Rodent Models of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis

 ,
,  ,
, 
Abstract
:1. Introduction
2. Identification of Articles on Rodent Models of NAFLD/NASH
3. Classification of Rodent Models of NAFLD/NASH
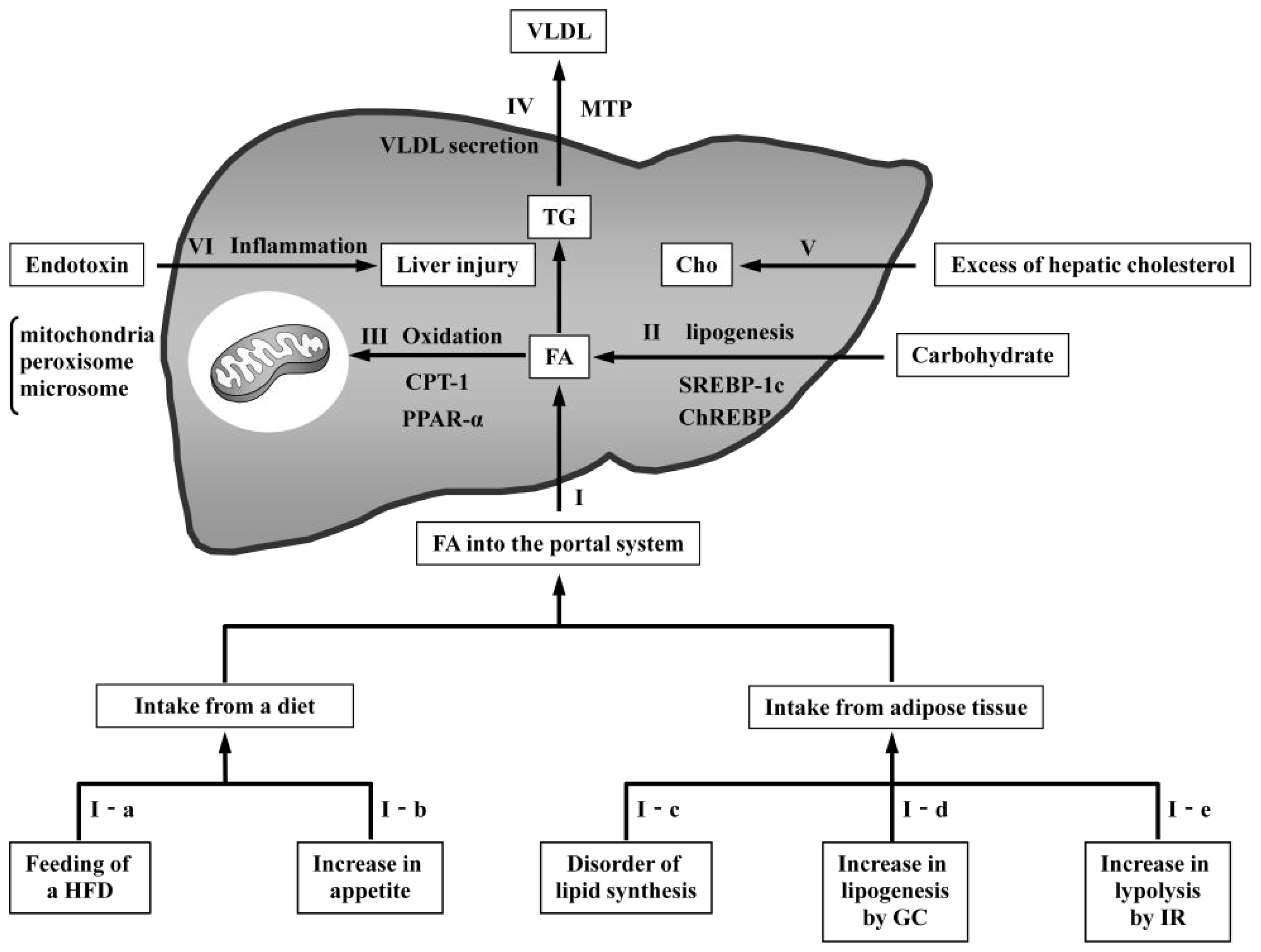
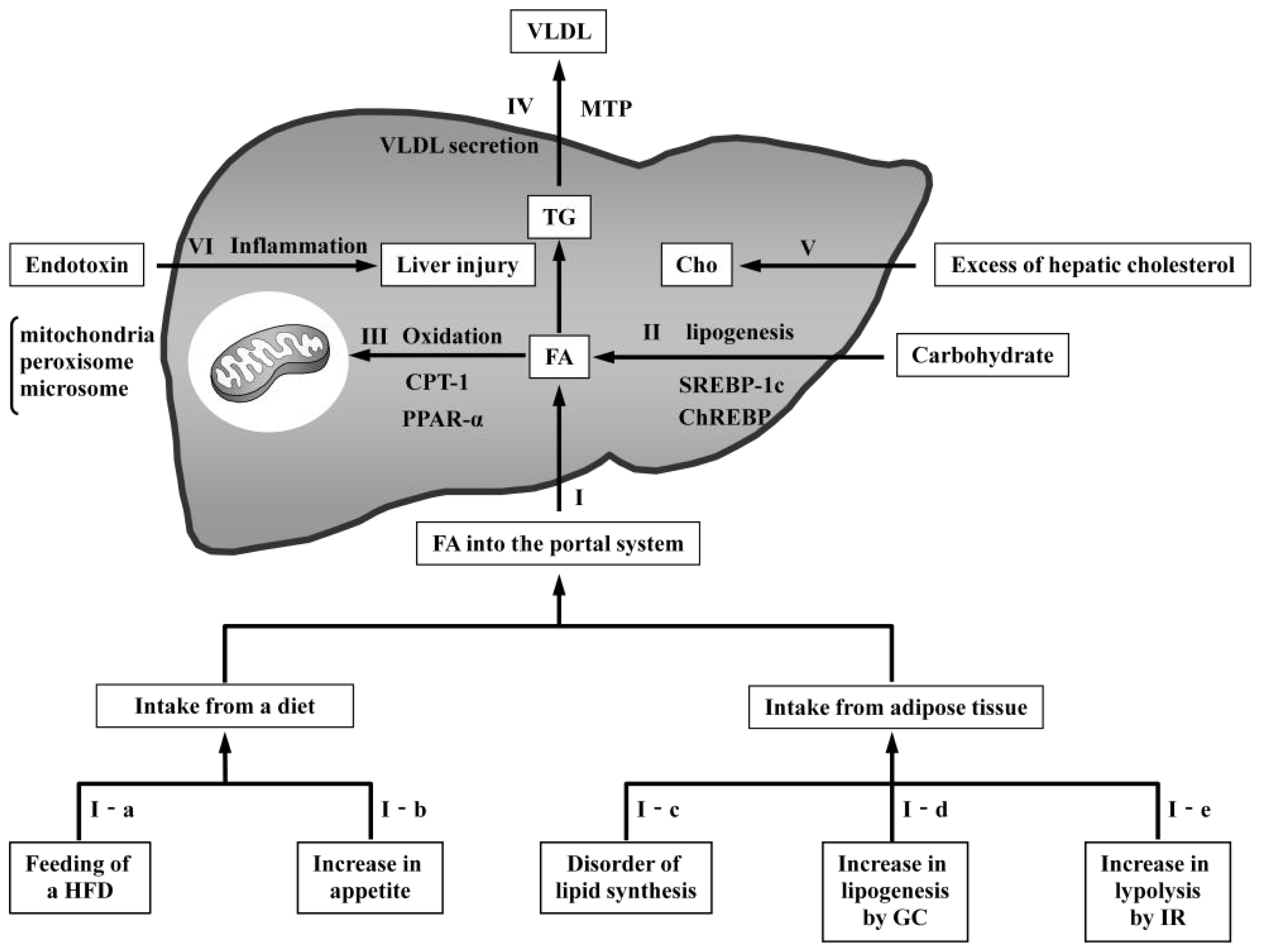
3.1. Models with Increased FA Delivery to the Liver
3.1.1. Models Involving Feeding of a High-Fat Diet
3.1.1.1. Mice with Chronic Exposure to a HFD
3.1.1.2. Mice with Intragastric Overfeeding of a HFD
3.1.1.3. Mice Fed a Combination of Fructose and a HFD
3.1.2. Models with Increased Appetite
3.1.2.1. Leptin-Deficient Mice (ob/ob Mice)
3.1.2.2. Leptin-Resistant Mice (db/db Mice)
3.1.2.3. Zucker Fatty (fa/fa) Rats
3.1.2.4. Melanocortin 4 Receptor (MC4R)-Deficient Mice
3.1.2.5. Other Genetic Models with Increased Appetite
3.1.3. Models with Disordered Lipid Synthesis in Adipose Tissue
3.1.3.1. aP2-nSREBP-1c-Overexpressing Mice
3.1.3.2. A-ZIP/F-1-Overexpressing Mice
3.1.3.3. CD36-Deficient Mice
3.1.3.4. Other Models with Increased FA from Peripheral Adipose Tissue
3.1.4. Models with Increased Glucocorticoids
3.1.4.1. 11β-HSD1-Overexpressing Mice and 7B2-Deficient Mice
3.1.5. Models with Increased Insulin Resistance
3.1.5.1. Insulin I-IGF-II-Overexpressing Mice
3.1.5.2. STAT5B-Deficient Mice
3.2. Models with Increased De Novo Lipogenesis
3.2.1. Hepatocyte-Specific PTEN-Deficient Mice
3.2.2. IDPc-Overexpressing Mice
3.2.3. ChREBP-Deficient Mice
3.2.4. Other Models of De Novo Lipogenesis
3.3. Models with Reduced Oxidation
3.3.1. Hepatocyte-Specific RARα Dominant-Negative Overexpressing Mice
3.3.2. AOX-Deficient Mice
3.3.3. PPARα-Deficient Mice
3.3.4. Other Models of Reduced Oxidation
3.4. Models with Reduced VLDL Secretion
3.4.1. Mice Fed a MCD Diet
3.4.2. Hepatocyte-Specific MTP-Deficient Mice
3.4.3. PITPα-Deficient Mice
3.4.4. Other Models of Reduced VLDL Secretion
3.5. Models with Increased Hepatic Cholesterol
3.5.1. IL-1 Ra-Deficient Mice
3.5.2. Rabbit Fed a High-Fat and High-Cholesterol Diet
3.6. Model with Endotoxin-Induced Liver Inflammation
3.6.1. Mice with Intraperitoneal Injection of Low-Dose LPS
4. Conclusions


{kind=link}
{kind=link}
| FA | Obese | IR | Inflammation | Fibrosis | Carcinoma | |
|---|---|---|---|---|---|---|
| I Models with increased FA delivery into the liver | ||||||
| I-a Models involving feeding of a HFD | ||||||
| Mice with chronic exposure to a HFD | Y | Y | Y | Y | Y | N |
| Mice with intragastric overfeeding of a HFD | Y | Y | Y | Y | Y | N |
| Mice fed a combination of fructose and a HFD | Y | Y | Y | Y | Y | N |
| I-b Models with increased appetite | ||||||
| ob/ob mice | Y | Y | Y | N | N | N |
| db/db mice | Y | Y | Y | N | N | N |
| Zucker fatty (fa/fa) rat | Y | Y | Y | N | N | N |
| MC4R knockout mice | Y | Y | Y | Y | Y | Y |
| LRbS1138/1138 knockin mice | Y | Y | Y | N | N | N |
| Nestin-Cre STAT3 knockout mice | Y | Y | Y | N | N | N |
| CRE3/Shp2 knockout mice | Y | Y | Y | N | N | N |
| KKAy mice | Y | Y | Y | Y | Y | N |
| FLS-ob mice | Y | Y | Y | Y | Y | Y |
| OLEFT rat | Y | Y | Y | Y | N | N |
| I-c Models with disordered lipid synthesis in adipose tissue | ||||||
| aP2-nSREBP-1c overexpressing mice | Y | N | Y | Y | N | N |
| A-ZIP/F-1 overexpressing mice | Y | N | Y | N | N | N |
| CD36 knockout mice | Y | N | N | Y | N | N |
| aP2-diphtheria toxin overexpressing mice | Y | N | Y | N | N | N |
| PPARγ hypomorphic mice | Y | N | Y | N | N | N |
| I-d Models with increased GC | ||||||
| aP2-11β-HSD1 overexpressing mice | Y | Y | Y | N | N | N |
| 7B2 knockout mice | Y | Y | Y | Y | N | N |
| I-e Models with increased IR | ||||||
| insulin I-IGF-II overexpressing mice | Y | Y | Y | N | N | N |
