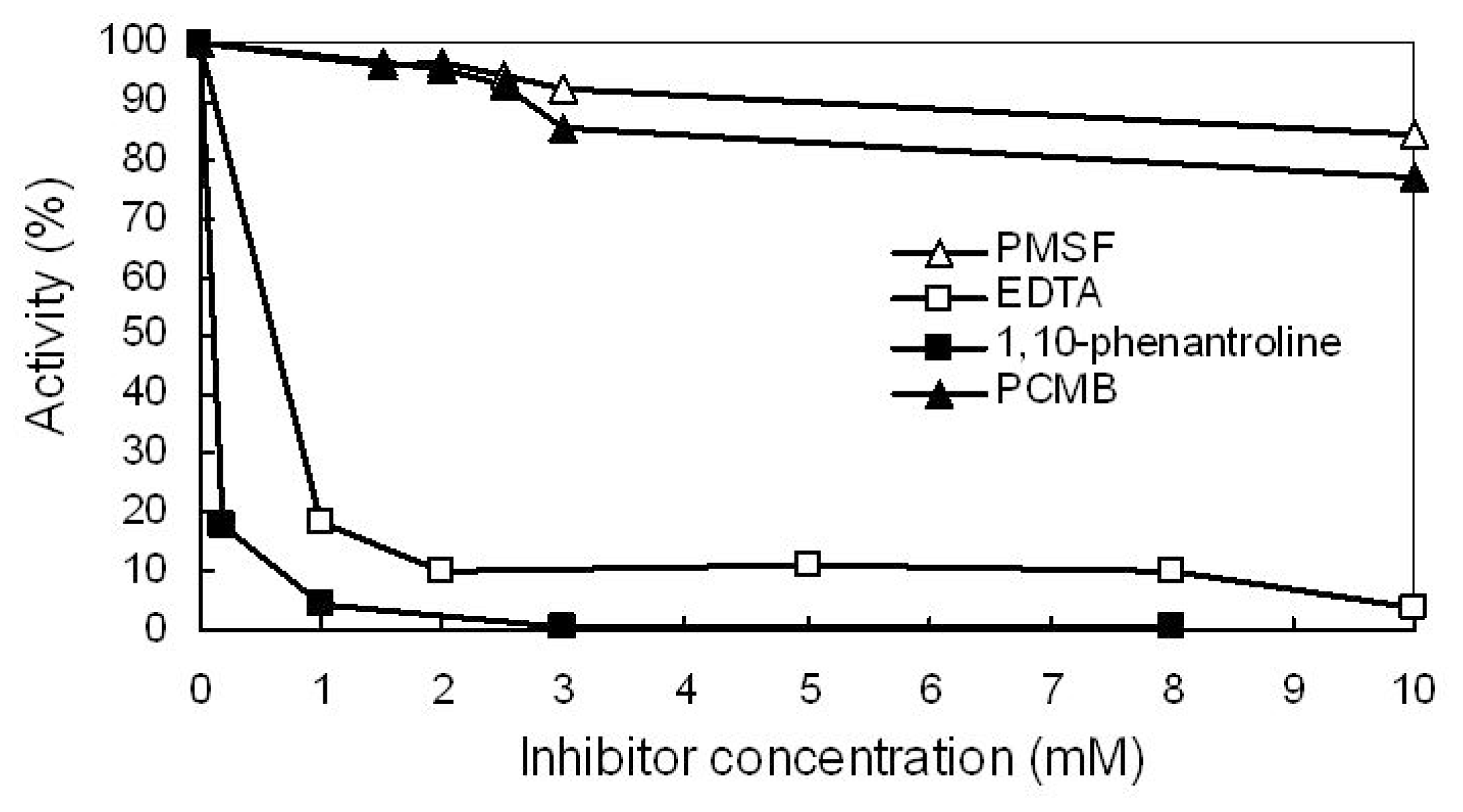
2.7. Effect of Protease Inhibitors on Enzyme Activity
2.8. Insect Toxicity Assay
3. Experimental Section
3.1. Media, Growth Conditions and Protease Activity Assay
3.2. Screening for Protease Activity
3.3. Strain Identification
3.4. Purification of Protease from P. luminescens
3.5. SDS-PAGE and Zymograms
3.6. Isoelectric Focusing (IEF)
3.7. N-terminal Amino Acid Sequence and Mass Spectrometry
3.8. Effect of pH on Enzyme Activity and Stability
3.9. Effect of Temperature on Enzyme Activity and Stability
3.10. Effect of Protease Inhibitors on Enzyme Activity
3.11. Insect Toxicity Assay
4. Discussion
5. Conclusions
Acknowledgments
References
- Cabral, C.M.; Cherqui, A.; Pereira, A.; Simoes, N. Purification and characterization of two distinct metalloproteases secreted by the entomopathogenic bacterium Photorhabdus sp. strain Az29. Appl. Environ. Microbiol 2004, 70, 3831–3838. [Google Scholar]
- Waterfield, N.R.; Ciche, T.; Clark, D. Photorhabdus and a host of hosts. Annu. Rev. Microbiol 2009, 63, 557–574. [Google Scholar]
- Ffrench-Constant, R.H.; Waterfield, N.; Daborn, P.; Joyce, S.; Bennett, H.; Au, C.; Dowling, A.; Boundy, S.; Reynolds, S.; Clarke, D. Photorhabdus: Towards a functional genomic analysis of a symbiont and pathogen. FEMS Microbiol. Rev 2003, 26, 433–456. [Google Scholar]
- Bowen, D.; Blackburn, M.; Rocheleau, T.; Grutzmacher, C.; Ffrench-Constant, R.H. Secreted proteases from Photorhabdus luminescens: Separation of the extracellular proteases from the insecticidal Tc toxin complexes. Insect Biochem. Mol. Biol 2000, 30, 69–74. [Google Scholar]
- Bowen, D.J.; Rocheleau, T.A.; Grutzmacher, C.K.; Meslet, L.; Valens, M.; Marble, D.; Dowling, A.; Ffrench-Constant, R.H.; Blight, M.A. Genetic and biochemical characterization of PrtA, an RTX-like metalloprotease from Photorhabdus. Microbiology 2003, 149, 1581–1591. [Google Scholar]
- Harrison, R.L.; Bonning, B.C. Proteases as insecticidal agents. Toxins 2010, 2, 935–953. [Google Scholar]
- Miyoshi, S.; Shinoda, S. Microbial metalloproteases and pathogenesis. Microbes Infect 2000, 2, 91–98. [Google Scholar]
