Twist and miR-34a Are Involved in the Generation of Tumor-Educated Myeloid-Derived Suppressor Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
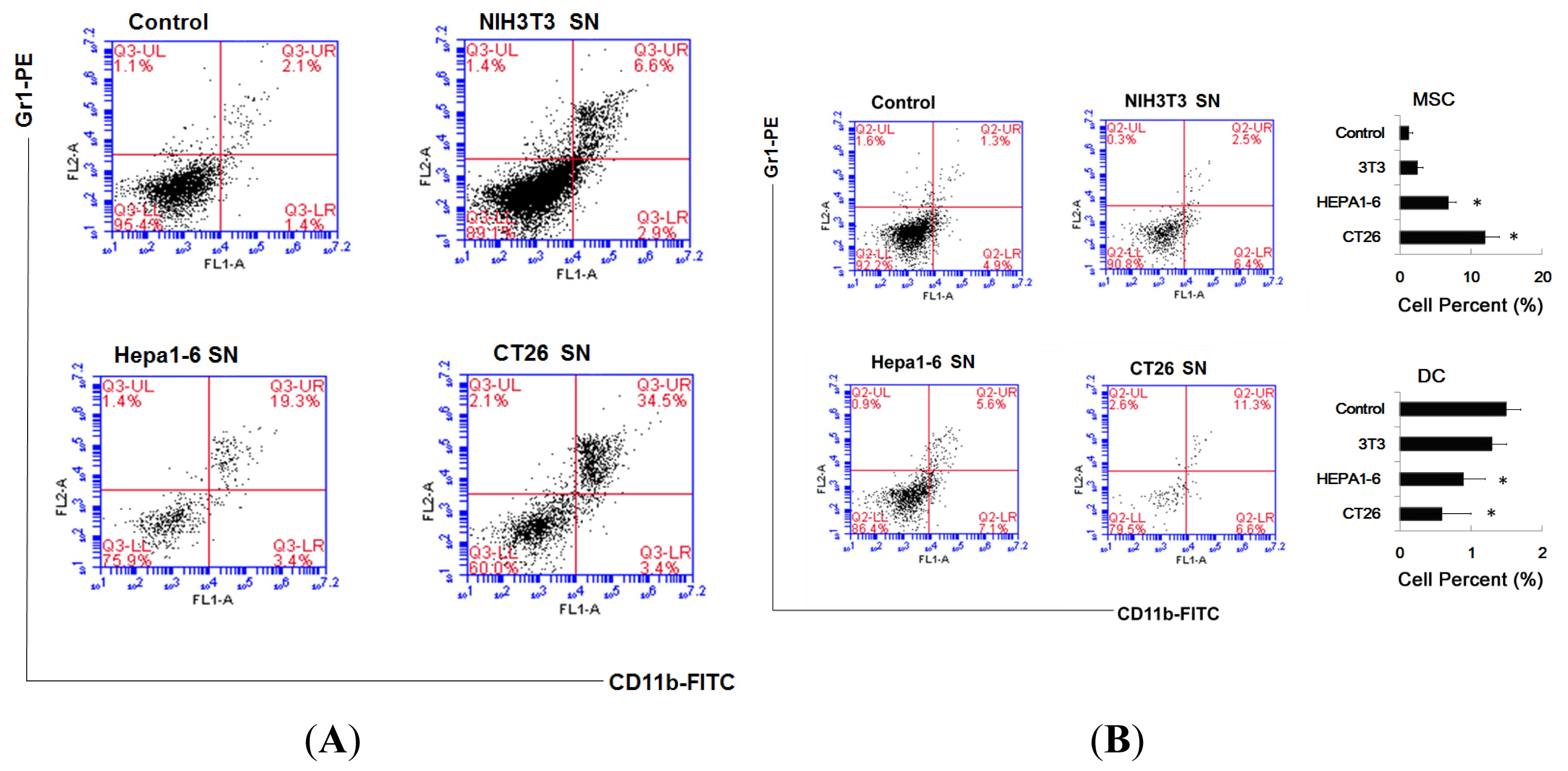
2.1. Tumor Culture Supernatants Disturb the Development of the Bone Marrow-Derived Dendritic Cell
2.2. Supernatants of Tumor Cells Inhibit the DC Differentiation from Myeloid Precursor Cells, and Promote MDSC Accumulation
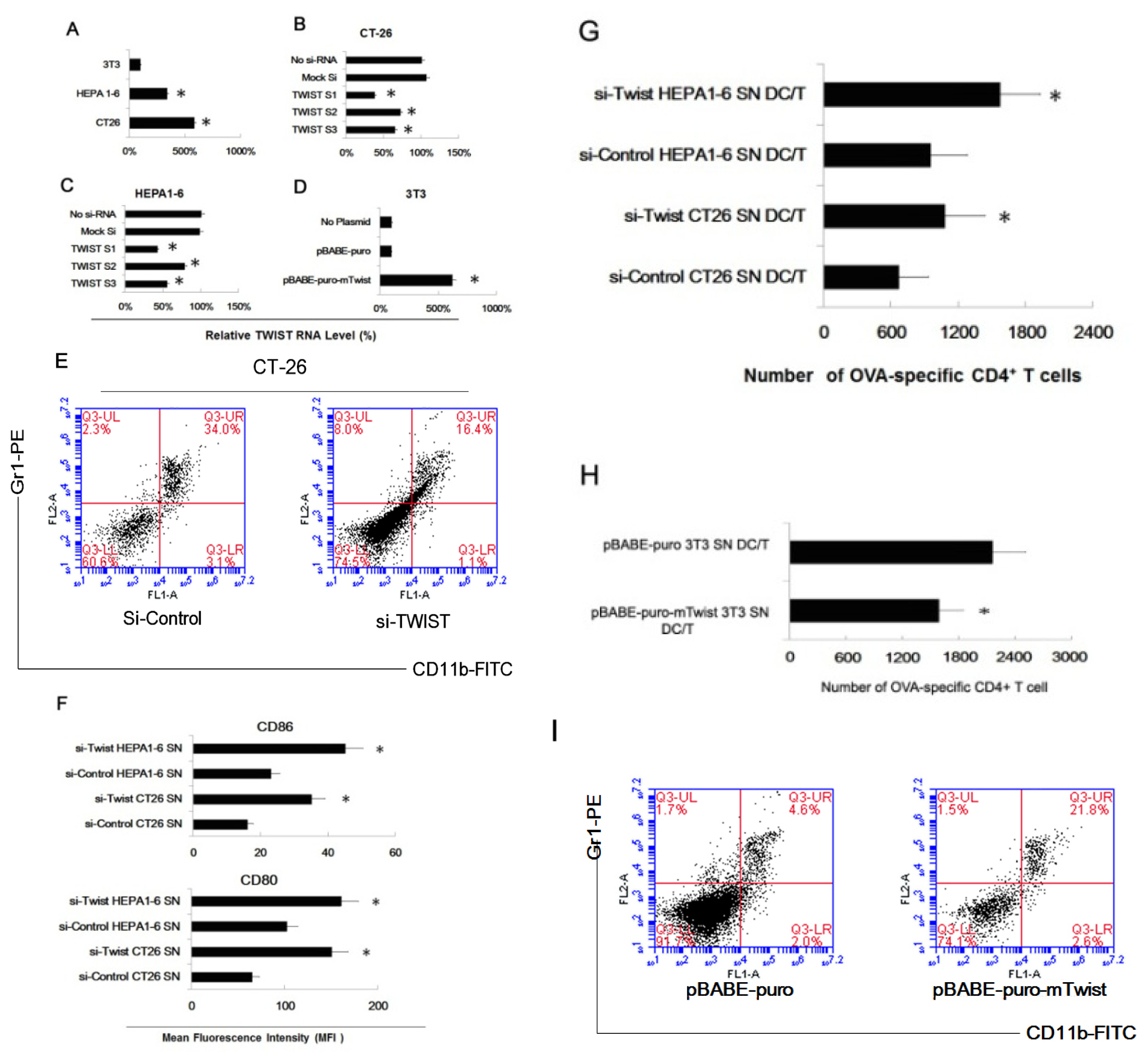
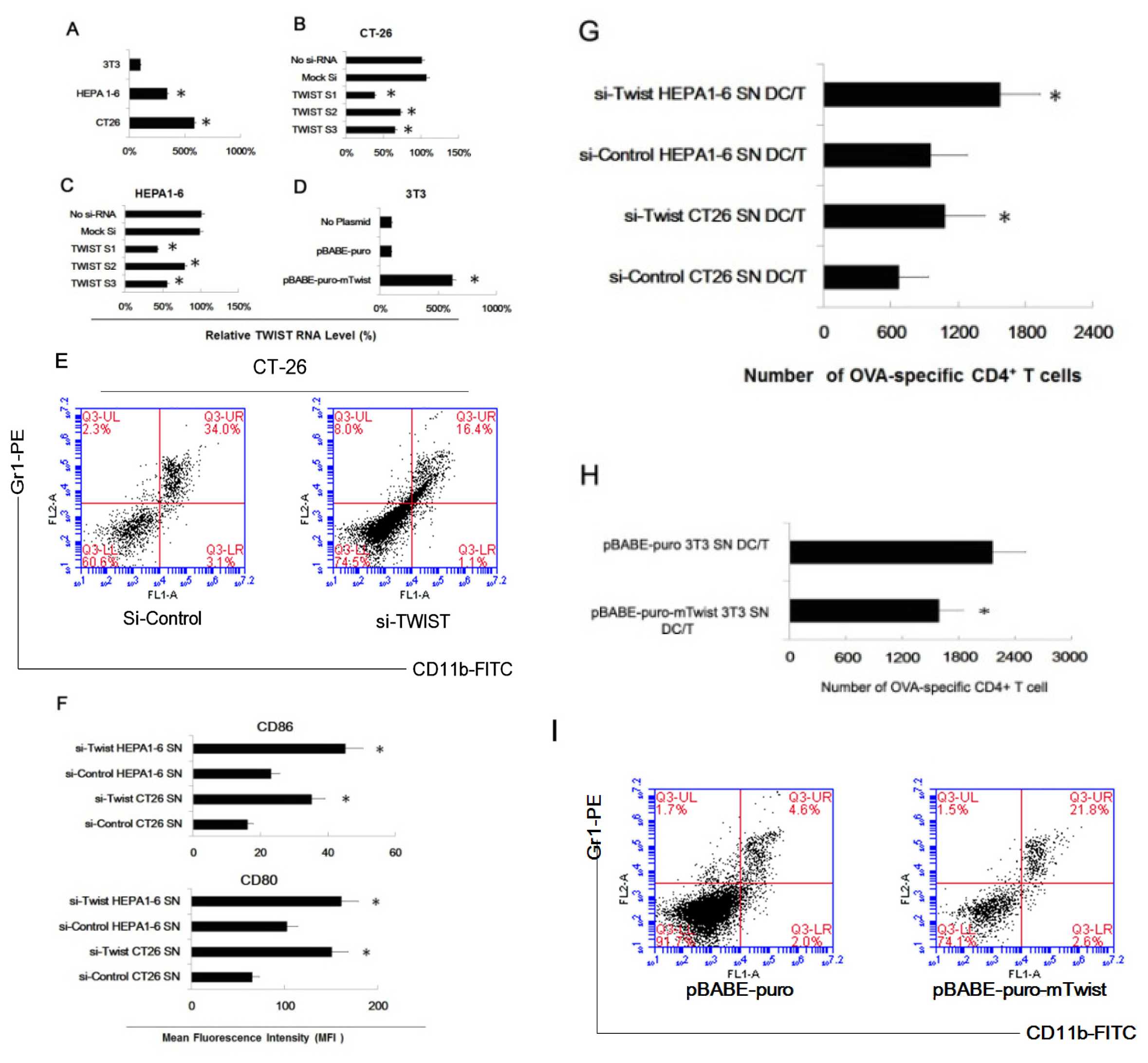
2.3. Twist Regulated the Effect of Tumor Cells of Inducing Myeloid Precursor Cells into MDSC
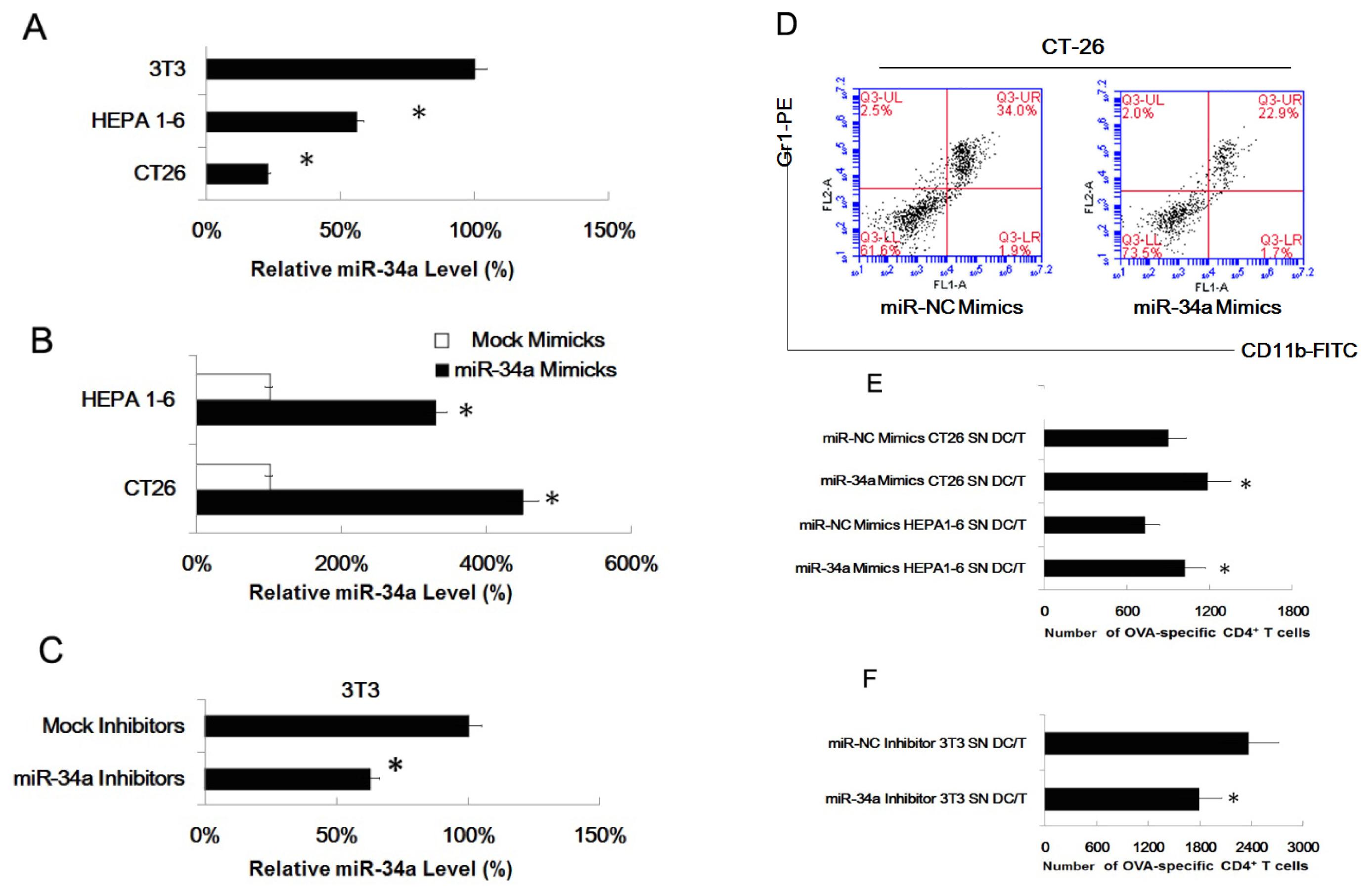
2.4. MiR-34a Regulated the Effect of Tumor Cells of Inducing Myeloid Precursor Cells into MDSC
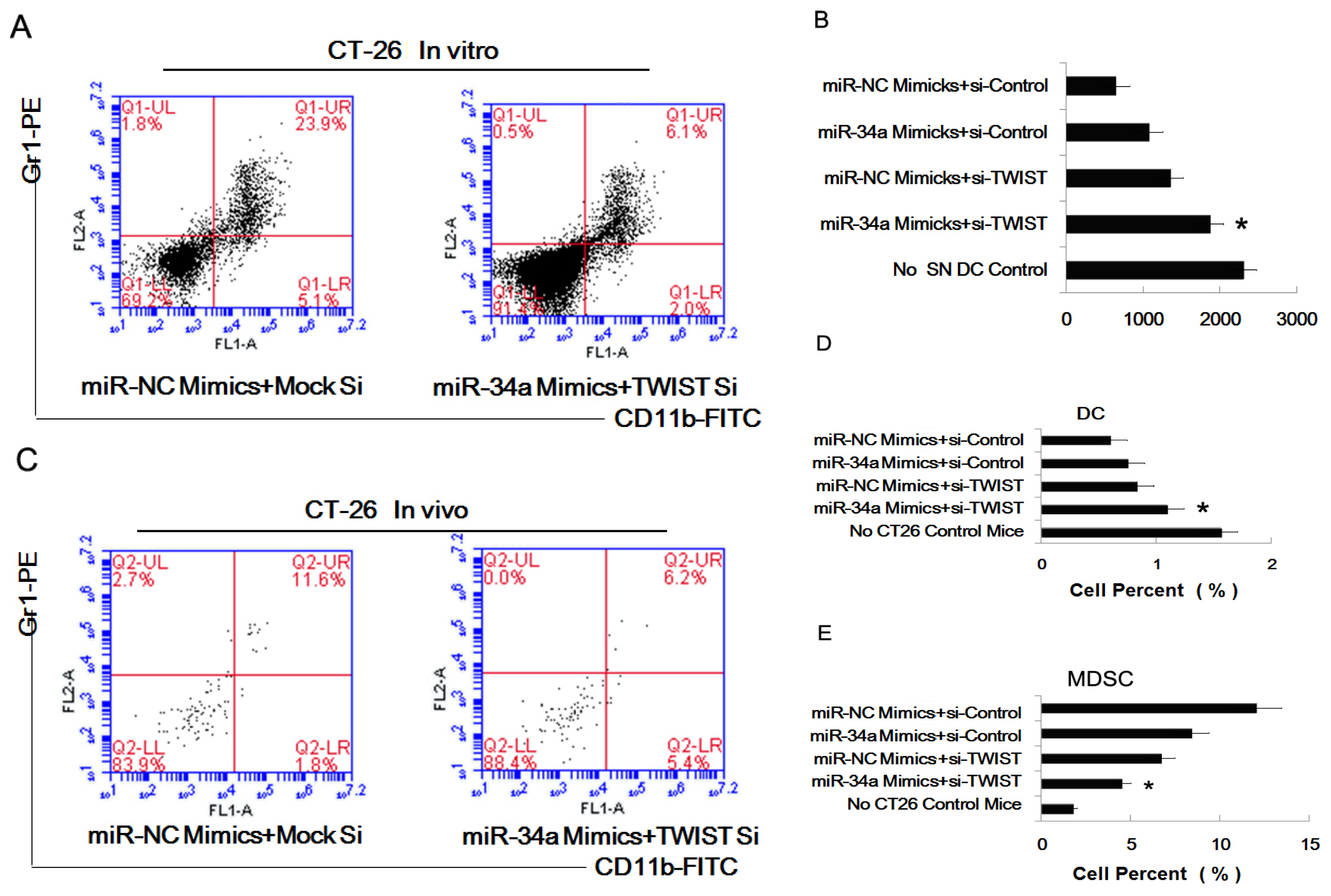
2.5. The Combined Effect of Twist and miR-34a on Inducing MDSC
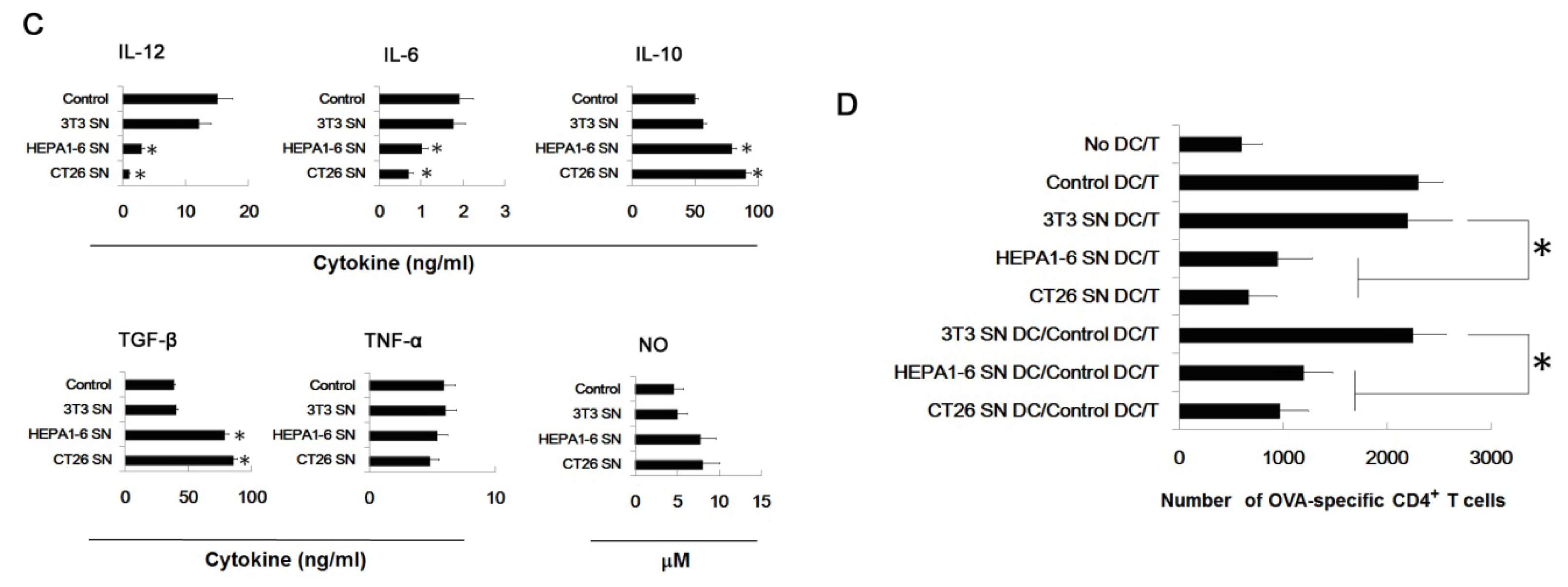
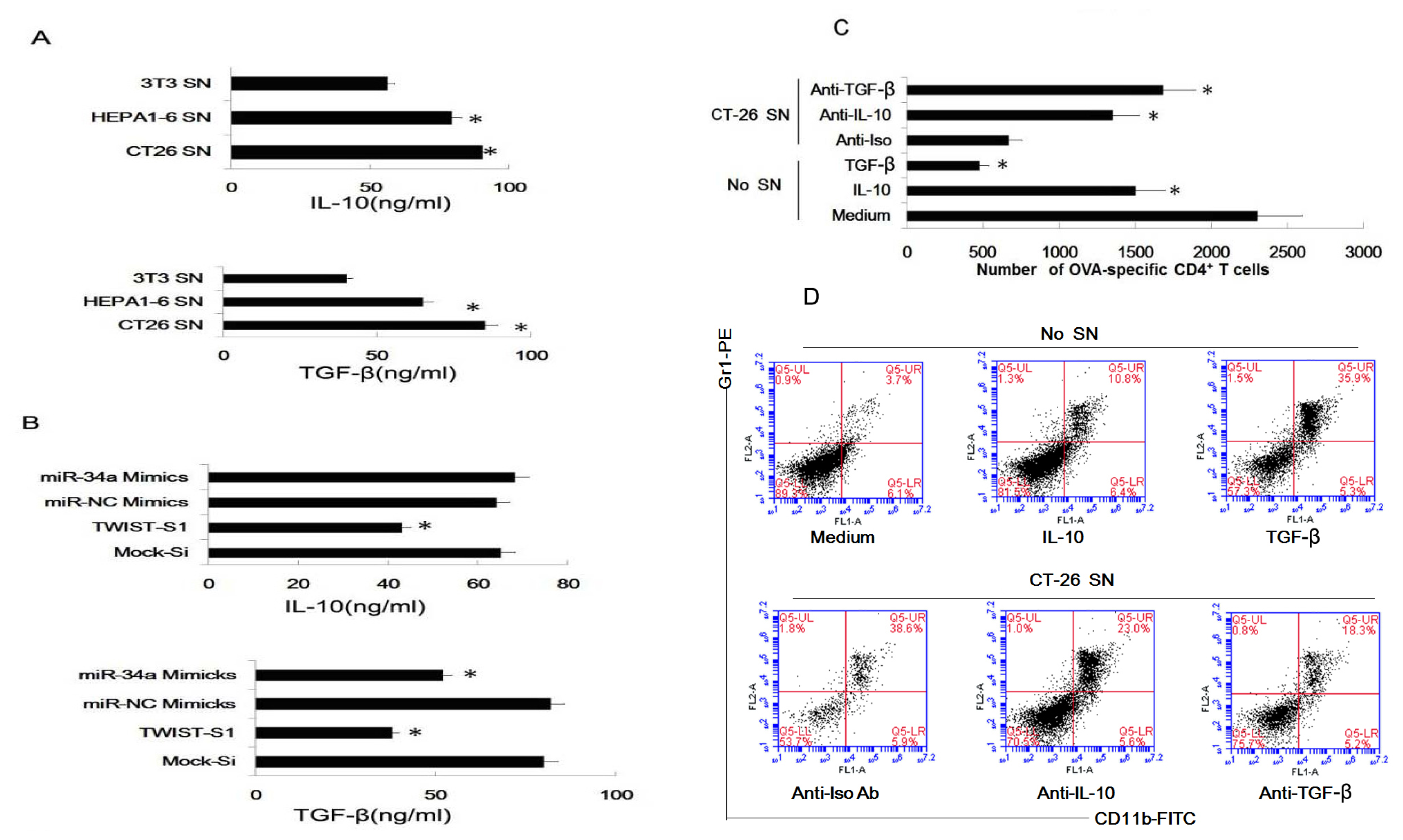
2.6. Tumor-Derived IL-10 and TGF-β Are Responsible for the Differentiation of MDSC
2.7. Discussion
3. Experimental Section
3.1. Mice and Treatment
3.2. Reagents
3.3. Preparation of Tumor Supernatants Treated Myeloid Precursor Cells
3.4. Flow Cytometry
3.5. Tumor Model, Preparation of DC and Spleen Cells
3.6. Assay for Cytokines and NO
3.7. Assays for Ag-Specific CD4+ T Cell Response
3.8. Assay for Percent of MDSC and DC In Vivo
3.9. Real-Time RT-PCR
3.10. Twist Plasmids and siRNA Transfection
3.11. Transfection of miR-34 Mimics and Combined Transfection
3.12. Statistical Analysis
4. Conclusions






Acknowledgments
Conflicts of Interest
References
- Kim, R.; Emi, M.; Tanabe, K.; Arihiro, K. Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res 2006, 66, 5527–5536. [Google Scholar]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol 2007, 25, 267–296. [Google Scholar]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar]