| STAT5B knockout mice | Y | Y | Y | N | N | N |
| II Models with increased de novo lipogenesis | ||||||
| Hepatocyte-specific PTEN knockout mice | N | N | Y | Y | Y | Y |
| IDPc overexpressing mice | Y | Y | Y | ND | N | N |
| ChREBP knockout mice | Y | N | Y | N | N | N |
| PEPCK-nSREBP1α overexpressing mice | N | N | Y | Y | N | N |
| III Models with reduced oxidation | ||||||
| Hepatocyte-specific RARα dominant negative overexpressing mice | ND | N | N | Y | N | Y |
| AOX knockout mice | N | N | ND | Y | N | Y |
| PPARα knockout mice | Y | N | N | N | N | N |
| aromatase knockout mice | Y | Y | Y | N | N | N |
| JVS mice | N | N | N | N | N | N |
| ADK knockout mice | N | N | N | N | N | N |
| CBS knockout mice | Y | Y | ND | Y | Y | N |
| Alms1 knockout mice fed a HFD | Y | Y | Y | Y | Y | ND |
| IV Models with decreased VLDL secretion | ||||||
| Mice fed a MCD diet | N | N | N | Y | Y | Y |
| Hepatocyte-specific MTP knockout mice | N | N | Y | N | N | N |
| PITPα knockout mice | N | N | N | N | N | N |
| PEMT knockout mice | N | N | N | Y | ND | N |
| ApoE knockout mice | N | N | ND | N | N | N |
| V Models with increased intrahepatic cholesterol | ||||||
| IL-1 Ra knockout mice with an atherogenic diet | N | N | ND | Y | Y | N |
| A rabbit fed a HFD and high-cholesterol diet | Y | Y | Y | Y | Y | N |
| VI Models with endotoxin-induced liver inflammation | ||||||
| Mice with intraperitoneal injection of low-dose lipopolysaccharide | Y | Y | Y | Y | Y | N |
Acknowledgments
Conflicts of Interest
References
- Sanyal, A.J. AGA technical review on nonalcoholic fatty liver disease. Gastroenterology 2002, 123, 1705–1725. [Google Scholar]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerrelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Melchionda, N.; et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003, 37, 917–923. [Google Scholar]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med 2002, 346, 1221–1231. [Google Scholar]
- Clark, J.M.; Brancati, F.L.; Diehl, A.M. Nonalcoholic fatty liver disease. Gastroenterology 2002, 122, 1649–1657. [Google Scholar]
- Neuschwander-Tetri, B.A.; Caldwell, S.H. Nonalcoholic steatohepatitis, summary of an AASLD single topic conference. Hepatology 2003, 37, 1202–1219. [Google Scholar]
- Day, C.P. Non-alcoholic steatohepatitis (NASH), where are we now and where are we going? Gut 2002, 50, 585–588. [Google Scholar]
- Fan, J.-G.; Li, F.; Cai, X.-B.; Peng, Y.-D.; Ao, Q.-H.; Gao, Y. Effects of nonalcoholic fatty liver disease on the development of metabolic disorders. J. Gastroenterol. Hepatol 2007, 22, 1086–1091. [Google Scholar]
- Harrison, S.A.; Torgerson, S.; Hayashi, P.H. The natural history of nonalcoholic fatty liver disease, a clinical histopathological study. Am. J. Gastroenterol 2003, 98, 2042–2047. [Google Scholar]
- Day, C.P.; Saksena, S. Non-alcoholic steatohepatitis, definitions and pathogenesis. J. Gastroenterol. Hepatol 2002, 17, 377–384. [Google Scholar]
- Powell, E.E.; Cooksley, W.G.; Hanson, R.; Searle, J.; Halliday, J.W.; Powell, L.W. The natural history of nonalcoholic steatohepatitis, a follow-up study of forty-two patients for up to 21 years. Hepatology 1990, 11, 74–80. [Google Scholar]
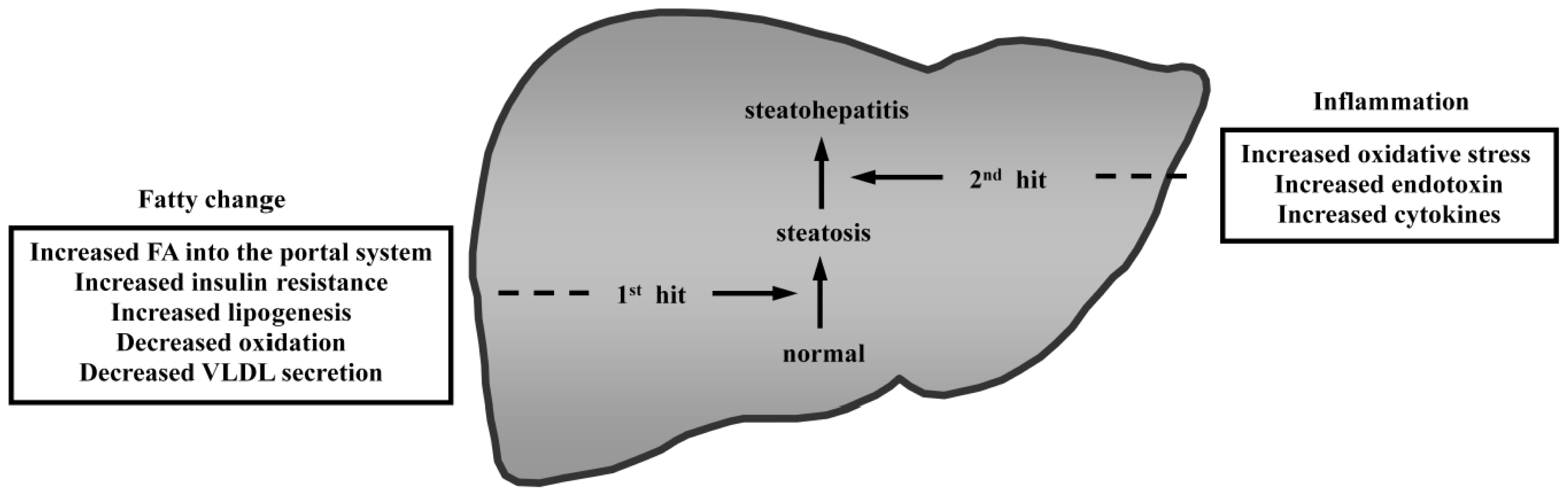
- Day, C.P.; James, O.F. Steatohepatitis, a tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar]
- Day, C.P. Pathogensis of steatohepatitis. Best Pract. Res. Clin. Gastroenterol 2002, 16, 663–678. [Google Scholar]
- London, R.M.; George, J. Pathogenesis of NASH, animal models. Clin. Liver Dis 2007, 11, 55–74. [Google Scholar]
