Oxidative Stress and Neurodegenerative Disorders

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
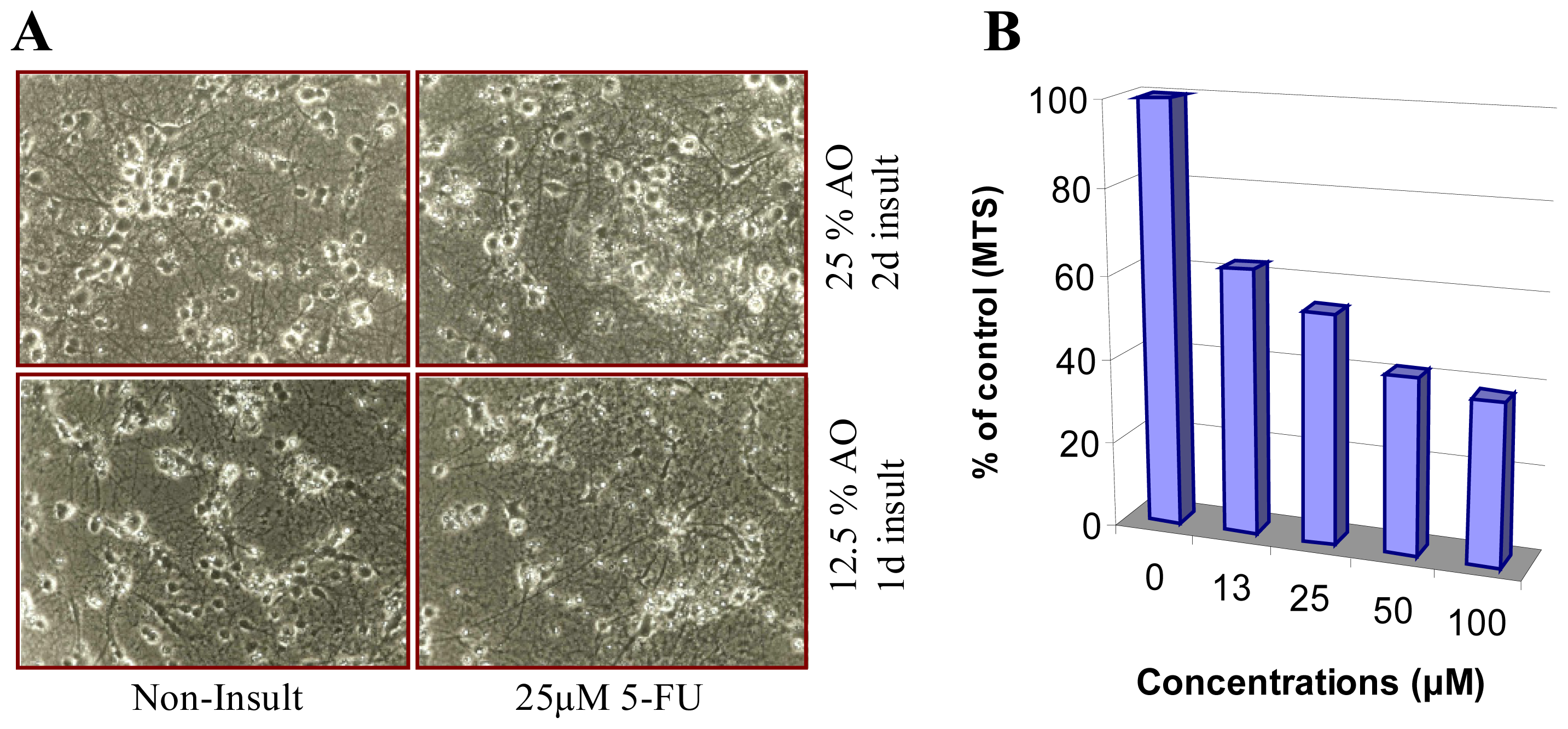{kind=link}
1. Introduction
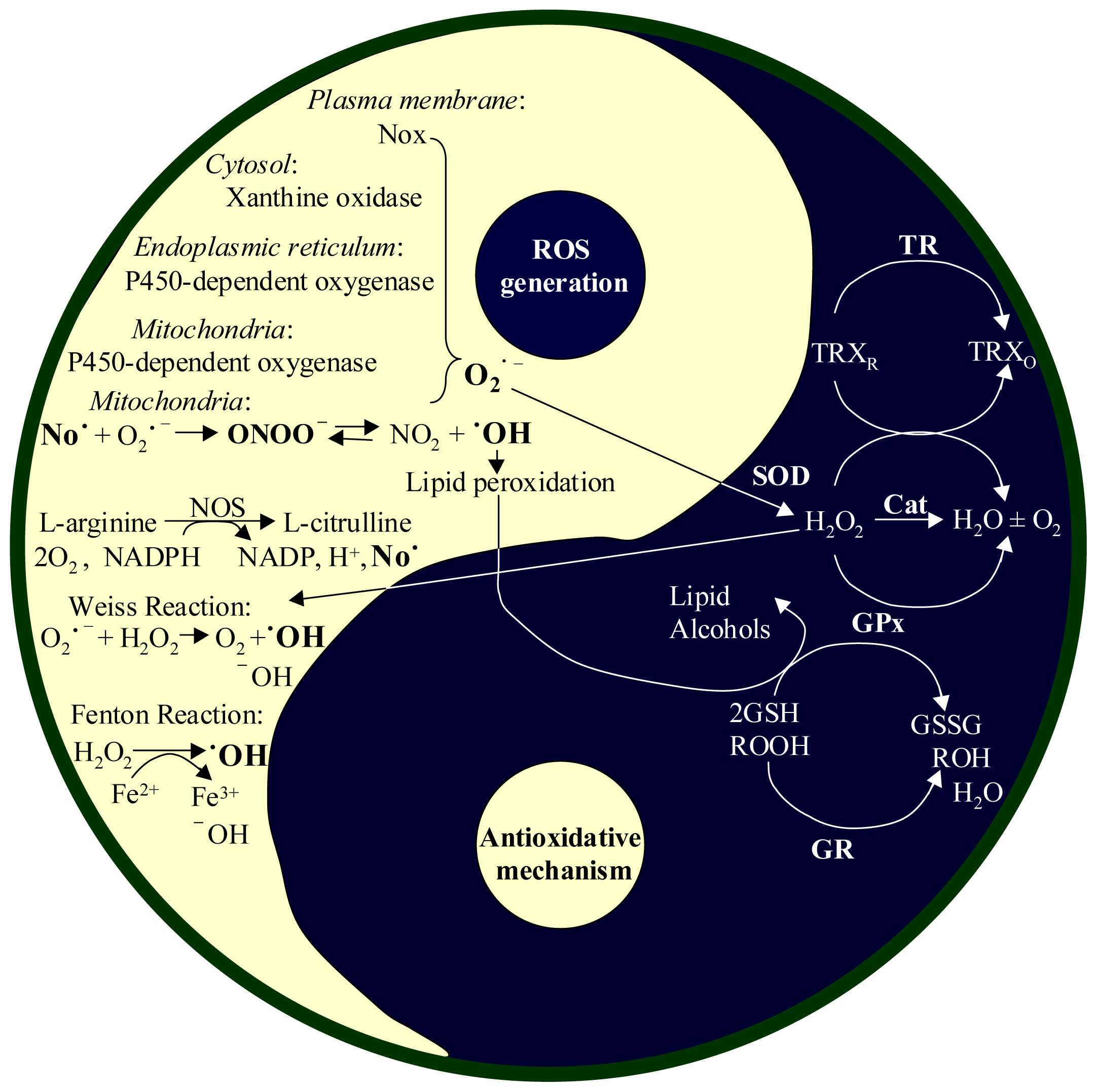
2. Reactive Oxygen Species (ROS)
2.1. Productions of ROS and RNI
2.2. Antioxidant Systems in the Body
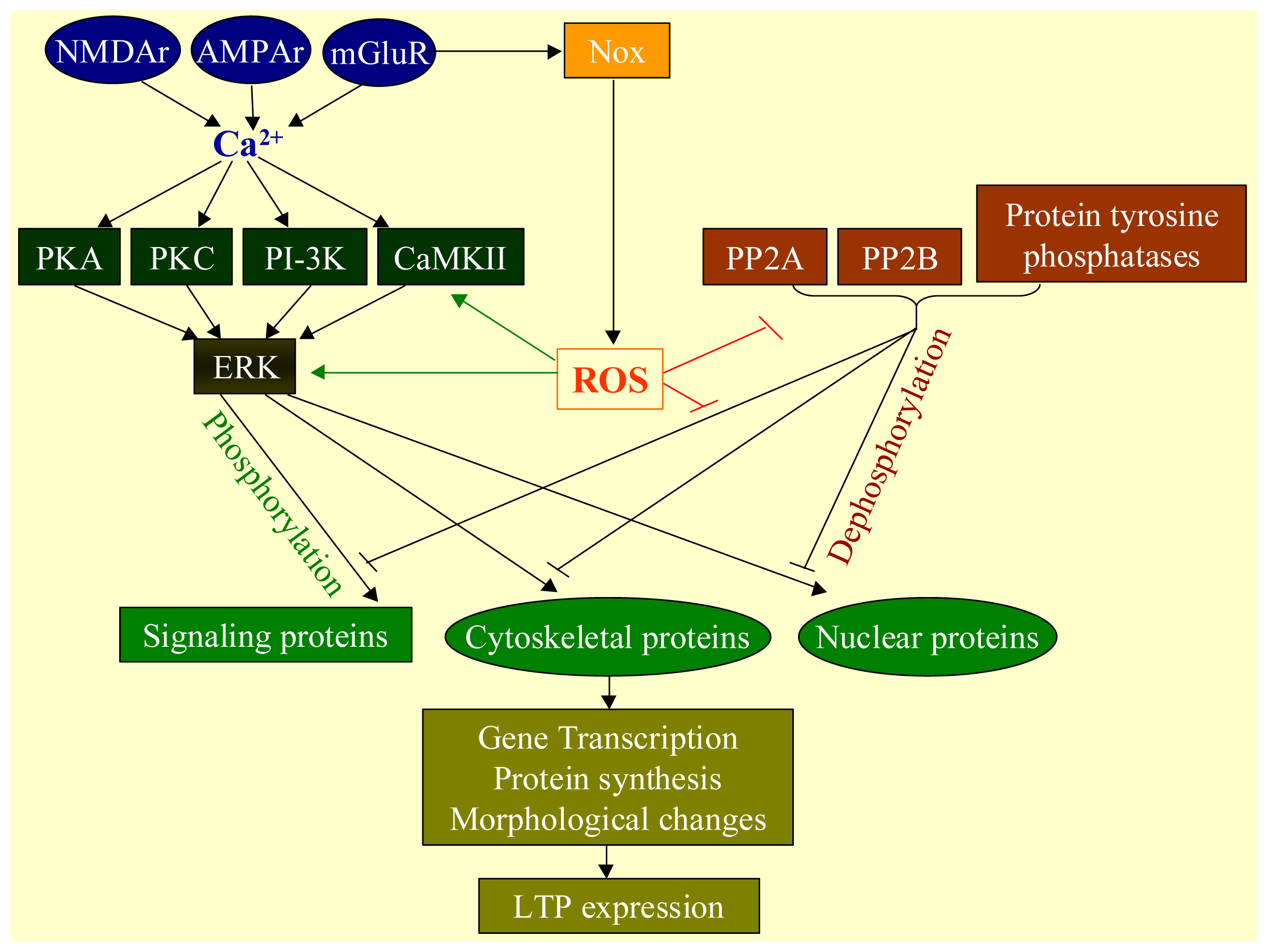
2.3. Physiological Functions of ROS
2.4. Oxidative Stress
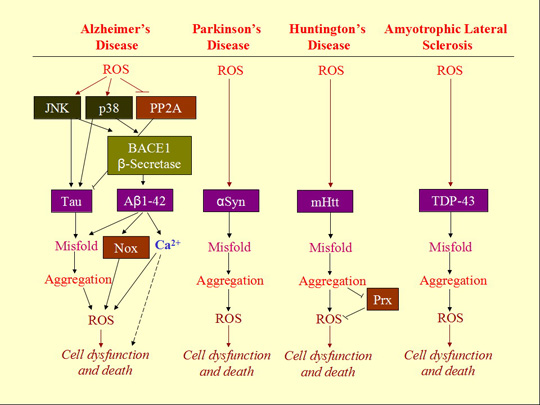
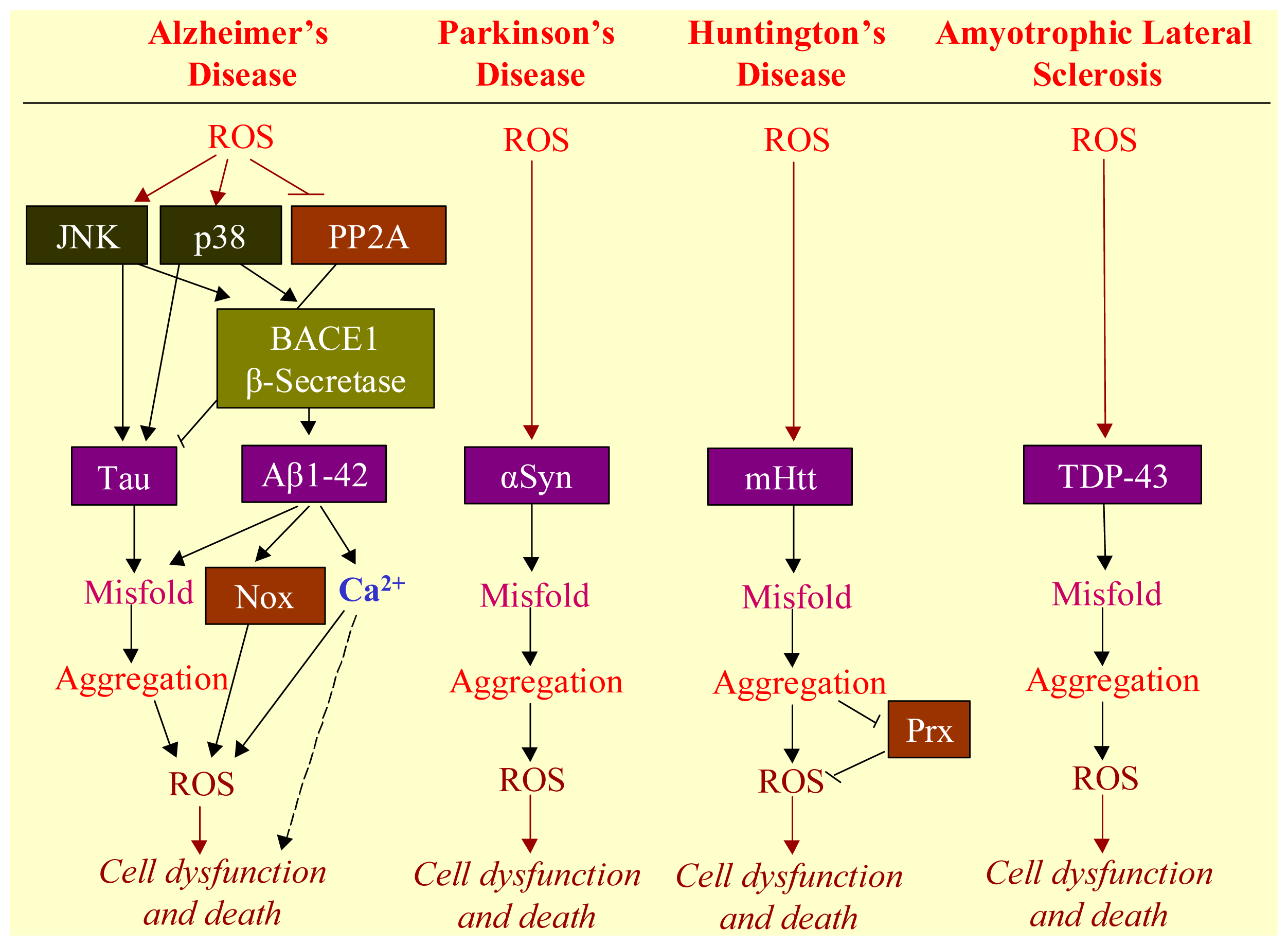
3. ROS Mediates Neurodegenerative Diseases
3.1. Neurodegenerative Diseases
3.2. Aggregation of Misfolded Proteins as the Hallmark of Neurodegenerative Diseases
3.3. Involvement of ROS in Misfolded Protein Formation
3.4. ROS-Bridging Misfolded Proteins and Cell Death
3.5. ER Stress Bridging Misfolded Proteins and Cell Death
4. ROS Mediated Chemotherapy-Induced Neuropathy
4.1. The Role of ROS in Chemobrain
4.2. The Role of ROS in Chemotheray-Induced Peripheral Neuropathy (CIPN)
5. Antioxidation Is a Strategy to Block the Progress of Neurodegenerative Diseases and Chemotherapy-Induced Chemobrain and Peripheral Neuropathy
5.1. Antioxidative Treatment for Neurodegenerative Diseases
5.2. Antioxidative Treatment for Treatment of Chemobrain and CIPN
6. Methods to Determine Cellular Redox Status and Screen Antioxidants
6.1. Methods to Evaluate the Involvement of ROS and/or RNS in Physiological or Pathophysiological Processes
6.2. Methods to Determine ROS and RNS Generations
6.2.1. Fluorescent and Chemiluminescent Probes for ROS
6.2.2. Chromatography without or with Mass Spectrometry
6.2.3. Electron Paramagnetic Resonance with Spin Trap Technique
6.2.4. Fluorescent and Chemiluminescent Probes for RNS
6.2.5. Activity of Enzymes in RNS Generation
6.3. Methods to Measure the Strength of Antioxidant Systems
6.3.1. Fluorescent Probes for Glutathione
6.3.2. Activity Measurement of Enzymatic Antioxidants
6.4. Method for Identification of Free Radical and Nitric Oxide Scavenger
6.4.1. Method for Identification of Free Radical Scavenger
6.4.2. Method for Identification of Nitric Oxide Scavenger
6.5. Method for Identification of Oxidized or Nitrated Large Molecules
6.5.1. Method for Identification of Oxidized Large Molecules
6.5.2. Method for Identification of Nitrated Large Molecules
7. Concluding Remarks and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol 2003, 552, 335–344. [Google Scholar]
- Coon, M.J.; Ding, X.; Pernecky, S.J.; Vaz, A.D.N. Cytochrome P450: Progress and predictions. FASEB J 1992, 6, 669–673. [Google Scholar]
- Reed, J.R.; Backes, W.L. Formation of P450·P450 complexes and their effect on P450 function. Pharmacol. Ther 2012, 133, 299–310. [Google Scholar]
- DeLeo, F.R.; Quinn, M.T. Assembly of the phagocyte NADPH oxidase: Molecular interaction of oxidase proteins. J. Leukoc. Biol 1996, 60, 677–691. [Google Scholar]
- Finkel, T. Redox-dependent signal transduction. FEBS Lett 2000, 476, 52–54. [Google Scholar]
- Gandhi, S.; Abramov, A.Y. Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell. Longev 2012, 2012, 428010. [Google Scholar]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev 1979, 59, 527–605. [Google Scholar]
- Packer, M.A.; Porteous, C.M.; Murphy, M.P. Superoxide production by mitochondria in the presence of nitric oxide forms peroxynitrite. Biochem. Mol. Biol. Int 1996, 40, 527–534. [Google Scholar]
- Bringold, U.; Ghafourifar, P.; Richter, C. Peroxynitrite formed by mitochondrial NO synthase promotes mitochondrial Ca2+ release. Free Radic. Biol. Med 2000, 29, 343–348. [Google Scholar]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol 2007, 39, 44–84. [Google Scholar]
- Stanley, B.A.; Sivakumaran, V.; Shi, S.; McDonald, I.; Lloyd, D.; Watson, W.H.; Aon, M.A.; Paolocci, N. Thioredoxin reductase-2 is essential for keeping low levels of H2O2 emission from isolated heart mitochondria. J. Biol. Chem 2011, 286, 33669–33677. [Google Scholar]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med 2013. [Google Scholar] [CrossRef]
- Massaad, C.A.; Klann, E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid. Redox Signal 2011, 14, 2013–2054. [Google Scholar]
- Rao, R.K.; Clayton, L.W. Regulation of protein phosphatase 2A by hydrogen peroxide and glutathionylation. Biochem. Biophys. Res. Commun 2002, 293, 610–616. [Google Scholar]
