Advanced Glycation End Product (AGE)-AGE Receptor (RAGE) System Upregulated Connexin43 Expression in Rat Cardiomyocytes via PKC and Erk MAPK Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
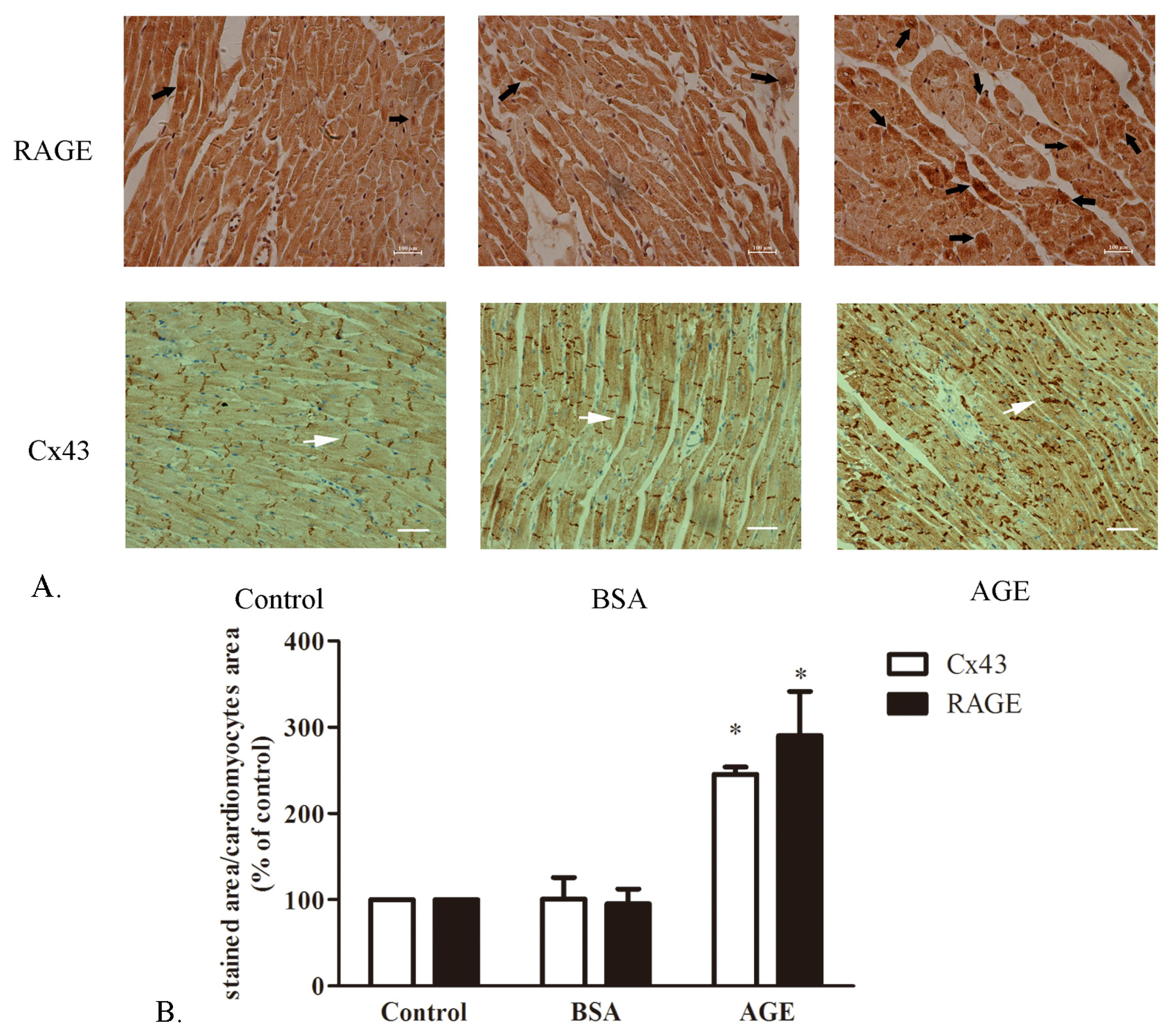
2.1.1. The Cardiac RAGE and Cx43 Expressions in AGE-Infused Rats
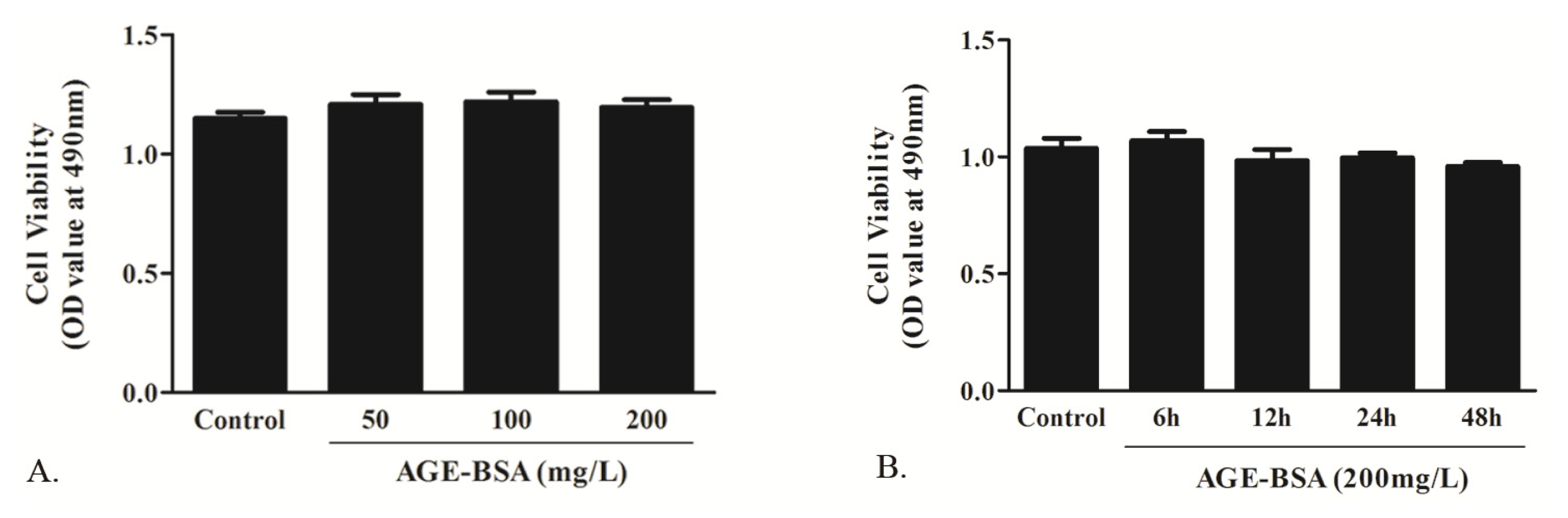