- Wanless, I.R.; Shiota, K. The pathogenesis of nonalcoholic steatohepatitis and other fatty liver diseases, a four-step model including the role of lipid release and hepatic venular obstruction in the progression to cirrhosis. Semin. Liver Dis 2004, 24, 99–106. [Google Scholar]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease, from steatosis to cirrhosis. Hepatology 2006, 43, 99–112. [Google Scholar]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar]
- Fan, J.G.; Xu, Z.J.; Wang, G.L. Effect of lactulose on establishment of a rat non-alcoholic steatohepatitis model. World J. Gastroenterol 2005, 11, 5053–5056. [Google Scholar]
- Larter, C.Z. Not all models of fatty liver are created equal, understanding mechanisms of steatosis development is important. J. Gastroenterol. Hepatol 2007, 22, 1353–1354. [Google Scholar]
- Koteish, A.; Diehl, A.M. Animal models of steatosis. Semin. Liver Dis 2001, 21, 89–104. [Google Scholar]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig 2005, 115, 1343–1351. [Google Scholar]
- Nehra, V.; Angulo, P.; Buchman, A.L.; Lindor, K.D. Nutritional and metabolic considerations in the etiology of nonalcoholic steatohepatitis. Dig. Dis. Sci 2001, 46, 2347–2352. [Google Scholar]
- Bradbury, M.W. Lipid metabolism and liver inflammation. Hepatic fatty acid uptake-possible role in steatosis. Am. J. Physiol. Gastrointest. Liver Physiol 2006, 290, 194–198. [Google Scholar]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Lehrke, M.; Hendler, R.E.; Shulman, G.I. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 2005, 54, 603–608. [Google Scholar]
- Previs, S.F.; Withers, D.J.; Ren, J.M.; White, M.F.; Shulman, G.I. Contrasting effects of IRS-1 versus IRS-2 gene disruption on carbohydrate and lipid metabolism in vivo Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J. Biol. Chem 2000, 275, 38990–38904. [Google Scholar]
- Utzschneider, K.M.; Kahn, S.E. The role of insulin resistance in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab 2006, 91, 4753–4761. [Google Scholar]
- Abdelmalek, M.F.; Diehl, A.M. Nonalcoholic fatty liver disease as a complication of insulin resistance. Med. Clin. N. Am 2007, 91, 1125–1149. [Google Scholar]
- Kim, J.K.; Michael, M.D.; Previs, S.F.; Peroni, O.D.; Mauvais-Jarvis, F.; Neschen, S.; Kahn, B.B.; Kahn, C.R.; Shulman, G.I. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J. Clin. Investig 2000, 105, 1791–1797. [Google Scholar]
- Koch, L.; Wunderlich, F.T.; Seibler, J.; Konner, A.C.; Hampel, B.; Irlenbusch, S.; Brabant, G.; Kahn, C.R.; Schwenk, F.; Brüning, J.C. Central insulin action regulates peripheral glucose and fat metabolism in mice. J. Clin. Investig 2008, 118, 2132–2147. [Google Scholar]
- Blüher, M.; Michael, M.D.; Peroni, O.D.; Ueki, K.; Carter, N.; Kahn, B.B.; Kahn, C.R. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev. Cell 2002, 3, 25–38. [Google Scholar]
- Reddy, J.K.; Rao, M.S. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am. J. Physiol. Gastrointest. Liver Physiol 2006, 290, 852–858. [Google Scholar]
- Ito, M.; Suzuki, J.; Sasaki, M.; Watanabe, K.; Tsujioka, S.; Takahashi, Y.; Gomori, A.; Hirose, H.; Ishihara, A.; Iwaasa, H.; et al. Development of nonalcoholic steatohepatitis model through combination of high-fat diet and tetracycline with morbid obesity in mice. Hepatol. Res 2006, 34, 92–98. [Google Scholar]
- Ito, M.; Suzuki, J.; Tsujioka, S.; Sasaki, M.; Gomori, A.; Shirakura, T.; Hirose, H.; Ito, M.; Ishihara, A.; Iwaasa, H.; et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol. Res 2007, 37, 50–57. [Google Scholar]
- Deng, Q.G.; She, H.; Cheng, J.H.; French, S.W.; Koop, D.R.; Xiong, S.; Tsukamoto, H. Steatohepatitis Induced by Intragastric overfeeding in mice. Hepatology 2005, 42, 905–914. [Google Scholar]