- Wang, Y.; Ma, Y.; Fang, Y.; Wu, S.; Liu, L.; Fu, D.; Shen, X. Regulatory T cell: A protection for tumour cells. J. Cell Mol. Med 2012, 16, 425–436. [Google Scholar]
- Shurin, M.R.; Shurin, G.V.; Lokshin, A.; Yurkovetsky, Z.R.; Gutkin, D.W.; Chatta, G.; Zhong, H.; Han, B.; Ferris, R.L. Intratumoral cytokines/chemokines/growth factors and tumor infiltrating dendritic cells: Friends or enemies? Cancer Metastasis Rev 2006, 25, 333–356. [Google Scholar]
- Kusmartsev, S.; Gabrilovich, D.I. Effect of tumor-derived cytokines and growth factors on differentiation and immune suppressive features of myeloid cells in cancer. Cancer Metastasis Rev 2006, 25, 323–331. [Google Scholar]
- Lin, W.W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Invest 2007, 117, 1175–1183. [Google Scholar]
- Liu, Q.; Zhang, C.; Sun, A.; Zheng, Y.; Wang, L.; Cao, X. Tumor-educated CD11bhighIalow regulatory dendritic cells suppress T cell response through arginase I. J. Immunol 2009, 182, 6207–6216. [Google Scholar]
- Liu, C.; Zhang, C.; Lu, H.; Cai, J.; Wang, Z.; Chen, J.; Liu, F.; Wu, Z.; Liu, X.; Sun, W. Poly(I:C) induce bone marrow precursor cells into myeloid-derived suppressor cells. Mol. Cell. Biochem 2011, 358, 317–323. [Google Scholar]
- Greifenberg, V.; Ribechini, E.; Rossner, S.; Lutz, M.B. Myeloid-derived suppressor cell activation by combined LPS and IFN-gamma treatment impairs DC development. Eur. J. Immunol 2009, 39, 2865–2876. [Google Scholar]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol 2009, 9, 162–174. [Google Scholar]
- Bronte, V.; Zanovello, P. Regulation of immune responses by l-arginine metabolism. Nat. Rev. Immunol 2005, 5, 641–654. [Google Scholar]
- Rodriguez, P.C.; Ochoa, A.C. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: Mechanisms and therapeutic perspectives. Immunol. Rev 2008, 222, 180–191. [Google Scholar]
- Youn, J.I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol 2008, 181, 5791–5802. [Google Scholar]