- O’Loghlen, A.; Pérez-Morgado, M.I.; Salinas, M.; Martín, M.E. Reversible inhibition of the protein phosphatase 1 by hydrogen peroxide. Potential regulation of eIF2 alpha phosphorylation in differentiated PC12 cells. Arch. Biochem. Biophys 2003, 417, 194–202. [Google Scholar]
- Groeger, G.; Quiney, C.; Cotter, T.G. Hydrogen peroxide as a cell-survival signaling molecule. Antioxid. Redox Signal 2009, 11, 2655–2671. [Google Scholar]
- Gamou, S.; Shimizu, N. Hydrogen peroxide preferentially enhances the tyrosine phosphorylation of epidermal growth factor receptor. FEBS Lett 1995, 357, 161–164. [Google Scholar]
- Goldkorn, T.; Balaban, N.; Matsukuma, K.; Chea, V.; Gould, R.; Last, J.; Chan, C.; Chavez, C. EGF-Receptor phosphorylation and signaling are targeted by H2O2 redox stress. Am. J. Respir. Cell. Mol. Biol 1998, 19, 786–798. [Google Scholar]
- Catarzi, S.; Romagnoli, C.; Marcucci, G.; Favilli, F.; Iantomasi, T.; Vincenzini, M.T. Redox regulation of ERK1/2 activation induced by sphingosine 1-phosphate in fibroblasts: Involvement of NADPH oxidase and platelet-derived growth factor receptor. Biochim. Biophys. Acta 2011, 1810, 446–456. [Google Scholar]
- McCubrey, J.A.; LaHair, M.M.; Franklin, R.A. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid. Redox Signal 2006, 8, 1775–1789. [Google Scholar]
- Whisler, R.L.; Goyette, M.A.; Grants, I.S.; Newhouse, Y.G. Sublethal levels of oxidant stress stimulate multiple serine/threonine kinases and suppress protein phosphatases in Jurkat T cells. Arch. Biochem. Biophys 1995, 319, 23–25. [Google Scholar]
- Brookmeyer, R.; Gray, S.; Kawas, C. Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am. J. Public Health 1998, 88, 1337–1342. [Google Scholar]
- Choi, T.G.; Lee, J.; Ha, J.; Kim, S.S. Apoptosis signal-regulating kinase 1 is an intracellular inducer of p38 MAPK-mediated myogenic signalling in cardiac myoblasts. Biochim. Biophys. Acta 2011, 1813, 1412–1421. [Google Scholar]
- Zhou, Y.; Yan, H.; Guo, M.; Zhu, J.; Xiao, Q.; Zhang, L. Reactive oxygen species in vascular formation and development. Oxid. Med. Cell. Longev 2013, 2013, 374963. [Google Scholar]
- Keisari, Y.; Braun, L.; Flescher, E. The oxidative burst and related phenomena in mouse macrophages elicited by different sterile inflammatory stimuli. Immunobiology 1983, 165, 78–89. [Google Scholar]
- Weisskopf, M.G.; Castillo, P.E.; Zalutsky, R.A.; Nicoll, R.A. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science 1994, 265, 1878–1882. [Google Scholar]
- Ito, M. Long term depression. Ann. Rev. Neurosci 1989, 12, 85–102. [Google Scholar]
- Mulkey, R.M.; Malenka, R.C. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron 1992, 9, 967–975. [Google Scholar]
- Villers, A.; Godaux, E.; Ris, L. Long-lasting LTP requires neither repeated trains for its induction nor protein synthesis for its development. PLoS One 2012, 7, e40823. [Google Scholar]
- Lau, C.G.; Takeuchi, K.; Rodenas-Ruano, A.; Takayasu, Y.; Murphy, J.; Bennett, M.V.; Zukin, R.S. Regulation of NMDA receptor Ca2+ signalling and synaptic plasticity. Biochem. Soc. Trans 2009, 37, 1369–1374. [Google Scholar]
- Kiritoshi, T.; Ikeda, H.; Murase, K. Long-term potentiation of neuronal excitation in the central nucleus of the rat amygdala revealed by imaging with a voltage-sensitive dye. Brain Res 2010, 1349, 32–40. [Google Scholar]
- Klann, E.; Chen, S.-J.; Sweatt, J.D. Persistent protein kinase activation in the maintenance phase of long-term potentiation. J. Biol. Chem 1991, 266, 24253–24256. [Google Scholar]
- Klann, E.; Chen, S.-J.; Sweatt, J.D. Mechanism of protein kinase C activation during the induction and maintenance of long-term potentiation probed using a novel peptide substrate. Proc. Natl. Acad. Sci. USA 1993, 90, 8337–8341. [Google Scholar]
- Sacktor, T.C.; Osten, P.; Valsamis, H.; Jiang, X.; Naik, M.U.; Sublette, E. Persistent activation of the ζ isoform of proten kinase C in the maintenance of long-term potentiation. Proc. Natl. Acad. Sci. USA 1993, 90, 8342–8346. [Google Scholar]
- Fukanaga, K.; Stoppini, L.; Miyamoto, E.; Muller, D. Long-term potentiation is associated with an increased activity of Ca2+/calmodulindependent protein kinase II. J. Biol. Chem 1993, 268, 7863–7867. [Google Scholar]
- Barria, A.; Muller, D.; Derkach, V.; Griffith, L.C.; Soderling, T.R. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science 1997, 276, 2042–2045. [Google Scholar]