- Tetri, L.H.; Basaranoglu, M.; Brunt, E.M.; Yerian, L.M.; Neuschwander-Tetri, B.A. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am. J. Physiol. Gastrointest. Liver Physiol 2008, 295, 987–995. [Google Scholar]
- Tsuchiya, H.; Ebata, Y.; Sakabe, T. High-fat, high-fructose diet induces hepatic iron overload via a hepcidin-independent mechanism prior to the onset of liver steatosis and insulin resistance in mice. Metabolism 2013, 62, 62–69. [Google Scholar]
- Bergheim, I.; Weber, S.; Vos, M. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol 2008, 48, 983–992. [Google Scholar]
- Spruss, A.; Kanuri, G.; Wagnerberger, S.; Haub, S.; Bischoff, S.C.; Bergheim, I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009, 50, 1094–1104. [Google Scholar]
- Kanuri, G.; Spruss, A.; Wagnerberger, S. Role of tumor necrosis factor alpha (TNFalpha) in the onset of fructose-induced nonalcoholic fatty liver disease in mice. J. Nutr. Biochem 2011, 22, 527–534. [Google Scholar]
- Tappy, L.; Le, K.A.; Tran, C. Fructose and metabolic diseases: New findings, new questions. Nutrition 2010, 26, 1044–1049. [Google Scholar]
- Ingalls, A.M.; Dickie, M.M.; Snell, G.D. Obese, a new mutation in the house mouse. J. Hered 1950, 41, 317–318. [Google Scholar]
- Mayer, J.; Russell, R.E.; bates, M.W.; dickie, M.M. Metabolic, nutritional and endocrine studies of the hereditary obesity-diabetes syndrome of mice and mechanism of its development. Metabolism 1953, 2, 9–21. [Google Scholar]
- Garthwaite, T.L.; Martinson, D.R.; Tseng, L.F.; Hagen, T.C.; Menahan, L.A. A longitudinal hormonal profile of the genetically obese mouse. Endocrinology 1980, 107, 671–676. [Google Scholar]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homolog. Nat. Dec 1994, 372, 425–432. [Google Scholar]
- Pelleymounter, M.A.; Cullen, M.J.; Baker, M.B.; Hecht, R.; Winters, D.; Boone, T.; Collins, F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science 1995, 269, 540–543. [Google Scholar]
- Halaas, J.L.; Gajiwala, K.S.; Maffei, M.; Cohen, S.L.; Chait, B.T.; Rabinowitz, D.; Lallone, R.L.; Burley, S.K.; Friedman, J.M. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995, 269, 543–546. [Google Scholar]
- Lindström, P. The physiology of obese-Hyperglycemic mice (ob/ob mice). Sci. World J 2007, 29, 666–685. [Google Scholar]
- Hummel, K.P.; Dickie, M.M.; Coleman, D.L. Diabetes, a new mutation in the mouse. Science 1966, 153, 1127–1128. [Google Scholar]
- Tartaglia, L.A.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.J.; Campfield, L.A.; Clark, F.T.; Deeds, J. Identification and expression cloning of a leptin receptor. OB-R. Cell 1995, 83, 1263–1271. [Google Scholar]
- Wortham, M.; He, L.; Gyamfi, M.; Copple, B.L.; Wan, Y.J. The transition from fatty liver to NASH associates with SAMe depletion in db/db mice fed a methionine choline-deficient diet. Dig. Dis. Sci 2008, 53, 2761–2774. [Google Scholar]
- Takaya, K.; Ogawa, Y.; Isse, N.; Okazaki, T.; Satoh, N.; Masuzaki, H.; Mori, K.; Tamura, N.; Hosoda, K.; Nakao, K. Molecular cloning of rat leptin receptor isoform complementary DNAs-identification of a missense mutation in zucker fatty (fa/fa) rats. Biochem. Biophys. Res. Commun 1996, 225, 75–83. [Google Scholar]
- Balthasar, N.; Dalgaard, L.T.; Lee, C.E. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 2005, 123, 493–505. [Google Scholar]
- Itoh, M.; Suganami, T.; Nakagawa, N. Melanocortin 4 receptor-deficient mice as a novel mouse model of nonalcoholic steatohepatitis. Am. J. Pathol 2011, 179, 2454–2463. [Google Scholar]
- Vaisse, C.; Clement, K.; Durand, E. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J. Clin. Investig 2000, 106, 253–262. [Google Scholar]
- Jiang, L.; You, J.; Yu, X.; Gonzalez, L.; Yu, Y.; Wang, Q.; Yang, G.; Li, W.; Li, C.; Liu, Y. Tyrosine-dependent and -independent actions of leptin receptor in control of energy balance and glucose homeostasis. EPUB 2008, 105, 18619–18624. [Google Scholar]