- Yamanaka, S.; Hagiwara, A.; Nishimura, Y.; Tanabe, H.; Ishibashi, N. Biochemical and physiological characteristics of Xenorhabdus species, symbiotically associated with entomopathogenic nematodes including Steinernema kushidai and their pathogenicity against Spodoptera litura (Lepidoptera: Noctuidae). Arch. Microbiol 1992, 158, 387–393. [Google Scholar]
- Daborn, P.J.; Waterfield, N.; Blight, M.A.; Ffrench-Constant, R.H. Measuring virulence factor expression by the pathogenic bacterium Photorhabdus luminescens in culture and during insect infection. J. Bacteriol 2001, 183, 5834–5839. [Google Scholar]
- Hsieh, F.C.; Tzeng, C.Y.; Tseng, J.T.; Tsai, Y.S.; Meng, M.; Kao, S.S. Isolation and characterization of the native entomopathogenic nematode, Heterorhabditis brevicaudis, and its symbiotic bacteria from Taiwan. Curr. Microbiol 2009, 58, 564–570. [Google Scholar]
- Schmidt, T.M.; Bleakley, B.; Nealson, K.H. Characterization of an extracellular protease from the insect pathogen Xenorhabdus luminescens. Appl. Environ. Microbiol 1988, 54, 2793–2797. [Google Scholar]
- Shelton, A.M.; Kroening, M.K.; Wilsey, W.T.; Eigenbrode, S.D. Comparative analysis of two rearing procedures for diamondback moth, Plutella xylostella (Lepidoptera: Plutellidae). J. Entomol. Sci 1991, 26, 17–26. [Google Scholar]
- Finney, D.J. Probit Analysis: A Statistical Treatment of the Sigmoid Response Curve, 2nd ed; Cambridge University Press: New York, NY, USA, 1952; pp. 236–245. [Google Scholar]
- Felfoldi, G.; Marokhazi, J.; Kepiro, M.; Venekei, I. Identification of natural target proteins indicates functions of a serralysin-type metalloprotease, PrtA, in anti-immune mechanisms. Appl. Environ. Microbiol 2009, 75, 3120–3126. [Google Scholar]
- Gomis-Ruth, F.X. Structural aspects of the metzincin clan of metalloendopeptidases. Mol. Biotechnol 2003, 24, 157–202. [Google Scholar]
- Massaoud, M.K.; Marokhazi, J.; Fodor, A.; Venekei, I. Proteolytic enzyme production by strains of the insect pathogen Xenorhabdus and characterization of an early-log-phase-secreted protease as a potential virulence factor. Appl. Environ. Microbiol 2010, 76, 6901–6909. [Google Scholar]