- Ouyang, Y.; Kantor, D.; Harris, K.M.; Schuman, E.M.; Kennedy, M.B. Visualization of the distribution of autophosphorylated calcium/calmodulin-dependent protein kinase II after tetanic stimulation in the CA1 area of the hippocampus. J. Neurosci 1997, 17, 5416–5427. [Google Scholar]
- Blitzer, R.D.; Connor, J.H.; Brown, G.P.; Wong, T.; Shenolikar, S.; Iyengar, R.; Landau, E.M. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science 1998, 280, 1940–1942. [Google Scholar]
- Malenka, R.C.; Bear, M.F. LTP and LTD: An embarrassment of riches. Neuron 2004, 44, 5–21. [Google Scholar]
- Serrano, P.; Yao, Y.; Sacktor, T. Persistent phosphorylation by protein kinase Mzeta maintains late-phase long-term potentiation. J. Neurosci 2005, 25, 1979–1984. [Google Scholar]
- Pastalkova, E.; Serrano, P.; Pinkhasova, D.; Wallace, E.; Fenton, A.; Sacktor, T. Storage of spatial information by the maintenance mechanism of LTP. Science 2006, 313, 1141–1144. [Google Scholar]
- Anwyl, R. Metabotropic glutamate receptor-dependent long-term potentiation. Neuropharmacology 2009, 56, 735–740. [Google Scholar]
- Skeberdis, V.A.; Lan, J.; Opitz, T.; Zheng, X.; Bennett, M.V.; Zukin, R.S. mGluR1-mediated potentiation of NMDA receptors involves a rise in intracellular calcium and activation of protein kinase C. Neuropharmacology 2001, 40, 856–865. [Google Scholar]
- Bindokas, V.P.; Jordan, J.; Lee, C.C.; Miller, R.J. Superoxide production in rat hippocampal neurons: Selective imaging with hydroethidine. J. Neurosci 1996, 16, 1324–1336. [Google Scholar]
- Klann, E. Cell-permeable scavengers of superoxide prevent long-term potentiation in hippocampal area CA1. J. Neurophysiol 1998, 80, 452–457. [Google Scholar]
- Kishida, K.T.; Hoeffer, C.A.; Hu, D.; Pao, M.; Holland, S.M.; Klann, E. Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol. Cell. Biol 2006, 26, 5908–5920. [Google Scholar]
- Klann, E.; Roberson, E.D.; Knapp, L.T.; Sweatt, J.D. A role for superoxide in protein kinase C activation and the induction of long-term potentiation. J. Biol. Chem 1998, 273, 4516–4522. [Google Scholar]
- Klann, E.; Thiels, E. Modulation of protein kinases and protein phosphatases by reactive oxygen species: Implications for hippocampal synaptic plasticity. Prog. Neuropsychopharmacol. Biol. Psychiatry 1999, 23, 359–376. [Google Scholar]
- Knapp, L.T.; Klann, E. Potentiation of hippocampal synaptic transmission by superoxide requires the oxidative activation of protein kinase C. J. Neurosci 2002, 22, 674–683. [Google Scholar]
- Huddleston, A.T.; Tang, W.; Takeshima, H.; Hamilton, S.L.; Klann, E. Superoxide-induced potentiation in the hippocampus requires activation of ryanodine receptor type 3 and ERK. J. Neurophysiol 2008, 99, 1565–1571. [Google Scholar]
- Perry, T.L.; Godin, D.V.; Hansen, S. Parkinson’s disease: A disorder due to nigral glutathione deficiency? Neurosci. Lett 1982, 33, 305–310. [Google Scholar]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol 1994, 36, 348–355. [Google Scholar]
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother 2003, 57, 145–155. [Google Scholar]
- Lü, J.M.; Lin, P.H.; Yao, Q.; Chen, C. Chemical and molecular mechanisms of antioxidants: Experimental approaches and model systems. J. Cell. Mol. Med 2010, 14, 840–860. [Google Scholar]
- Dizdaroglu, M. Quantitative determination of oxidative base damage in DNA by stable isotope-dilution mass spectrometry. FEBS Lett 1993, 315, 1–6. [Google Scholar]
- Cooke, M.S.; Olinski, R.; Evans, M.D. Does measurement of oxidative damage to DNA have clinical significance? Clin. Chim. Acta 2006, 365, 30–49. [Google Scholar]
- Hu, J.; de Souza-Pinto, N.C.; Haraguchi, K.; Hogue, B.A.; Jaruga, P.; Greenberg, M.M.; Dizdaroglu, M.; Bohr, V.A. Repair of formamidopyrimidines in DNA involves different glycosylases: Role of the OGG1, NTH1, and NEIL1 enzymes. J. Biol. Chem 2005, 280, 40544–40551. [Google Scholar]
- Castellani, R.J.; Nunomura, A.; Rolston, R.K.; Moreira, P.I.; Takeda, A.; Perry, G.; Smith, M.A. Sublethal RNA oxidation as a mechanism for neurodegenerative disease. Int. J. Mol. Sci 2008, 9, 789–806. [Google Scholar]
- Katzman, R. Alzheimer’s disease. N. Engl. J. Med 1986, 314, 964–973. [Google Scholar]
- Smith, M.A. Alzheimer’s disease. Int. Rev. Neurobiol 1998, 42, 1–54. [Google Scholar]
- The World Health Report 2000—Health systems: Improving performance. Available online: http://www.who.int/whr/2000/en/ (accessed on 8 January 2009).