- Piper, M.L.; Unger, E.K.; Myers, M.G., Jr.; Xu, A.W. Specific physiological roles for signal transducer and activator of transcription 3 in leptin receptor-expressing neurons. Mol. Endocrinol 2008, 22, 751–759. [Google Scholar]
- Bates, S.H.; Kulkarni, R.N.; Seifert, M.; Myers, M.G., Jr. Roles for leptin receptor/STAT3-dependent and -independent signals in the regulation of glucose homeostasis. Cell Metab 2005, 1, 169–178. [Google Scholar]
- Okumura, K.; Ikejima, K.; Kon, K.; Abe, W.; Yamashina, S.; Enomoto, N.; Takei, Y.; Sato, N. Exacerbation of dietary steatohepatitis and fibrosis in obese, diabetic KKAy mice. Hepatol. Res 2006, 36, 217–228. [Google Scholar]
- Soga, M.; Hashimoto, S.; Kishimoto, Y.; Hirasawa, T.; Makino, S.; Inagaki, S. Insulin resistance, steatohepatitis, and Hepatocellular carcinoma in a New Congenic Strain of fatty liver Shionogi (FLS) with the Lepob gene. Exp. Anim 2010, 59, 407–419. [Google Scholar]
- Kawano, K.; Hirashima, T.; Mori, S.; Saitoh, Y.; Kurosumi, M.; Natori, T. Spontaneous long-term hyperglycemic rat with diabetic complications. Otsuka Long-Evans Tokushima Fatty (OLETF) strain. Diabetes 1992, 42, 1422–1428. [Google Scholar]
- Nielsen, S.; Guo, Z.; Johnson, C.M.; Hensrud, D.D.; Jensen, M.D. Splanchnic lipolysis in human obesity. J. Clin. Investig 2004, 113, 1582–1588. [Google Scholar]
- Shimomura, I.; Hammer, R.E.; Richardson, J.A.; Ikemoto, S.; Bashmakov, Y.; Goldstein, J.L.; Brown, M.S. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue, model for congenital generalized lipodystrophy. Genes Dev 1998, 12, 3182–3194. [Google Scholar]
- Moitra, J.; Mason, M.M.; Olive, M.; Krylov, D.; Gavrilova, O.; Marcus-Samuels, B.; Feigenbaum, L.; Lee, E.; Aoyama, T.; Eckhaus, M. Life without white fat, a transgenic mouse. Genes Dev 1998, 12, 3168–3181. [Google Scholar]
- Coburn, C.T.; Knapp, F.F., Jr.; Febbraio, M.; Beets, A.L.; Silverstein, R.L.; Abumrad, N.A. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J. Biol. Chem 2000, 275, 32523–32529. [Google Scholar]
- Ross, S.R.; Graves, R.A.; Spiegelman, B.M. Targeted expression of a toxin gene to adipose tissue, transgenic mice resistant to obesity. Genes Dev 1993, 7, 1318–1324. [Google Scholar]
- Koutnikova, H.; Cock, T.A.; Watanabe, M.; Houten, S.M.; Champy, M.F.; Dierich, A.; Auwerx, J. Compensation by the muscle limits the metabolic consequences of lipodystrophy in PPAR hypomorphic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 14457–14462. [Google Scholar]
- Tomlinson, J.W.; Walker, E.A.; Bujalska, I.J.; Draper, N.; Lavery, G.G.; Cooper, M.S.; Hewison, M.; Stewart, P.M. 11beta-hydroxysteroid dehydrogenase type 1, a tissue-specific regulator of glucocorticoid response. Endocr. Rev 2004, 25, 831–866. [Google Scholar]
- Morton, N.M.; Seckl, J.R. 11beta-hydroxysteroid dehydrogenase type 1 and obesity. Front. Horm. Res 2008, 36, 146–164. [Google Scholar]
- Laurent, V.; Kimble, A.; Peng, B.; Zhu, P.; Pintar, J.E.; Steiner, D.F.; Lindberg, I. Mortality in 7B2 null mice can be rescued by adrenalectomy, involvement of dopamine in ACTH hypersecretion. Proc. Natl. Acad. Sci. USA 2002, 99, 3087–3092. [Google Scholar]
- Gilham, D.; Perreault, K.R.; Holmes, C.F.; Brindley, D.N.; Vance, D.E.; Lehner, R. Insulin, glucagon and fatty acid treatment of hepatocytes does not result in phosphorylation or changes in activity of triacylglycerol hydrolase. Biochim. Biophys. Acta 2005, 1736, 189–199. [Google Scholar]
- Rossetti, L.; Barzilai, N.; Chen, W.; Harris, T.; Yang, D.; Rogler, C.E. Hepatic overexpression of insulin-like growth factor-II in adulthood increases basal and insulin-stimulated glucose disposal in conscious mice. J. Biol. Chem 1996, 271, 203–208. [Google Scholar]