- Kusmartsev, S.; Nefedova, Y.; Yoder, D.; Gabrilovich, D.I. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol 2004, 172, 989–999. [Google Scholar]
- Schmielau, J.; Finn, O.J. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of T-cell function in advanced cancer patients. Cancer Res 2001, 61, 4756–4760. [Google Scholar]
- Kusmartsev, S.; Nagaraj, S.; Gabrilovich, D.I. Tumor-associated CD8+ T cell tolerance induced by bone marrow-derived immature myeloid cells. J. Immunol 2005, 175, 4583–4592. [Google Scholar]
- Szuster-Ciesielska, A.; Hryciuk-Umer, E.; Stepulak, A.; Kupisz, K.; Kandefer-Szerszen, M. Reactive oxygen species production by blood neutrophils of patients with laryngeal carcinoma and antioxidative enzyme activity in their blood. Acta Oncol 2004, 43, 252–258. [Google Scholar]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog 2006, 5, 14. [Google Scholar]
- Mantovani, G.; Maccio, A.; Madeddu, C.; Mura, L.; Gramignano, G.; Lusso, M.R.; Massa, E.; Mocci, M.; Serpe, R. Antioxidant agents are effective in inducing lymphocyte progression through cell cycle in advanced cancer patients: Assessment of the most important laboratory indexes of cachexia and oxidative stress. J. Mol. Med 2003, 81, 664–673. [Google Scholar]
- Agostinelli, E.; Seiler, N. Non-irradiation-derived reactive oxygen species (ROS) and cancer: Therapeutic implications. Amino Acids 2006, 31, 341–355. [Google Scholar]
- Yang, R.; Cai, Z.; Zhang, Y.; Yutzy, W.H.T.; Roby, K.F.; Roden, R.B. CD80 in immune suppression by mouse ovarian carcinoma-associated Gr-1+CD11b+ myeloid cells. Cancer Res 2006, 66, 6807–6815. [Google Scholar]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res 2006, 66, 1123–1131. [Google Scholar]
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol 1991, 9, 271–296. [Google Scholar]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar]
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. [Google Scholar]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar]