- Obeso, J.A.; Rodríguez-Oroz, M.C.; Benitez-Temino, B.; Blesa, F.J.; Guridi, J.; Marin, C.; Rodriguez, M. Functional organization of the basal ganglia: Therapeutic implications for Parkinson’s disease. Mov. Disord 2008, 3, S548–S559. [Google Scholar]
- Aylward, E.H.; Sparks, B.F.; Field, K.M.; Yallapragada, V.; Shpritz, B.D.; Rosenblatt, A.; Brandt, J.; Gourley, L.M.; Liang, K.; Zhou, H.; et al. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology 2004, 63, 66–72. [Google Scholar]
- Kipps, C.M.; Duggins, A.J.; Mahant, N.; Gomes, L.; Ashburner, J.; McCusker, E.A. Progression of structural neuropathology in preclinical Huntington’s disease: A tensor based morphometry study. J. Neurol. Neurosurg. Psychiatry 2005, 76, 650–655. [Google Scholar]
- Neymotin, A.; Petri, S.; Calingasan, N.Y.; Wille, E.; Schafer, P.; Stewart, C.; Hensley, K.; Beal, M.F.; Kiaei, M. Lenalidomide (Revlimid) administration at symptom onset is neuroprotective in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol 2009, 220, 191–197. [Google Scholar]
- Hashimoto, M.; Rockenstein, E.; Crews, L.; Masliah, E. Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer’s and Parkinson’s diseases. Neuromol. Med 2003, 4, 21–36. [Google Scholar]
- Priller, C.; Bauer, T.; Mitteregger, G.; Krebs, B.; Kretzschmar, H.A.; Herms, J. Synapse formation and function is modulated by the amyloid precursor protein. J. Neurosci 2006, 26, 7212–7221. [Google Scholar]
- Turner, P.R.; O’Connor, K.; Tate, W.P.; Abraham, W.C. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog. Neurobiol 2003, 70, 1–32. [Google Scholar]
- Hooper, N.M. Roles of proteolysis and lipid rafts in the processing of the amyloid precursor protein and prion protein. Biochem. Soc. Trans 2005, 33, 335–338. [Google Scholar]
- Tiraboschi, P.; Hansen, L.A.; Thal, L.J.; Corey-Bloom, J. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology 2004, 62, 1984–1989. [Google Scholar]
- Ohnishi, S.; Takano, K. Amyloid fibrils from the viewpoint of protein folding. Cell. Mol. Life Sci 2004, 61, 511–524. [Google Scholar]
- Xiang, W.; Schlachetzki, J.C.; Helling, S.; Bussmann, J.C.; Berlinghof, M.; Schäffer, T.E.; Marcus, K.; Winkler, J.; Klucken, J.; Becker, C.M. Oxidative stress-induced posttranslational modifications of alpha-synuclein: Specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol. Cell. Neurosci 2013, 54, 71–83. [Google Scholar]
- Zhou, M.; Xu, S.; Mi, J.; Uéda, K.; Chan, P. Nuclear translocation of alpha-synuclein increases susceptibility of MES23.5 cells to oxidative stress. Brain Res 2013, 1500, 19–27. [Google Scholar]
- De Vos, K.J.; Grierson, A.J.; Ackerley, S.; Miller, C.C. Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci 2008, 31, 151–173. [Google Scholar]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar]
- Su, B.; Wang, X.; Lee, H.G.; Tabaton, M.; Perry, G.; Smith, M.A.; Zhu, X. Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci. Lett 2010, 468, 267–271. [Google Scholar]
- Gómez-Ramos, A.; Díaz-Nido, J.; Smith, M.A.; Perry, G.; Avila, J. Effect of the lipid peroxidation product acrolein on tau phosphorylation in neural cells. J. Neurosci. Res 2003, 71, 863–870. [Google Scholar]
- Perez, M.; Cuadros, R.; Smith, M.A.; Perry, G.; Avila, J. Phosphorylated, but not native, tau protein assembles following reaction with the lipid peroxidation product, 4-hydroxy-2-nonenal. FEBS Lett 2000, 486, 270–274. [Google Scholar]
- Wataya, T.; Nunomura, A.; Smith, M.A.; Siedlak, S.L.; Harris, P.L.R.; Shimohama, S.; Szweda, L.I.; Kaminski, M.A.; Avila, J.; Price, D.L.; et al. High molecular weight neurofilament proteins are physiological substrates of adduction by the lipid peroxidation product hydroxynonenal. J. Biol. Chem 2002, 277, 4644–4648. [Google Scholar]
- Liu, Q.; Smith, M.A.; Avilá, J.; DeBernardis, J.; Kansal, M.; Takeda, A.; Zhu, X.; Nunomura, A.; Honda, K.; Moreira, P.I.; et al. Alzheimer-specific epitopes of tau represent lipid peroxidation-induced conformations. Free Radic. Biol. Med 2005, 38, 746–754. [Google Scholar]
- Misonou, H.; Morishima-Kawashima, M.; Ihara, Y. Oxidative stress induces intracellular accumulation of amyloid beta-protein (Abeta) in human neuroblastoma cells. Biochemistry 2000, 39, 6951–6959. [Google Scholar]
- Gabuzda, D.; Busciglio, J.; Chen, L.B.; Matsudaira, P.; Yankner, B.A. Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J. Biol. Chem 1994, 269, 13623–13628. [Google Scholar]
- Apelt, J.; Bigl, M.; Wunderlich, P.; Schliebs, R. Agingrelated increase in oxidative stress correlates with developmental pattern of beta-secretase activity and beta-amyloid plaque formation in transgenic Tg2576 mice with Alzheimerlike pathology. Int. J. Dev. Neurosci 2004, 22, 475–484. [Google Scholar]
- Ghiso, J.; Frangione, B. Cerebral amyloidosis, amyloid angiopathy, and their relationship to stroke and dementia. J. Alzheimers Dis 2001, 3, 65–73. [Google Scholar]
- Coma, M.; Guix, F.X.; Ill-Raga, G.; Uribesalgo, I.; Alameda, F.; Valverde, M.A.; Muñoz, F.J. Oxidative stress triggers the amyloidogenic pathway in human vascular smooth muscle cells. Neurobiol. Aging 2008, 29, 969–980. [Google Scholar]
- Nekooki-Machida, Y.; Kurosawa, M.; Nukina, N.; Ito, K.; Oda, T.; Tanaka, M. Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 9679–9684. [Google Scholar]
- Mitomi, Y.; Nomura, T.; Kurosawa, M.; Nukina, N.; Furukawa, Y. Post-aggregation oxidation of mutant huntingtin controls the interactions between aggregates. J. Biol. Chem 2012, 287, 34764–34775. [Google Scholar]
- Goswami, A.; Dikshit, P.; Mishra, A.; Mulherkar, S.; Nukina, N.; Jana, N.R. Oxidative stress promotes mutant huntingtin aggregation and mutant huntingtin-dependent cell death by mimicking proteasomal malfunction. Biochem. Biophys. Res. Commun 2006, 342, 184–190. [Google Scholar]
- Sorolla, M.A.; Reverter-Branchat, G.; Tamarit, J.; Ferrer, I.; Ros, J.; Cabiscol, E. Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic. Biol. Med 2008, 45, 667–678. [Google Scholar]
- Cohen, T.J.; Hwang, A.W.; Unger, T.; Trojanowski, J.Q.; Lee, V.M. Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. EMBO J 2012, 31, 1241–1252. [Google Scholar]
- Chang, C.K.; Chiang, M.H.; Toh, E.K.; Chang, C.F.; Huang, T.H. Molecular mechanism of oxidation-induced TDP-43 RRM1 aggregation and loss of function. FEBS Lett 2013, 587, 575–582. [Google Scholar]
- Behl, C.; Davies, J.B.; Lesley, R.; Schubert, D. Hydrogen peroxide mediates amyloid protein toxicity. Cell 1994, 77, 817–827. [Google Scholar]
- Bush, A.I.; Masters, C.L.; Tanzi, R.E. Copper, beta-amyloid, and Alzheimer’s disease: Tapping a sensitive connection. Proc. Natl. Acad. Sci. USA 2003, 100, 11193–11194. [Google Scholar]
- Behl, C.; Moosmann, B. Oxidative nerve cell death in Alzheimer’s disease and stroke: Antioxidants as neuroprotective compounds. Biol. Chem 2002, 383, 521–536. [Google Scholar]
- Lynch, T.; Cherny, R.A.; Bush, A.I. Oxidative processes in Alzheimer’s disease: The role of abeta-metal interactions. Exp. Gerontol 2000, 35, 445–451. [Google Scholar]
- Mark, R.J.; Hensley, K.; Butterfield, D.A.; Mattson, M.P. Amyloid beta-peptide impairs ion-motive ATPase activities: Evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J. Neurosci 1995, 15, 6239–6249. [Google Scholar]
- Mattson, M.P.; Chan, S.L. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium 2003, 34, 385–397. [Google Scholar]
- Kawahara, M.; Kuroda, Y. Molecular mechanism of neurodegeneration induced by Alzheimer’s beta-amyloid protein: Channel formation and disruption of calcium homeostasis. Brain Res. Bull 2000, 53, 389–397. [Google Scholar]