2.1.2. Effects of AGE on Cell Viability
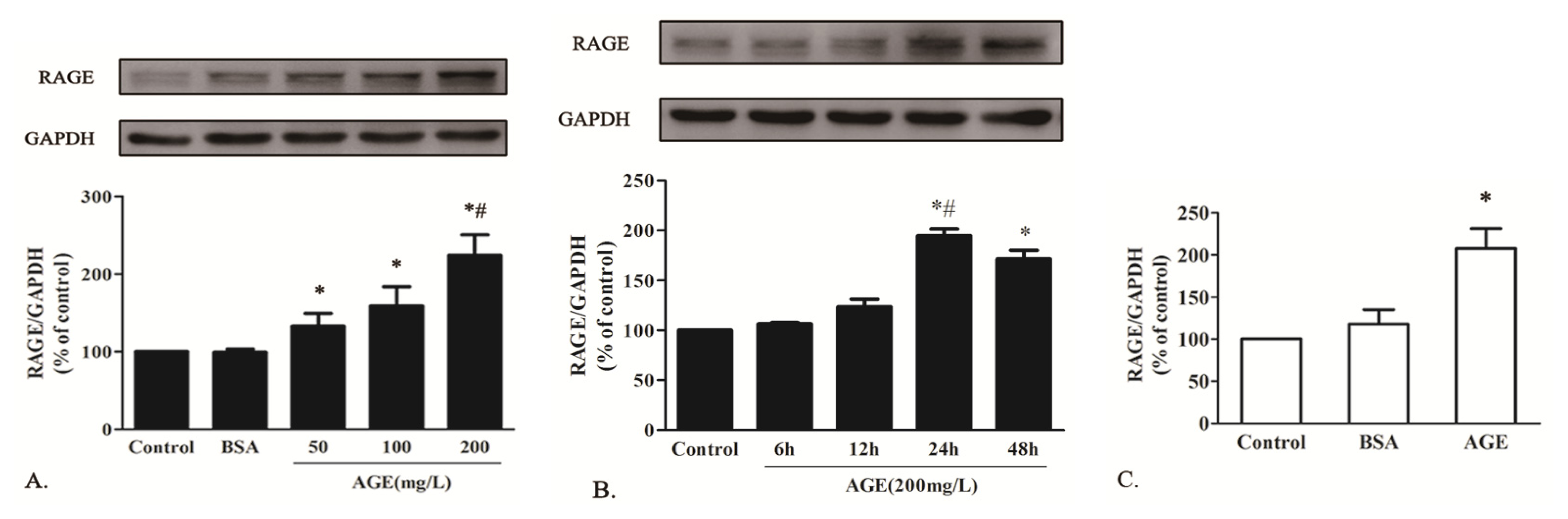
2.1.3. AGE Upregulated RAGE Expression in Cardiomyocytes
2.1.4. The Effects of AGE on Cx43 Expression and GJIC Function in Cardiomyocytes
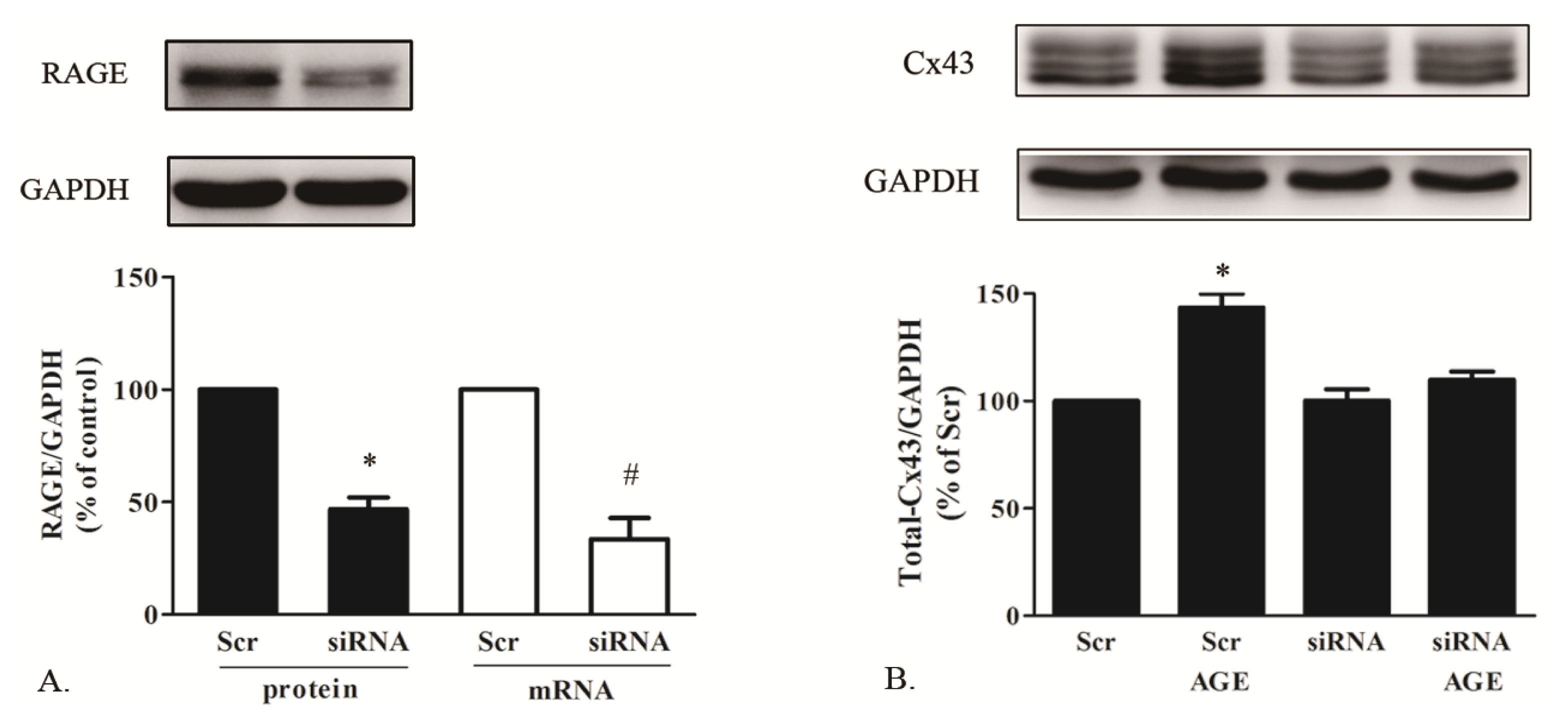
2.1.5. AGE Increased Cx43 Expression via AGE-RAGE Pathway
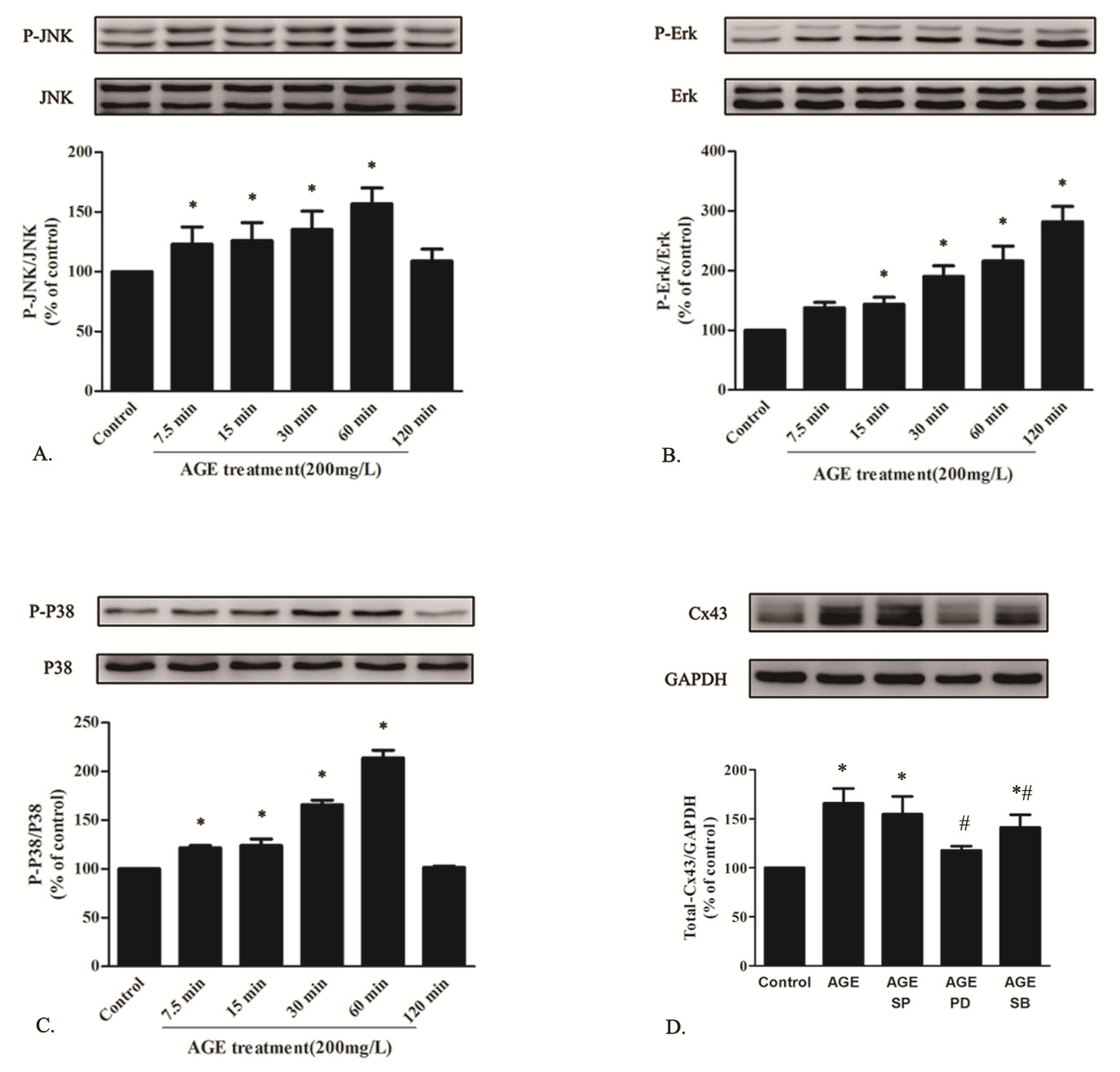