- Lee, T.K.; Poon, R.T.; Yuen, A.P.; Ling, M.T.; Kwok, W.K.; Wang, X.H.; Wong, Y.C.; Guan, X.Y.; Man, K.; Chau, K.L.; et al. Twist overexpression correlates with hepatocellular carcinoma metastasis through induction of epithelial-mesenchymal transition. Clin. Cancer Res 2006, 12, 5369–5376. [Google Scholar]
- Yang, M.H.; Chen, C.L.; Chau, G.Y.; Chiou, S.H.; Su, C.W.; Chou, T.Y.; Peng, W.L.; Wu, J.C. Comprehensive analysis of the independent effect of twist and snail in promoting metastasis of hepatocellular carcinoma. Hepatology 2009, 50, 1464–1474. [Google Scholar]
- Kwok, W.K.; Ling, M.T.; Lee, T.W.; Lau, T.C.; Zhou, C.; Zhang, X.; Chua, C.W.; Chan, K.W.; Chan, F.L.; Glackin, C.; et al. Up-regulation of TWIST in prostate cancer and its implication as a therapeutic target. Cancer Res 2005, 65, 5153–5162. [Google Scholar]
- Yuen, H.F.; Chua, C.W.; Chan, Y.P.; Wong, Y.C.; Wang, X.; Chan, K.W. Significance of TWIST and E-cadherin expression in the metastatic progression of prostatic cancer. Histopathology 2007, 50, 648–658. [Google Scholar]
- Yuen, H.F.; Chan, Y.P.; Wong, M.L.; Kwok, W.K.; Chan, K.K.; Lee, P.Y.; Srivastava, G.; Law, S.Y.; Wong, Y.C.; Wang, X.; et al. Upregulation of Twist in oesophageal squamous cell carcinoma is associated with neoplastic transformation and distant metastasis. J. Clin. Pathol 2007, 60, 510–514. [Google Scholar]
- Sasaki, K.; Natsugoe, S.; Ishigami, S.; Matsumoto, M.; Okumura, H.; Setoyama, T.; Uchikado, Y.; Kita, Y.; Tamotsu, K.; Sakamoto, A.; et al. Significance of Twist expression and its association with E-cadherin in esophageal squamous cell carcinoma. J. Exp. Clin. Cancer Res 2009, 28, 158. [Google Scholar]
- Zhang, Z.; Xie, D.; Li, X.; Wong, Y.C.; Xin, D.; Guan, X.Y.; Chua, C.W.; Leung, S.C.; Na, Y.; Wang, X. Significance of TWIST expression and its association with E-cadherin in bladder cancer. Hum. Pathol 2007, 38, 598–606. [Google Scholar]