- Kagan, B.L.; Hirakura, Y.; Azimov, R.; Azimova, R.; Lin, M.C. The channel hypothesis of Alzheimer’s disease: Current status. Peptides 2002, 23, 1311–1315. [Google Scholar]
- Ma, T.; Hoeffer, C.A.; Wong, H.; Massaad, C.A.; Zhou, P.; Iadecola, C.; Murphy, M.P.; Pautler, R.G.; Klann, E. Amyloid β-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. J. Neurosci 2011, 31, 5589–5595. [Google Scholar]
- Dumont, M.; Wille, E.; Stack, C.; Calingasan, N.Y.; Beal, M.F.; Lin, M.T. Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer’s disease. FASEB J 2009, 23, 2459–2466. [Google Scholar]
- Massaad, C.A.; Washington, T.M.; Pautler, R.G.; Klann, E. Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 13576–13581. [Google Scholar]
- Pitts, A.; Dailey, K.; Newington, J.T.; Chien, A.; Arseneault, R.; Cann, T.; Thompson, L.M.; Cumming, R.C. Dithiol-based compounds maintain expression of antioxidant protein peroxiredoxin 1 that counteracts toxicity of mutant huntingtin. J. Biol. Chem 2012, 287, 22717–22729. [Google Scholar]
- Braun, R.J.; Sommer, C.; Carmona-Gutierrez, D.; Khoury, C.M.; Ring, J.; Büttner, S.; Madeo, F. Neurotoxic 43-kDa TAR DNA-binding protein (TDP-43) triggers mitochondrion-dependent programmed cell death in yeast. J. Biol. Chem 2011, 286, 19958–19972. [Google Scholar]
- Duan, W.; Li, X.; Shi, J.; Guo, Y.; Li, Z.; Li, C. Mutant TAR DNA-binding protein-43 induces oxidative injury in motor neuron-like cell. Neuroscience 2010, 169, 1621–1629. [Google Scholar]
- Hashimoto, M.; Hsu, L.J.; Xia, Y.; Takeda, A.; Sisk, A.; Sundsmo, M.; Masliah, E. Oxidative stress induces amyloid-like aggregate formation of NACP/alpha-synuclein in vitro. Neuroreport 1999, 10, 717–721. [Google Scholar]
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett 2001, 297, 191–194. [Google Scholar]
- Deleidi, M.; Maetzler, W. Protein clearance mechanisms of alpha-synuclein and amyloid-beta in lewy body disorders. Int. J. Alzheimers Dis 2012, 2012, 391438. [Google Scholar]
- Harry, G.J. Microglia during development and aging. Pharmacol. Ther 2013, 139, 313–326. [Google Scholar]
- Kanemoto, S.; Wang, H. Roles of endoplasmic reticulum stress in neurodegenerative diseases. Transl. Med 2012, 2, 1000e108. [Google Scholar]
- Bhandary, B.; Marahatta, A.; Kim, H.R.; Chae, H.J. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int. J. Mol. Sci 2012, 14, 434–456. [Google Scholar]
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol 2013, 5, a013169. [Google Scholar]
- Doyle, K.M.; Kennedy, D.; Gorman, A.M.; Gupta, S.; Healy, S.J.; Samali, A. Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. J. Cell. Mol. Med 2011, 15, 2025–2039. [Google Scholar]
- Newton, H.B. Neurological complications of chemotherapy to the central nervous system. Handb. Clin. Neurol 2012, 105, 903–916. [Google Scholar]
- Van Dam, F.S.; Schagen, S.B.; Muller, M.J.; Boogerd, W.; vd Wall, E.; Droogleever Fortuyn, M.E.; Rodenhuis, S. Impairment of cognitive function in women receiving adjuvant treatment for high-risk breast cancer: high-dose versus standarddose chemotherapy. J. Natl. Cancer Inst 1998, 90, 210–218. [Google Scholar]
- Castellon, S.A.; Ganz, P.A.; Bower, J.E.; Petersen, L.; Abraham, L.; Greendale, G.A. Neurocognitive performance in breast cancer survivors exposed to adjuvant chemotherapy and tamoxifen. J. Clin. Exp. Neuropsychol 2004, 26, 955–969. [Google Scholar]
- Vardy, J.; Wefel, J.S.; Ahles, T.; Tannock, I.F.; Schagen, S.B. Cancer and cancer therapy related cognitive dysfunction: An international perspective from the Venice cognitive workshop. Ann. Oncol 2008, 91, 623–629. [Google Scholar]
- Vardy, J.; Tannock, I. Cognitive function after chemotherapy in adults with solid tumours. Crit. Rev. Oncol. Hematol 2007, 63, 183–202. [Google Scholar]
- Joly, F.; Rigal, O.; Noal, S.; Giffard, B. Cognitive dysfunction and cancer: Which consequences in terms of disease management? Psychooncology 2011, 20, 1251–1258. [Google Scholar]
- Schagen, S.B.; Muller, M.J.; Boogerd, W.; Mellenbergh, G.J.; van Dam, F.S. Change in cognitive function after chemotherapy: A prospective longitudinal study in breast cancer patients. J. Natl. Cancer Inst 2006, 98, 1742–1745. [Google Scholar]
- Ahles, T.A.; Saykin, A.J.; McDonald, B.C.; Li, Y.; Furstenberg, C.T.; Hanscom, B.S.; Mulrooney, T.J.; Schwartz, G.N.; Kaufman, P.A. Longitudinal assessment of cognitive changes associated with adjuvant treatment for breast cancer: Impact of age and cognitive reserve. J. Clin. Oncol 2010, 28, 4434–4440. [Google Scholar]
- Kreukels, B.P.; Schagen, S.B.; Ridderinkhof, K.R.; Boogerd, W.; Hamburger, H.L.; Muller, M.J.; van Dam, F.S. Effects of high-dose and conventional-dose adjuvant chemotherapy on long-term cognitive sequelae in patients with breast cancer: an electrophysiologic study. Clin. Breast Cancer 2006, 7, 67–78. [Google Scholar]
- De Ruiter, M.B.; Reneman, L.; Boogerd, W.; Veltman, D.J.; van Dam, F.S.; Nederveen, A.J.; Boven, E.; Schagen, S.B. Cerebral hyporesponsiveness and cognitive impairment 10 years after chemotherapy for breast cancer. Hum. Brain Mapp 2011, 32, 1206–1219. [Google Scholar]
- Ahles, T.A.; Saykin, A.J.; McDonald, B.C.; Furstenberg, C.T.; Cole, B.F.; Hanscom, B.S.; Mulrooney, T.J.; Schwartz, G.N.; Kaufman, P.A. Cognitive function in breast cancer patients prior to adjuvant treatment. Breast Cancer Res. Treat 2008, 110, 143–152. [Google Scholar]
- Wefel, J.S.; Saleeba, A.K.; Buzdar, A.U.; Meyers, C.A. Acute and late onset cognitive dysfunction associated with chemotherapy in women with breast cancer. Cancer 2010, 116, 3348–3356. [Google Scholar]
- Tangpong, J.; Cole, M.P.; Sultana, R.; Joshi, G.; Estus, S.; Vore, M.; St Clair, W.; Ratanachaiyavong, S.; St Clair, D.K.; Butterfield, D.A. Adriamycin-induced, TNF-alpha-mediated central nervous system toxicity. Neurobiol. Dis 2006, 23, 127–139. [Google Scholar]
- Joshi, G.; Hardas, S.; Sultana, R.; St Clair, D.K.; Vore, M.; Butterfield, D.A. Glutathione elevation by gamma-glutamyl cysteine ethyl ester as a potential therapeutic strategy for preventing oxidative stress in brain mediated by in vivo administration of adriamycin: Implication for chemobrain. J. Neurosci. Res 2007, 85, 497–503. [Google Scholar]
- Seigers, R.; Timmermans, J.; van der Horn, H.J.; de Vries, E.F.; Dierckx, R.A.; Visser, L.; Schagen, S.B.; van Dam, F.S.; Koolhaas, J.M.; Buwalda, B. Methotrexate reduces hippocampal blood vessel density and activates microglia in rats but does not elevate central cytokine release. Behav. Brain Res 2010, 207, 265–272. [Google Scholar]
- Seigers, R.; Schagen, S.B.; Beerling, W.; Boogerd, W.; van Tellingen, O.; van Dam, F.S.; Koolhaas, J.M.; Buwalda, B. Long-lasting suppression of hippocampal cell proliferation and impaired cognitive performance by methotrexate in the rat. Behav. Brain Res 2008, 186, 168–175. [Google Scholar]
- Mustafa, S.; Walker, A.; Bennett, G.; Wigmore, P.M. 5-Fluorouracil chemotherapy affects spatial working memory and newborn neurons in the adult rat hippocampus. Eur. J. Neurosci 2008, 28, 323–330. [Google Scholar]
- Madhyastha, S.; Somayaji, S.N.; Rao, M.S.; Nalini, K.; Bairy, K.L. Hippocampal brain amines in methotrexate-induced learning and memory deficit. Can. J. Physiol. Pharmacol 2002, 80, 1076–1084. [Google Scholar]
- Seigers, R.; Fardell, J.E. Neurobiological basis of chemotherapy-induced cognitive impairment: A review of rodent research. Neurosci. Biobehav. Rev 2011, 35, 729–741. [Google Scholar]
- Antioxidant Supplementation in Cancer: Potential Interactions with Conventional Chemotherapy and Radiation Therapy. Available online: http://jdc.jefferson.edu/jmbcim/12/ (accessed on 1 May 2001).