- Zhou, Y.C.; Davey, H.W.; McLachlan, M.J.; Xie, T.; Waxman, D.J. Elevated basal expression of liver peroxisomal beta-oxidation enzymes and CYP4A microsomal fatty acid omega-hydroxylase in STAT5b−/− mice, cross-talk in vivo between peroxisome proliferator-activated receptor and signal transducer and activator of transcription signaling pathways. Toxicol. Appl. Pharmacol 2002, 182, 1–10. [Google Scholar]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig 2004, 114, 147–152. [Google Scholar]
- Foufelle, F.; Ferre, P. New perspectives in the regulation of hepatic glycolytic and lipogenic genes by insulin and glucose, a role for the transcription factor sterol regulatory element binding protein-1c. Biochem. J 2002, 366, 377–391. [Google Scholar]
- Trumble, G.; Smith, M.; Winder, W. Purification and characterization of rat skeletal muscle acetyl-CoA carboxylase. Eur. J. Biochem 1995, 231, 192–198. [Google Scholar]
- Nosadini, R.; Ursini, F.; Tessari, P.; Tiengo, A.; Gregolin, C. Perfused liver carnitine palmitoyl-transferase activity and ketogenesis in streptozotocin treated and genetic hyperinsulinemic rats. Effect of glucagon. Horm. Metab. Res 1979, 11, 661–664. [Google Scholar]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem 1998, 273, 13375–13378. [Google Scholar]
- Horie, Y.; Suzuki, A.; Kataoka, E.; Sasaki, T.; Hamada, K.; Sasaki, J.; Mizuno, K.; Hasegawa, G.; Kishimoto, H.; Iizuka, M.; et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Investig 2004, 113, 1774–1783. [Google Scholar] [Green Version]
- Koh, H.J.; Lee, S.M.; Son, B.G.; Lee, S.H.; Ryoo, Z.Y.; Chang, K.T.; Park, J.-W.; Park, D.-C.; Song, B.J.; Veech, R.L.; et al. Cytosolic NADP+-dependent isocitrate dehydrogenase plays a key role in lipid metabolism. J. Biol. Chem 2004, 279, 39968–39974. [Google Scholar]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar]
- Takahashi, A.; Shimano, H.; Nakagawa, Y.; Yamamoto, T.; Motomura, K.; Matsuzaka, T.; Sone, H.; Suzuki, H.; Toyoshima, H.; Yamada, N.; et al. Transgenic mice overexpressing SREBP-1a under the control of the PEPCK promoter exhibit insulin resistance, but not diabetes. Biochim. Biophys. Acta 2005, 1740, 427–433. [Google Scholar]
- Bennett, M.J. Pathophysiology of fatty acid oxidation disorders. J. Inherit. Metab. Dis 2009, 33, 533–537. [Google Scholar]
- McGarry, J.D.; Brown, N.F. The mitochondrial carnitine palmitoyltransferase system, from concept to molecular analysis. Eur. J. Biochem 1997, 244, 1–6. [Google Scholar]
- Yanagitani, A.; Yamada, S.; Yasui, S.; Shimomura, T.; Murai, R.; Murawaki, Y.; Hashiguchi, K.; Kanbe, T.; Saeki, T.; Ichiba, M.; et al. Retinoic acid receptor alpha dominant negative form causes steatohepatitis and liver tumors in transgenic mice. Hepatology 2004, 40, 366–375. [Google Scholar]
- Fan, C.Y.; Pan, J.; Usuda, N.; Yeldandi, A.V.; Rao, M.S.; Reddy, J.K. Steatohepatitis, spontaneous peroxisome proliferation and liver tumors in mice lacking peroxisomal fatty acyl-CoA oxidase. Implications for peroxisome proliferator-activated receptor alpha natural ligand metabolism. J. Biol. Chem 1998, 273, 15639–15645. [Google Scholar]
- Aoyama, T.; Peters, J.M.; Iritani, N.; Nakajima, T.; Furihata, K.; Hashimoto, T.; Gonzalez, F.J. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J. Biol. Chem 1998, 273, 5678–5684. [Google Scholar]
- Nemoto, Y.; Toda, K.; Ono, M.; Adachi, K.F.; Saibara, T.; Onishi, S.; Enzan, H.; Okada, T.; Shizuta, Y. Altered expression of fatty acid-metabolizing enzymes in aromatase deficient mice. J. Clin. Investig 2000, 105, 1819–1825. [Google Scholar]