- Wallerand, H.; Robert, G.; Pasticier, G.; Ravaud, A.; Ballanger, P.; Reiter, R.E.; Ferriere, J.M. The epithelial-mesenchymal transition-inducing factor TWIST is an attractive target in advanced and/or metastatic bladder and prostate cancers. Urol. Oncol 2010, 28, 473–479. [Google Scholar]
- Satoh, K.; Hamada, S.; Kimura, K.; Kanno, A.; Hirota, M.; Umino, J.; Fujibuchi, W.; Masamune, A.; Tanaka, N.; Miura, K.; et al. Up-regulation of MSX2 enhances the malignant phenotype and is associated with twist 1 expression in human pancreatic cancer cells. Am. J. Pathol 2008, 172, 926–939. [Google Scholar]
- Qin, Q.; Xu, Y.; He, T.; Qin, C.; Xu, J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res 2012, 22, 90–106. [Google Scholar]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar]
- Chang, T.C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 2007, 26, 745–752. [Google Scholar]
- Bommer, G.T.; Gerin, I.; Feng, Y.; Kaczorowski, A.J.; Kuick, R.; Love, R.E.; Zhai, Y.; Giordano, T.J.; Qin, Z.S.; Moore, B.B.; et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol 2007, 17, 1298–1307. [Google Scholar]
- He, X.; He, L.; Hannon, G.J. The guardian’s little helper: MicroRNAs in the p53 tumor suppressor network. Cancer Res 2007, 67, 11099–11101. [Google Scholar]
- Zhang, M.; Tang, H.; Guo, Z.; An, H.; Zhu, X.; Song, W.; Guo, J.; Huang, X.; Chen, T.; Wang, J.; et al. Splenic stroma drives mature dendritic cells to differentiate into regulatory dendritic cells. Nat. Immunol 2004, 5, 1124–1133. [Google Scholar]
- Tang, H.; Guo, Z.; Zhang, M.; Wang, J.; Chen, G.; Cao, X. Endothelial stroma programs hematopoietic stem cells to differentiate into regulatory dendritic cells through IL-10. Blood 2006, 108, 1189–1197. [Google Scholar]
- Xia, S.; Guo, Z.; Xu, X.; Yi, H.; Wang, Q.; Cao, X. Hepatic microenvironment programs hematopoietic progenitor differentiation into regulatory dendritic cells, maintaining liver tolerance. Blood 2008, 112, 3175–3185. [Google Scholar]