- Pirzada, N.A.; Ali, I.I.; Dafer, R.M. Fluorouracil-induced neurotoxicity. Ann. Pharmacother 2000, 34, 35–38. [Google Scholar]
- Han, R.; Yang, Y.M.; Dietrich, J.; Luebke, A.; Mayer-Pröschel, M.; Noble, M. Systemic 5-fluorouracil treatment causes a syndrome of delayed myelin destruction in the central nervous system. J. Biol 2008, 7, 12. [Google Scholar]
- Lamberti, M.; Porto, S.; Marra, M.; Zappavigna, S.; Grimaldi, A.; Feola, D.; Pesce, D.; Naviglio, S.; Spina, A.; Sannolo, N.; et al. 5-Fluorouracil induces apoptosis in rat cardiocytes through intracellular oxidative stress. J. Exp. Clin. Cancer Res 2012, 31, 60. [Google Scholar]
- Baba, Y.; Sonoda, J.I.; Hayashi, S.; Tosuji, N.; Sonoda, S.; Makisumi, K.; Nakajo, M. Reduction of oxidative stress in liver cancer patients by oral green tea polyphenol tablets during hepatic arterial infusion chemotherapy. Exp. Ther. Med 2012, 4, 452–458. [Google Scholar]
- Numazawa, S.; Sugihara, K.; Miyake, S.; Tomiyama, H.; Hida, A.; Hatsuno, M.; Yamamoto, M.; Yoshida, T. Possible involvement of oxidative stress in 5-fluorouracil-mediated myelosuppression in mice. Basic Clin. Pharmacol. Toxicol 2011, 108, 40–45. [Google Scholar]
- Miketova, P.; Kaemingk, K.; Hockenberry, M.; Pasvogel, A.; Hutter, J.; Krull, K.; Moore, I.M. Oxidative changes in cerebral spinal fluid phosphatidylcholine during treatment for acute lymphoblastic leukemia. Biol. Res. Nurs 2005, 6, 187–195. [Google Scholar]
- Caron, J.E.; Krull, K.R.; Hockenberry, M.; Jain, N.; Kaemingk, K.; Moore, I.M. Oxidative stress and executive function in children receiving chemotherapy for acute lymphoblastic leukemia. Pediatr. Blood Cancer 2009, 53, 551–556. [Google Scholar]
- Hildebrand, J. Neurological complications of cancer chemotherapy. Curr. Opin. Oncol 2006, 18, 321–324. [Google Scholar]
- NCI Report. Available online: http://www.cancer.gov/aboutnci/ncicancerbulletin/archive/2010/022310/page6 (accessed on 30 September 2013).
- Postma, T.J.; Aaronson, N.K.; Heimans, J.J.; Muller, M.J.; Hildebrand, J.G.; Delattre, J.Y.; Hoang-Xuan, K.; Lantéri-Minet, M.; Grant, R.; Huddart, R.; et al. The development of an EORTC quality of life questionnaire to assess chemotherapy-induced peripheral neuropathy: The QLQ-CIPN20. Eur. J. Cancer 2005, 41, 1135–1139. [Google Scholar]
- Tofthagen, C. Patient perceptions associated with chemotherapy-induced peripheral neuropathy. Clin. J. Oncol. Nurs 2010, 14, E22–E28. [Google Scholar]
- Raffa, R.B.; Tallarida, R.J. Effects on the visual system might contribute to some of the cognitive deficits of cancer chemotherapy-induced “chemo-fog”. J. Clin. Pharm. Ther 2010, 35, 249–255. [Google Scholar]
- Jonas, C.R.; Puckett, A.B.; Jones, D.P.; Griffith, D.P.; Szeszycki, E.E.; Bergman, G.F.; Furr, C.E.; Tyre, C.; Carlson, J.L.; Galloway, J.R.; et al. Plasma antioxidant status after high-dose chemotherapy: A randomized trial of parenteral nutrition in bone marrow transplantation patients. Am. J. Clin. Nutr 2000, 72, 181–189. [Google Scholar]
- Di Cesare Mannelli, L.; Zanardelli, M.; Failli, P.; Ghelardini, C. Oxaliplatin-induced neuropathy: Oxidative stress as pathological mechanism. Protective effect of silibinin. J. Pain 2012, 13, 276–284. [Google Scholar]
- Valério, D.A.; Georgetti, S.R.; Magro, D.A.; Casagrande, R.; Cunha, T.M.; Vicentini, F.T.; Vieira, S.M.; Fonseca, M.J.; Ferreira, S.H.; Cunha, F.Q.; et al. Quercetin reduces inflammatory pain: Inhibition of oxidative stress and cytokine production. J. Nat. Prod 2009, 72, 1975–1979. [Google Scholar]
- Jiang, Y.; Guo, C.; Vasko, M.R.; Kelley, M.R. Implications of apurinic/apyrimidinic endonuclease in reactive oxygen signaling response after cisplatin treatment of dorsal root ganglion neurons. Cancer Res 2008, 68, 6425–6434. [Google Scholar]
- Minami, S.B.; Sha, S.H.; Schacht, J. Antioxidant protection in a new animal model of cisplatin-induced ototoxicity. Hear. Res 2004, 198, 137–143. [Google Scholar]
- Waissbluth, S.; Daniel, S.J. Cisplatin-induced ototoxicity: Transporters playing a role in cisplatin toxicity. Hear. Res 2013, 299, 37–45. [Google Scholar]
- Rahman, K. Studies on free radicals, antioxidants, and co-factors. Clin. Interv. Aging 2007, 2, 219–236. [Google Scholar]
- Conte, V.; Uryu, K.; Fujimoto, S.; Yao, Y.; Rokach, J.; Longhi, L.; Trojanowski, J.Q.; Lee, V.M.; McIntosh, T.K.; Praticò, D. Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. J. Neurochem 2004, 90, 758–764. [Google Scholar]
- Sung, S.; Yao, Y.; Uryu, K.; Yang, H.; Lee, V.M.; Trojanowski, J.Q.; Praticò, D. Early vitamin E supplementation in young but not aged mice reduces Abeta levels and amyloid deposition in a transgenic model of Alzheimer’s disease. FASEB J 2004, 18, 323–325. [Google Scholar]
- Niki, E. Mechanisms and dynamics of antioxidant action of ubiquinol. Mol. Aspects Med 1997, 18, S63–S70. [Google Scholar]
- Yang, L.; Calingasan, N.Y.; Wille, E.J.; Cormier, K.; Smith, K.; Ferrante, R.J.; Beal, M.F. Combination therapy with coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson’s and Huntington’s diseases. J. Neurochem 2009, 109, 1427–1439. [Google Scholar]
- Pandya, R.S.; Zhu, H.; Li, W.; Bowser, R.; Friedlander, R.M.; Wang, X. Therapeutic neuroprotective agents for amyotrophic lateral sclerosis. Cell. Mol. Life Sci 2013. [Google Scholar] [CrossRef]
- Gopal, K.V.; Wu, C.; Shrestha, B.; Campbell, K.C.; Moore, E.J.; Gross, G.W. d-Methionine protects against cisplatin-induced neurotoxicity in cortical networks. Neurotoxicol. Teratol 2012, 34, 495–504. [Google Scholar]
- Cloven, N.G.; Re, A.; McHale, M.T.; Burger, R.A.; DiSaia, P.J.; Rose, G.S.; Campbell, K.C.; Fan, H. Evaluation of D-methionine as a cytoprotectant in cisplatin treatment of an animal model for ovarian cancer. Anticancer Res 2000, 20, 4205–4209. [Google Scholar]
- Lorito, G.; Hatzopoulos, S.; Laurell, G.; Campbell, K.C.; Petruccelli, J.; Giordano, P.; Kochanek, K.; Sliwa, L.; Martini, A.; Skarzynski, H. Dose-dependent protection on cisplatin-induced ototoxicity—An electrophysiological study on the effect of three antioxidants in the Sprague-Dawley rat animal model. Med. Sci. Monit 2011, 17, BR179–BR186. [Google Scholar]
- Gulec, M.; Oral, E.; Dursun, O.B.; Yucel, A.; Hacimuftuoglu, A.; Akcay, F.; Suleyman, H. Mirtazapine protects against cisplatin-induced oxidative stress and DNA damage in the rat brain. Psychiatry Clin. Neurosci 2013, 67, 50–58. [Google Scholar]
- Trevisan, G.; Materazzi, S.; Fusi, C.; Altomare, A.; Aldini, G.; Lodovici, M.; Patacchini, R.; Geppetti, P.; Nassini, R. Novel therapeutic strategy to prevent chemotherapy-induced persistent sensory neuropathy by TRPA1 blockade. Cancer Res 2013, 73, 3120–3131. [Google Scholar]
- CIPN: Treatment preservation and prevention are the goals. Available online: http://www.oncologynurseadvisor.com/cipn-treatment-preservation-and-prevention-are-the-goals/article/212533/ (accessed on 1 September 2011).