- Massaoud, M.K.; Marokhazi, J.; Venekei, I. Enzymatic characterization of a serralysin-like metalloprotease from the entomopathogen bacterium, Xenorhabdus. Biochim. Biophys. Acta 2011, 1814, 1333–1339. [Google Scholar]
- Silva, C.P.; Waterfield, N.R.; Daborn, P.J.; Dean, P.; Chilver, T.; Au, C.P.Y.; Sharma, S.; Potter, U.; Reynolds, S.E.; Ffrench-Constant, R.H. Bacterial infection of a model insect: Photorhabdus luminescens and Manduca sexta. Cell. Microbiol 2002, 4, 329–339. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration (ng/μL) | Total insects treated1 | Number of death | Average mortality rate2 (%) |
|---|---|---|---|
| 11.6 | 90 | 90 | 100 |
| 9.5 | 90 | 76 | 84 |
| 7.3 | 90 | 70 | 78 |
| 6.5 | 90 | 65 | 72 |
| 5.8 | 90 | 59 | 66 |
| 4.8 | 90 | 53 | 59 |
| 3.7 | 90 | 43 | 48 |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chang, Y.-T.; Hsieh, C.; Wu, L.-C.; Chang, H.C.; Kao, S.-S.; Meng, M.; Hsieh, F.-C. Purification and Properties of an Insecticidal Metalloprotease Produced by Photorhabdus luminescens Strain 0805-P5G, the Entomopathogenic Nematode Symbiont. Int. J. Mol. Sci. 2013, 14, 308-321. https://doi.org/10.3390/ijms14010308
Chang Y-T, Hsieh C, Wu L-C, Chang HC, Kao S-S, Meng M, Hsieh F-C. Purification and Properties of an Insecticidal Metalloprotease Produced by Photorhabdus luminescens Strain 0805-P5G, the Entomopathogenic Nematode Symbiont. International Journal of Molecular Sciences. 2013; 14(1):308-321. https://doi.org/10.3390/ijms14010308
Chicago/Turabian StyleChang, Yu-Tzu, Chienyan Hsieh, Li-Ching Wu, Hebron C. Chang, Suey-Sheng Kao, Menghsiao Meng, and Feng-Chia Hsieh. 2013. "Purification and Properties of an Insecticidal Metalloprotease Produced by Photorhabdus luminescens Strain 0805-P5G, the Entomopathogenic Nematode Symbiont" International Journal of Molecular Sciences 14, no. 1: 308-321. https://doi.org/10.3390/ijms14010308
APA StyleChang, Y.-T., Hsieh, C., Wu, L.-C., Chang, H. C., Kao, S.-S., Meng, M., & Hsieh, F.-C. (2013). Purification and Properties of an Insecticidal Metalloprotease Produced by Photorhabdus luminescens Strain 0805-P5G, the Entomopathogenic Nematode Symbiont. International Journal of Molecular Sciences, 14(1), 308-321. https://doi.org/10.3390/ijms14010308