- Ferrone, S.; Whiteside, T.L. Tumor microenvironment and immune escape. Surg. Oncol. Clin. N. Am 2007, 16, 755–774. [Google Scholar]
- Fricke, I.; Gabrilovich, D.I. Dendritic cells and tumor microenvironment: A dangerous liaison. Immunol. Invest 2006, 35, 459–483. [Google Scholar]
- Drake, C.G.; Jaffee, E.; Pardoll, D.M. Mechanisms of immune evasion by tumors. Adv. Immunol 2006, 90, 51–81. [Google Scholar]
- Hurwitz, A.A.; Watkins, S.K. Immune suppression in the tumor microenvironment: A role for dendritic cell-mediated tolerization of T cells. Cancer Immunol. Immunother 2012, 61, 289–293. [Google Scholar]
- Dhodapkar, M.V.; Dhodapkar, K.M.; Palucka, A.K. Interactions of tumor cells with dendritic cells: Balancing immunity and tolerance. Cell Death Differ 2008, 15, 39–50. [Google Scholar]
- Maestro, R.; Dei Tos, A.P.; Hamamori, Y.; Krasnokutsky, S.; Sartorelli, V.; Kedes, L.; Doglioni, C.; Beach, D.H.; Hannon, G.J. Twist is a potential oncogene that inhibits apoptosis. Genes Dev 1999, 13, 2207–2217. [Google Scholar]
- Valsesia-Wittmann, S.; Magdeleine, M.; Dupasquier, S.; Garin, E.; Jallas, A.C.; Combaret, V.; Krause, A.; Leissner, P.; Puisieux, A. Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells. Cancer Cell 2004, 6, 625–630. [Google Scholar]
- Ansieau, S.; Bastid, J.; Doreau, A.; Morel, A.P.; Bouchet, B.P.; Thomas, C.; Fauvet, F.; Puisieux, I.; Doglioni, C.; Piccinin, S.; et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 2008, 14, 79–89. [Google Scholar]
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.H. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res 2007, 67, 1979–1987. [Google Scholar]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar]
- Vesuna, F.; Lisok, A.; Kimble, B.; Raman, V. Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia 2009, 11, 1318–1328. [Google Scholar]