- Carmody, R.J.; McGowan, A.J.; Cotter, T.G. Reactive oxygen species as mediators of photoreceptor apoptosis in vitro. Exp. Cell. Res 1999, 248, 520–530. [Google Scholar]
- Friesen, C.; Fulda, S.; Debatin, K.M. Induction of CD95 ligand and apoptosis by doxorubicin is modulated by the redox state in chemosensitive- and drug-resistant tumor cells. Cell Death Differ 1999, 6, 471–480. [Google Scholar]
- Kirkland, R.A.; Franklin, J.L. Evidence for redox regulation of cytochrome C release during programmed neuronal death: Antioxidant effects of protein synthesis and caspase inhibition. J. Neurosci 2001, 21, 1949–1963. [Google Scholar]
- Jiang, Z.G.; Lu, X.C.; Nelson, V.; Yang, X.; Pan, W.; Chen, R.W.; Lebowitz, M.S.; Almassian, B.; Tortella, F.C.; Brady, R.O.; et al. A multifunctional cytoprotective agent that reduces neurodegeneration after ischemia. Proc. Natl. Acad. Sci. USA 2006, 103, 1581–1586. [Google Scholar]
- Kölker, S.; Ahlemeyer, B.; Krieglstein, J.; Hoffmann, G.F. Contribution of reactive oxygen species to 3-hydroxyglutarate neurotoxicity in primary neuronal cultures from chick embryo telencephalons. Pediatr. Res 2001, 50, 76–82. [Google Scholar]
- Shagirtha, K.; Muthumani, M.; Prabu, S.M. Melatonin abrogates cadmium induced oxidative stress related neurotoxicity in rats. Eur. Rev. Med. Pharmacol. Sci 2011, 15, 1039–1050. [Google Scholar]
- Boullerne, A.I.; Nedelkoska, L.; Benjamins, J.A. Synergism of nitric oxide and iron in killing the transformed murine oligodendrocyte cell line N20.1. J. Neurochem 1999, 72, 1050–1060. [Google Scholar]
- López-López, G.; Moreno, L.; Cogolludo, A.; Galisteo, M.; Ibarra, M.; Duarte, J.; Lodi, F.; Tamargo, J.; Perez-Vizcaino, F. Nitric oxide (NO) scavenging and NO protecting effects of quercetin and their biological significance in vascular smooth muscle. Mol. Pharmacol 2004, 65, 851–859. [Google Scholar]
- Zhang, Z.J.; Cheang, L.C.; Wang, M.W.; Lee, S.M. Quercetin exerts a neuroprotective effect through inhibition of the iNOS/NO system and pro-inflammation gene expression in PC12 cells and in zebrafish. Int. J. Mol. Med 2011, 27, 195–203. [Google Scholar]
- Hooper, D.C.; Scott, G.S.; Zborek, A.; Mikheeva, T.; Kean, R.B.; Koprowski, H.; Spitsin, S.V. Uric acid, a peroxynitrite scavenger, inhibits CNS inflammation, blood-CNS barrier permeability changes, and tissue damage in a mouse model of multiple sclerosis. FASEB J 2000, 14, 691–698. [Google Scholar]
- Kim, J.Y.; Jung, K.J.; Choi, J.S.; Chung, H.Y. Hesperetin: A potent antioxidant against peroxynitrite. Free Radic. Res 2004, 38, 761–769. [Google Scholar]
- Bass, D.A.; Parce, J.W.; Dechatelet, L.R.; Szejda, P.; Seeds, M.C.; Thomas, M. Flow cytometric studies of oxidative product formation by neutrophils: A graded response to membrane stimulation. J. Immunol 1983, 130, 1910–1917. [Google Scholar]
- Royall, J.A.; Ischiropoulos, H. Evaluation of 2′,7′-dichlorofluorescin and dihydrorhodamine 123 as fluorescent probes for intracellular H2O2 in cultured endothelial cells. Arch. Biochem. Biophys 1993, 302, 348–355. [Google Scholar]
- Rao, K.M.; Padmanabhan, J.; Kilby, D.L.; Cohen, H.J.; Currie, M.S.; Weinberg, J.B. Flow cytometric analysis of nitric oxide production in human neutrophils using dichlorofluorescein diacetate in the presence of a calmodulin inhibitor. J. Leukoc. Biol 1992, 51, 496–500. [Google Scholar]
- Myhre, O.; Andersen, J.M.; Aarnes, H.; Fonnum, F. Evaluation of the probes 2′,7′-dichlorofluorescin diacetate, luminol, and lucigenin as indicators of reactive species formation. Biochem. Pharmacol 2003, 65, 1575–1582. [Google Scholar]
- Keller, A.; Mohamed, A.; Dröse, S.; Brandt, U.; Fleming, I.; Brandes, R.P. Analysis of dichlorodihydrofluorescein and dihydrocalcein as probes for the detection of intracellular reactive oxygen species. Free Radic. Res 2004, 38, 1257–1267. [Google Scholar]
- Buxser, S.E.; Sawada, G.; Raub, T.J. Analytical and numerical techniques for evaluation of free radical damage in cultured cells using imaging cytometry and fluorescent indicators. Methods Enzymol 1999, 300, 256–275. [Google Scholar]
- Benov, L.; Sztejnberg, L.; Fridovich, I. Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic. Biol. Med 1998, 25, 826–831. [Google Scholar]
- Kohli, R.; Pan, X.; Malladi, P.; Wainwright, M.S.; Whitington, P.F. Mitochondrial reactive oxygen species signal hepatocyte steatosis by regulating the phosphatidylinositol 3-kinase cell survival pathway. J. Biol. Chem 2007, 282, 21327–21336. [Google Scholar]
- Kalyanaraman, B. Oxidative chemistry of fluorescent dyes: Implications in the detection of reactive oxygen and nitrogen species. Biochem. Soc. Trans 2011, 39, 1221–1225. [Google Scholar]
- Kalyanaraman, B.; Dranka, B.P.; Hardy, M.; Michalski, R.; Zielonka, J. HPLC-based monitoring of products formed from hydroethidine-based fluorogenic probes—The ultimate approach for intra- and extracellular superoxide detection. Biochim. Biophys. Acta 2013. [Google Scholar] [CrossRef]
- Daiber, A.; Oelze, M.; August, M.; Wendt, M.; Sydow, K.; Wieboldt, H.; Kleschyov, A.L.; Munzel, T. Detection of superoxide and peroxynitrite in model systems and mitochondria by the luminol analogue L-012. Free Radic. Res 2004, 38, 259–269. [Google Scholar]
- Faulkner, K.; Fridovich, I. Luminol and lucigenin as detectors for O2−. Free Radic. Biol. Med 1993, 15, 447–451. [Google Scholar]
- Spasojevic, I.; Liochev, S.I.; Fridovich, I. Lucigenin: Redox potential in aqueous media and redox cycling with O2− production. Arch. Biochem. Biophys 2000, 373, 447–450. [Google Scholar]
- Liochev, S.I.; Fridovich, I. Lucigenin as mediator of superoxide production: Revisited. Free Radic. Biol. Med 1998, 25, 926–928. [Google Scholar]
- Li, Y.; Zhu, H.; Kuppusamy, P.; Roubaud, V.; Zweier, J.L.; Trush, M.A. Validation of lucigenin (bis-N-methylacridinium) as a chemilumigenic probe for detecting superoxide anion radical production by enzymatic and cellular systems. J. Biol. Chem 1998, 273, 2015–2023. [Google Scholar]
- Zhao, H.; Joseph, J.; Fales, H.M.; Sokoloski, E.A.; Levine, R.L.; Vasquez-Vivar, J.; Kalyanaraman, B. Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proc. Natl. Acad. Sci. USA 2005, 102, 5727–5732. [Google Scholar]
- Kruk, I.; Bozdağ-Dündar, O.; Ertan, R.; Aboul-Enein, H.Y.; Michalska, T. Hydroxyl and superoxide radical scavenging abilities of chromonyl-thiazolidine-2,4-dione compounds. Luminescence 2009, 24, 96–101. [Google Scholar]
- Berczyński, P.; Kładna, A.; Kruk, I.; Piechowska, T.; Aboul-Enein, H.Y.; Bozdağ-Dündar, O.; Ceylan-Unlusoy, M. Antioxidant activities of some new chromonyl-2,4-thiazolidinediones and chromonyl-2,4-imidazolidinediones having chromone cores. J. Fluoresc 2013, 23, 1319–1327. [Google Scholar]
- Han, J.Y.; Takeshita, K.; Utsumi, H. Noninvasive detection of hydroxyl radical generation in lung by diesel exhaust particles. Free Radic. Biol. Med 2001, 30, 516–525. [Google Scholar]
- Utsumi, H.; Yamada, K. In vivo electron spin resonance-computed tomography/nitroxyl probe technique for non-invasive analysis of oxidative injuries. Arch. Biochem. Biophys 2003, 416, 1–8. [Google Scholar]
- Halliwell, B.; Whiteman, M. Measuring reactive species and oxidative damage in vivo and in cell culture: How should you do it and what do the results mean? Br. J. Pharmacol 2004, 142, 231–255. [Google Scholar]
- Kojima, H.; Nakatsubo, N.; Kikuchi, K.; Kawahara, S.; Kirino, Y.; Nagoshi, H.; Hirata, Y.; Nagano, T. Detection and imaging of nitric oxide with novel fluorescent indicators: Diaminofluoresceins. Anal. Chem 1998, 70, 2446–2453. [Google Scholar]
- Nakatsubo, N.; Kojima, H.; Kikuchi, K.; Nagoshi, H.; Hirata, Y.; Maeda, D.; Imai, Y.; Irimura, T.; Nagano, T. Direct evidence of nitric oxide production from bovine aortic endothelial cells using new fluorescence indicators: diaminofluoresceins. FEBS Lett 1998, 427, 263–266. [Google Scholar]
- Curtin, J.F.; Donovan, M.; Cotter, T.G. Regulation and measurement of oxidative stress in apoptosis. J. Immunol. Methods 2002, 265, 49–72. [Google Scholar]
- Ueno, T.; Urano, Y.; Kojima, H.; Nagano, T. Mechanism-based molecular design of highly selective fluorescence probes for nitrative stress. J. Am. Chem. Soc 2006, 128, 10640–10641. [Google Scholar]
- Zielonka, J.; Sikora, A.; Joseph, J.; Kalyanaraman, B. Peroxynitrite is the major species formed from different flux ratios of co-generated nitric oxide and superoxide: Direct reaction with boronate-based fluorescent probe. J. Biol. Chem 2010, 285, 14210–14216. [Google Scholar]
- Bredt, D.S.; Snyder, S.H. Isolation of nitric oxide synthetase, a calmodulinrequiring enzyme. Proc. Natl. Acad. Sci. USA 1990, 87, 682–685. [Google Scholar]