- Kuwajima, M.; Kono, N.; Horiuchi, M.; Imamura, Y.; Ono, A.; Inui, Y.; Kawata, S.; Koizumi, T.; Hayakawa, J.; Saheki, T.; et al. Animal model of systemic carnitine deficiency: Analysis in C3H-H-2 degrees strain of mouse associated with juvenile visceral steatosis. Biochem. Biophys. Res. Commun 1991, 174, 1090–1094. [Google Scholar]
- Boison, D.; Scheurer, L.; Zumsteg, V.; Rülicke, T.; Litynski, P.; Fowler, B.; Brandner, S.; Mohler, H. Neonatal hepatic steatosis by disruption of the adenosine kinase gene. Proc. Natl. Acad. Sci. USA 2002, 99, 6985–6990. [Google Scholar]
- Hamelet, J.; Demuth, K.; Paul, J.L.; Delabar, J.M.; Janel, N. Hyperhomocysteinemia due to cystathionine beta synthase deficiency induces dysregulation of genes involved in hepatic lipid homeostasis in mice. Hepatology 2007, 46, 151–159. [Google Scholar]
- Arsov, T.; Larter, C.Z.; Nolan, C.J.; Petrovsky, N.; Goodnow, C.C.; Teoh, N.C.; Yeh, M.M.; Farrell, G.C. Adaptive failure to high-fat diet characterizes steatohepatitis in Alms1 mutant mice. Biochem. Biophys. Res. Commun 2006, 342, 1152–1159. [Google Scholar]
- Julius, U. Influence of plasma free fatty acids on lipoprotein synthesis and diabetic dyslipidemia. Exp. Clin. Endocrinol. Diabetes 2003, 111, 246–250. [Google Scholar]
- Gordon, D.A. Recent advances in elucidating the role of the microsomal triglyceride transfer protein in apolipoprotein B lipoprotein assembly. Curr. Opin. Lipidol 1997, 8, 131–137. [Google Scholar]
- Wetterau, J.R.; Gregg, R.E.; Harrity, T.W.; Arbeeny, C.; Cap, M.; Connolly, F.; Chu, C.H.; George, R.J.; Gordon, D.A.; Jamil, H.; et al. An MTP inhibitor that normalizes atherogenic lipoprotein levels in WHHL rabbits. Science 1998, 282, 751–754. [Google Scholar]
- Rinella, M.E.; Elias, M.S.; Smolak, R.R.; Fu, T.; Borensztajn, J.; Green, R.M. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J. Lipid Res 2008, 49, 1068–1076. [Google Scholar]
- Raabe, M.; Véniant, M.M.; Sullivan, M.A.; Zlot, C.H.; Björkegren, J.; Nielsen, L.B.; Wong, J.S.; Hamilton, R.L.; Young, S.G. Analysis of the role of microsomal triglyceride transfer protein in the liver of tissue-specific knockout mice. J. Clin. Investig 1999, 103, 1287–1298. [Google Scholar]
- Helmkamp, G.M., Jr.; Harvey, M.S.; Wirtz, K.W.; Van Deenen, L.L. Phospholipid exchange between membranes. Purification of bovine brain proteins that preferentially catalyze the tranfer of phosphatidylinositol. J. Biol. Chem 1974, 249, 6382–6389. [Google Scholar]
- Alb, J.G., Jr.; Cortese, J.D.; Phillips, S.E.; Albin, R.L.; Nagy, T.R.; Hamilton, B.A.; Bankaitis, V.A. Mice lacking phosphatidylinositol transfer protein-alpha exhibit spinocerebellar degeneration, intestinal and hepatic steatosis, and hypoglycemia. J. Biol. Chem 2003, 278, 33501–33518. [Google Scholar]
- Zhu, X.; Song, J.; Mar, M.H.; Edwards, L.J.; Zeisel, S.H. Phosphatidylethanolamine N-methyltransferase (PEMT) knockout mice have hepatic steatosis and abnormal hepatic choline metabolite concentrations despite ingesting a recommended dietary intake of choline. Biochem. J 2003, 370, 987–993. [Google Scholar]
- Hussain, M.M.; Maxfield, F.R.; Más-Oliva, J.; Tabas, I.; Ji, Z.S.; Innerarity, T.L.; Mahley, R.W. Clearance of chylomicron remnants by the low density ipoprotein receptor-related protein/alpha 2-macroglobulin receptor. J. Biol. Chem 1991, 266, 13936–13940. [Google Scholar]
- Ma, K.L.; Ruan, X.Z.; Powis, S.H.; Chen, Y.; Moorhead, J.F.; Varghese, Z. Inflammatory stress exacerbates lipid accumulation in hepatic cells and fatty livers of apolipoprotein E knockout mice. Hepatology 2008, 48, 770–781. [Google Scholar]
- Vlahcevic, Z.R.; Stravitz, R.T.; Heuman, D.M.; Hylemon, P.B.; Pandak, W.M. Quantitative estimations of the contribution of different bile acid pathways to total bile acid synthesis in the rat. Gastroenterology 1997, 113, 1949–1957. [Google Scholar]