- Battula, V.L.; Evans, K.W.; Hollier, B.G.; Shi, Y.; Marini, F.C.; Ayyanan, A.; Wang, R.Y.; Brisken, C.; Guerra, R.; Andreeff, M.; et al. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells 2010, 28, 1435–1445. [Google Scholar]
- Fu, J.; Qin, L.; He, T.; Qin, J.; Hong, J.; Wong, J.; Liao, L.; Xu, J. The Twist/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res 2011, 21, 275–289. [Google Scholar]
- Qin, L.; Liu, Z.; Chen, H.; Xu, J. The steroid receptor coactivator-1 regulates twist expression and promotes breast cancer metastasis. Cancer Res 2009, 69, 3819–3827. [Google Scholar]
- Vernon, A.E.; LaBonne, C. Tumor metastasis: A new twist on epithelial-mesenchymal transitions. Curr. Biol 2004, 14, R719–R721. [Google Scholar]
- Karreth, F.; Tuveson, D.A. Twist induces an epithelial-mesenchymal transition to facilitate tumor metastasis. Cancer Biol. Ther 2004, 3, 1058–1059. [Google Scholar]
- Yang, J.; Mani, S.A.; Weinberg, R.A. Exploring a new twist on tumor metastasis. Cancer Res 2006, 66, 4549–4552. [Google Scholar]
- Pan, P.Y.; Wang, G.X.; Yin, B.; Ozao, J.; Ku, T.; Divino, C.M.; Chen, S.H. Reversion of immune tolerance in advanced malignancy: Modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood 2008, 111, 219–228. [Google Scholar]
- Sinha, P.; Clements, V.K.; Fulton, A.M.; Ostrand-Rosenberg, S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res 2007, 67, 4507–4513. [Google Scholar]
- Serafini, P.; Carbley, R.; Noonan, K.A.; Tan, G.; Bronte, V.; Borrello, I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res 2004, 64, 6337–6343. [Google Scholar]
- Bunt, S.K.; Yang, L.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res 2007, 67, 10019–10026. [Google Scholar]
- Gabrilovich, D.; Ishida, T.; Oyama, T.; Ran, S.; Kravtsov, V.; Nadaf, S.; Carbone, D.P. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood 1998, 92, 4150–4166. [Google Scholar]