- Mayer, B.; Klatt, P.; Werner, E.R.; Schmidt, K. Molecular mechanisms of inhibition of porcine brain nitric oxide synthase by the antinociceptive drug 7-nitroindazole. Neuropharmacology 1994, 33, 1253–1259. [Google Scholar]
- Ghafourifar, P.; Richter, C. Nitric oxide synthase activity in mitochondria. FEBS Lett 1997, 418, 291–296. [Google Scholar]
- Oda, T.; Sadakata, N.; Komatsu, N.; Muramatsu, T. Specific efflux of glutathione from the basolateral membrane domain in polarized MDCK cells during ricin-induced apoptosis. J. Biochem 1999, 126, 715–721. [Google Scholar]
- Franco, R.; Cidlowski, J.A. Glutathione efflux and cell death. Antioxid. Redox Signal 2012, 17, 1694–1713. [Google Scholar]
- Kosower, E.M.; Kosower, N.S. Bromobimane probes for thiols. Methods Enzymol 1995, 251, 133–148. [Google Scholar]
- Kaul, N.; Choi, J.; Forman, H.J. Transmembrane redox signaling activates NF-kappaB in macrophages. Free Radic. Biol. Med 1998, 24, 202–207. [Google Scholar]
- Tietze, F. Enzymatic method for quantification of total and oxidized glutathione: Applications to mammalian blood and other tissues. Anal. Biochem 1969, 27, 502–522. [Google Scholar]
- Li, Y.; Maher, P.; Schubert, D. Requirement for cGMP in nerve cell death caused by glutathione depletion. J. Cell Biol 1997, 139, 1317–1324. [Google Scholar]
- Peskin, A.V.; Winterbourn, C.C. A microtiter plate assay for superoxide dismutase using a water-soluble tetrazolium salt (WST-1). Clin. Chim. Acta 2000, 293, 157–166. [Google Scholar]
- Esposito, L.; Raber, J.; Kekonius, L.; Yan, F.; Yu, G.Q.; Bien-Ly, N.; Puoliväli, J.; Scearce-Levie, K.; Masliah, E.; Mucke, L. Reduction in mitochondrial superoxide dismutase modulates Alzheimer’s disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J. Neurosci 2006, 26, 5167–5179. [Google Scholar]
- Fujita, H.; Fujishima, H.; Chida, S.; Takahashi, K.; Qi, Z.; Kanetsuna, Y.; Breyer, M.D.; Harris, R.C.; Yamada, Y.; Takahashi, T. Reduction of renal superoxide dismutase in progressive diabetic nephropathy. J. Am. Soc. Nephrol 2009, 20, 1303–1313. [Google Scholar]
- Rousar, T.; Kucera, O.; Kriváková, P.; Lotková, H.; Kandár, R.; Muzáková, V.; Cervinková, Z. Evaluation of oxidative status in acetaminophen treated rat hepatocytes in culture. Physiol. Res 2009, 58, 239–246. [Google Scholar]
- Zemlan, F.P.; Thienhaus, O.J.; Bosmann, H.B. Superoxide dismutase activity in Alzheimer’s disease: Possible mechanism for paired helical filament formation. Brain Res 1989, 476, 160–162. [Google Scholar]
- Pappolla, M.A.; Omar, R.A.; Kim, K.S.; Robakis, N.K. Immunohistochemical evidence of oxidative [corrected] stress in Alzheimer’s disease. Am. J. Pathol 1992, 140, 621–628. [Google Scholar]
- Cohen, S.M.; Olin, K.L.; Feuer, W.J.; Hjelmeland, L.; Keen, C.L.; Morse, L.S. Low glutathione reductase and peroxidase activity in age-related macular degeneration. Br. J. Ophthalmol 1994, 78, 791–794. [Google Scholar]
- Paglia, D.E.; Valentine, W.N. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J. Lab. Clin. Med 1967, 70, 158–69. [Google Scholar]
- Braidy, N.; Selvaraju, S.; Essa, M.M.; Vaishnav, R.; Al-Adawi, S.; Al-Asmi, A.; Al-Senawi, H.; Abd Alrahman Alobaidy, A.; Lakhtakia, R.; Guillemin, G.J. Neuroprotective effects of a variety of pomegranate juice extracts against MPTP-induced cytotoxicity and oxidative stress in human primary neurons. Oxid. Med. Cell. Longev 2013, 2013, 685909. [Google Scholar]
- Johansson, L.H.; Borg, L.A. A spectrophotometric method for determination of catalase activity in small tissue samples. Anal. Biochem 1988, 174, 331–336. [Google Scholar]
- Reinholz, M.M.; Haggard, J.J.; Curran, G.L.; Poduslo, J.F. Plasma pharmacokinetics, nervous system biodistribution and biostability, and spinal cord permeability at the blood-brain barrier of putrescine-modified catalase in the adult rat. Exp. Neurol 1999, 159, 191–203. [Google Scholar]
- Kraus, R.L.; Pasieczny, R.; Lariosa-Willingham, K.; Turner, M.S.; Jiang, A.; Trauger, J.W. Antioxidant properties of minocycline: neuroprotection in an oxidative stress assay and direct radical-scavenging activity. J. Neurochem 2005, 94, 819–827. [Google Scholar]
- Banerjee, S.; Chanda, A.; Ghoshal, A.; Debnath, R.; Chakraborty, S.; Saha, R.; Das, A. Nitric Oxide scavenging activity study of ethanolic extracts of Ixora coccinea from two different areas of kolkata. Asian J. Exp. Biol. Sci 2011, 2, 595–599. [Google Scholar]
- Noda, Y.; Mori, A.; Liburdy, R.; Packer, L. Melatonin and its precursors scavenge nitric oxide. J. Pineal Res 1999, 27, 159–163. [Google Scholar]
- Králová, J.; Pekarová, M.; Drábiková, K.; Jančinová, V.; Nosál’, R.; Cíž, M.; Lojek, A. The effects of dithiaden on nitric oxide production by RAW 264.7 cells. Interdiscip. Toxicol 2008, 1, 214–217. [Google Scholar]
- Lang, J.; Celotto, C.; Esterbauer, H. Quantitative determination of the lipid peroxidation product 4-hydroxynonenal by high-performance liquid chromatography. Anal. Biochem 1985, 150, 369–378. [Google Scholar]
- Goldring, C.; Casini, A.F.; Maellaro, E.; Del Bello, B.; Comporti, M. Determination of 4-hydroxynonenal by high-performance liquid chromatography with electrochemical detection. Lipids 1993, 8, 141–145. [Google Scholar]
- Fucile, C.; Marini, V.; Zuccoli, M.L.; Leone, S.; Robbiano, L.; Martelli, A.; Mattioli, F. HPLC determination of malondialdehyde as biomarker for oxidative stress: application in patients with alcohol dependence. Clin. Lab 2013, 59, 837–841. [Google Scholar]
- Yoritaka, A.; Hattori, N.; Uchida, K.; Tanaka, M.; Stadtman, E.R.; Mizuno, Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc. Natl. Acad. Sci. USA 1996, 93, 2696–2701. [Google Scholar]
- Nelson, V.M.; Dancik, C.M.; Pan, W.; Jiang, Z.G.; Lebowitz, M.S.; Ghanbari, H.A. PAN-811 inhibits oxidative stress-induced cell death of human Alzheimer’s disease-derived and age-matched olfactory neuroepithelial cells via suppression of intracellular reactive oxygen species. J. Alzheimers Dis 2009, 17, 611–619. [Google Scholar]
- Zitnanová, I.; Sumegová, K.; Simko, M.; Maruniaková, A.; Chovanová, Z.; Chavko, M.; Duracková, Z. Protein carbonyls as a biomarker of foetal-neonatal hypoxic stress. Clin. Biochem 2007, 40, 567–570. [Google Scholar]
- Yang, H.; Zhang, Y.; Pöschl, U. Quantification of nitrotyrosine in nitrated proteins. Anal. Bioanal. Chem 2010, 397, 879–886. [Google Scholar]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-hydroxy-2′-deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health C 2009, 27, 120–139. [Google Scholar]
- Hofer, T.; Seo, A.Y.; Prudencio, M.; Leeuwenburgh, C. A method to determine RNA and DNA oxidation simultaneously by HPLC-ECD: Greater RNA than DNA oxidation in rat liver after doxorubicin administration. Biol. Chem 2006, 387, 103–111. [Google Scholar]
- Ohshima, H.; Sawa, T.; Akaike, T. 8-nitroguanine, a product of nitrative DNA damage caused by reactive nitrogen species: formation, occurrence, and implications in inflammation and carcinogenesis. Antioxid. Redox Signal 2006, 8, 1033–1045. [Google Scholar]
- Sawa, T.; Tatemichi, M.; Akaike, T.; Barbin, A.; Ohshima, H. Analysis of urinary 8-nitroguanine, a marker of nitrative nucleic acid damage, by high-performance liquid chromatography-electrochemical detection coupled with immunoaffinity purification: Association with cigarette smoking. Free Radic. Biol. Med 2006, 40, 711–720. [Google Scholar]
- Halliwell, B. What nitrates tyrosine? Is nitrotyrosine specific as a biomarker of peroxynitrite formation in vivo? FEBS Lett 1997, 411, 157–160. [Google Scholar]
- Tsikas, D.; Caidahl, K. Recent methodological advances in the mass spectrometric analysis of free and protein-associated 3-nitrotyrosine in human plasma. J. Chromatogr. B 2005, 814, 1–9. [Google Scholar]




© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, J.; O, W.; Li, W.; Jiang, Z.-G.; Ghanbari, H.A. Oxidative Stress and Neurodegenerative Disorders. Int. J. Mol. Sci. 2013, 14, 24438-24475. https://doi.org/10.3390/ijms141224438
Li J, O W, Li W, Jiang Z-G, Ghanbari HA. Oxidative Stress and Neurodegenerative Disorders. International Journal of Molecular Sciences. 2013; 14(12):24438-24475. https://doi.org/10.3390/ijms141224438
Chicago/Turabian StyleLi, Jie, Wuliji O, Wei Li, Zhi-Gang Jiang, and Hossein A. Ghanbari. 2013. "Oxidative Stress and Neurodegenerative Disorders" International Journal of Molecular Sciences 14, no. 12: 24438-24475. https://doi.org/10.3390/ijms141224438
APA StyleLi, J., O, W., Li, W., Jiang, Z.-G., & Ghanbari, H. A. (2013). Oxidative Stress and Neurodegenerative Disorders. International Journal of Molecular Sciences, 14(12), 24438-24475. https://doi.org/10.3390/ijms141224438