- Feng, B.; Yao, P.M.; Li, Y.; Devlin, C.M.; Zhang, D.; Harding, H.P.; Sweeney, M.; Rong, J.X.; Kuriakose, G.; Fisher, E.A.; et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol 2003, 5, 781–792. [Google Scholar]
- Devries-Seimon, T.; Li, Y.; Yao, P.M.; Stone, E.; Wang, Y.; Davis, R.J.; Flavell, R.; Tabas, I. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. J. Cell. Biol 2005, 171, 61–73. [Google Scholar]
- Bieghs, V.; Verheyen, F.; van Gorp, P.J.; Hendrikx, T.; Wouters, K.; Lütjohann, D.; Gijbels, M.J.; Febbraio, M.; Binder, C.J.; Hofker, M.H.; et al. Internalization of modified lipids by CD36 and SR-A leads to hepatic inflammation and lysosomal cholesterol storage in Kupffer cells. PLoS One 2012, 7, e34378. [Google Scholar]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar]
- Isoda, K.; Sawada, S.; Ayaori, M.; Matsuki, T.; Horai, R.; Kagata, Y.; Miyazaki, K.; Kusuhara, M.; Okazaki, M.; Matsubara, O. Deficiency of interleukin-1 receptor antagonist deteriorates fatty liver and cholesterol metabolism in hypercholesterolemic mice. J. Biol. Chem 2005, 280, 7002–7009. [Google Scholar]
- Otogawa, K.; Kawada, N. A rabbit model for the study of human NASH. Nippon Rinsho 2006, 64, 1043–1047. [Google Scholar]
- Matteoni, C.A.; Younossi, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.C.; McCullough, A.J. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419. [Google Scholar]
- Brunt, E.M.; Neuschwander-Tetri, B.A.; Oliver, D.; Wehmeier, K.R.; Bacon, B.R. Nonalcoholic steatohepatitis: Histologic features and clinical correlations with 30 blinded biopsy specimens. Hum. Pathol 2004, 35, 1070–1082. [Google Scholar]
- Burt, A.D.; Mutton, A.; Day, C.P. Diagnosis, interpretation of steatosis and steatohepatitis. Semin. Diagn. Pathol 1998, 15, 246–258. [Google Scholar]
- Yahagi, N.; Shimano, H.; Matsuzaka, T.; Sekiya, M.; Najima, Y.; Okazaki, S.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Inoue, N. p53 Involvement in the Pathogenesis of Fatty Liver Disease. J. Biol. Chem 2004, 279, 20571–20575. [Google Scholar]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig 2003, 112, 1821–1830. [Google Scholar]
- Imajo, K.; Fujita, K.; Yoneda, M.; Nozaki, Y.; Ogawa, Y.; Shinohara, Y.; Kato, S.; Mawatari, H.; Shibata, W.; Kitani, H.; et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab 2012, 16, 44–54. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Imajo, K.; Yoneda, M.; Kessoku, T.; Ogawa, Y.; Maeda, S.; Sumida, Y.; Hyogo, H.; Eguchi, Y.; Wada, K.; Nakajima, A. Rodent Models of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2013, 14, 21833-21857. https://doi.org/10.3390/ijms141121833
Imajo K, Yoneda M, Kessoku T, Ogawa Y, Maeda S, Sumida Y, Hyogo H, Eguchi Y, Wada K, Nakajima A. Rodent Models of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. International Journal of Molecular Sciences. 2013; 14(11):21833-21857. https://doi.org/10.3390/ijms141121833
Chicago/Turabian StyleImajo, Kento, Masato Yoneda, Takaomi Kessoku, Yuji Ogawa, Shin Maeda, Yoshio Sumida, Hideyuki Hyogo, Yuichiro Eguchi, Koichiro Wada, and Atsushi Nakajima. 2013. "Rodent Models of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis" International Journal of Molecular Sciences 14, no. 11: 21833-21857. https://doi.org/10.3390/ijms141121833
APA StyleImajo, K., Yoneda, M., Kessoku, T., Ogawa, Y., Maeda, S., Sumida, Y., Hyogo, H., Eguchi, Y., Wada, K., & Nakajima, A. (2013). Rodent Models of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. International Journal of Molecular Sciences, 14(11), 21833-21857. https://doi.org/10.3390/ijms141121833