- Kusmartsev, S.; Gabrilovich, D.I. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J. Immunol 2005, 174, 4880–4891. [Google Scholar]
- Movahedi, K.; Guilliams, M.; van den Bossche, J.; van den Bergh, R.; Gysemans, C.; Beschin, A.; de Baetselier, P.; van Ginderachter, J.A. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111, 4233–4244. [Google Scholar]
- Terabe, M.; Matsui, S.; Park, J.M.; Mamura, M.; Noben-Trauth, N.; Donaldson, D.D.; Chen, W.; Wahl, S.M.; Ledbetter, S.; Pratt, B.; et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: Abrogation prevents tumor recurrence. J. Exp. Med 2003, 198, 1741–1752. [Google Scholar]
- Ji, Q.; Hao, X.; Meng, Y.; Zhang, M.; Desano, J.; Fan, D.; Xu, L. Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumorspheres. BMC Cancer 2008, 8, 266. [Google Scholar]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 2005, 33, e179. [Google Scholar]
- Ji, Q.; Hao, X.; Zhang, M.; Tang, W.; Yang, M.; Li, L.; Xiang, D.; Desano, J.T.; Bommer, G.T.; Fan, D.; et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One 2009, 4, e6816. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, X.; Chang, X.; Zhuo, G.; Sun, M.; Yin, K. Twist and miR-34a Are Involved in the Generation of Tumor-Educated Myeloid-Derived Suppressor Cells. Int. J. Mol. Sci. 2013, 14, 20459-20477. https://doi.org/10.3390/ijms141020459
Wang X, Chang X, Zhuo G, Sun M, Yin K. Twist and miR-34a Are Involved in the Generation of Tumor-Educated Myeloid-Derived Suppressor Cells. International Journal of Molecular Sciences. 2013; 14(10):20459-20477. https://doi.org/10.3390/ijms141020459
Chicago/Turabian StyleWang, Xin, Xusheng Chang, Guangzuan Zhuo, Mingjuan Sun, and Kai Yin. 2013. "Twist and miR-34a Are Involved in the Generation of Tumor-Educated Myeloid-Derived Suppressor Cells" International Journal of Molecular Sciences 14, no. 10: 20459-20477. https://doi.org/10.3390/ijms141020459