2.1.6. AGE Activated MAPK Pathway
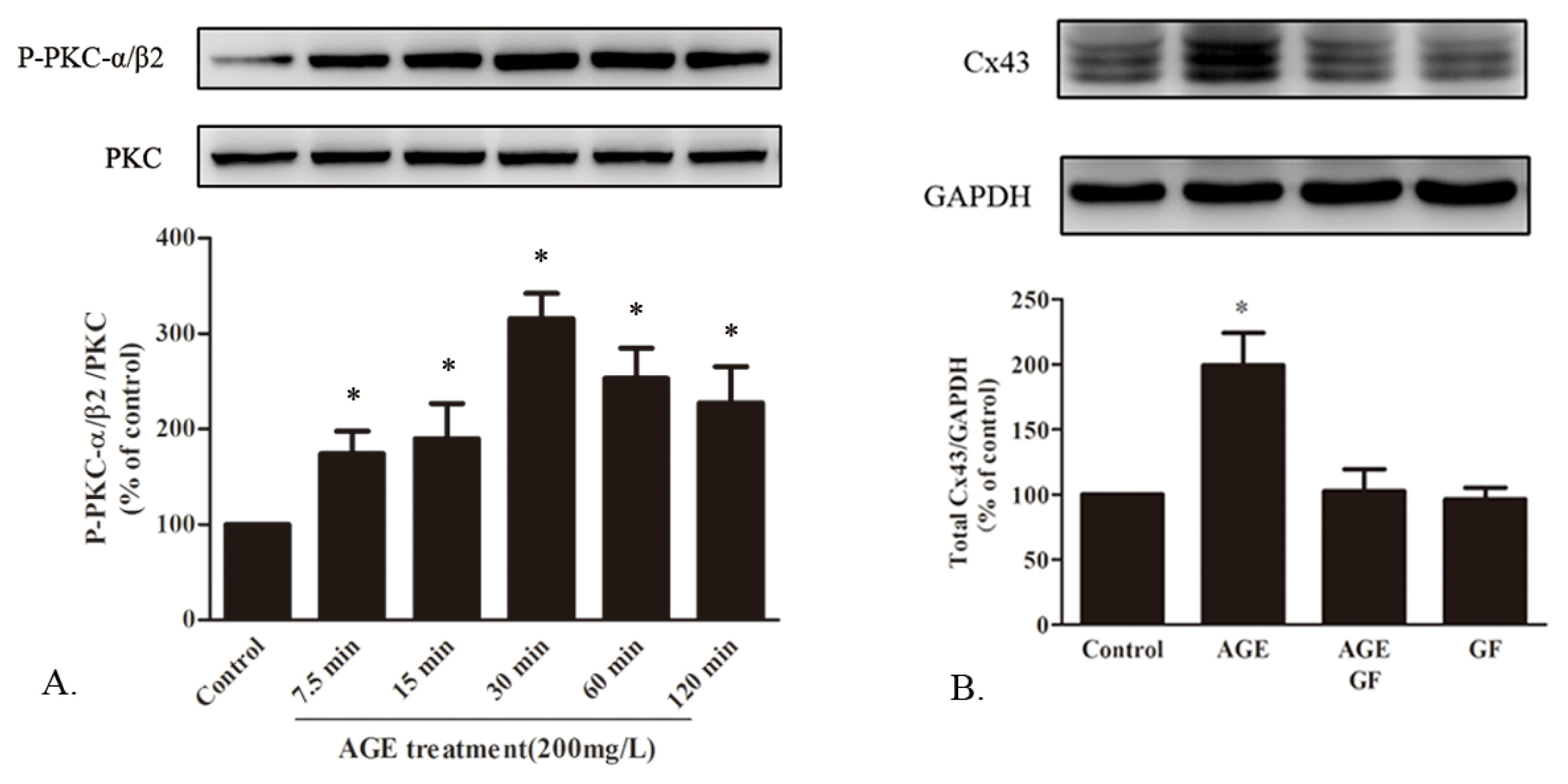
2.1.7. AGE Activated PKC Pathway
2.2. Discussion
3. Experimental Section
3.1. Chemicals and Reagents
3.2. AGE-Infused SD Rat Model
3.3. Immuohistochemisty
3.4. Cell Culture
3.5. Cell Viability Assay
3.6. Western Blot Assay
3.7. Real-Time Reverse Transcription-PCR (Real-Time RT-PCR)
3.8. Scrape Loading Dye Transfer Assay
3.9. Small Interference RNA Transfection
3.10. Statistical Analysis
4. Conclusions
Acknowledgments
References
- Howarth, F.C.; Shafiullah, M.; Adeghate, E.; Ljubisavljevic, M.; Jacobson, M. Heart rhythm disturbances in the neonatal alloxan-induced diabetic rat. Pathophysiology 2011, 18, 185–192. [Google Scholar]
- Shaw, R.M.; Rudy, Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and l-type calcium currents during reduced excitability and decreased gap junction coupling. Circ. Res 1997, 81, 727–741. [Google Scholar]
- Kleber, A.G.; Rudy, Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol. Rev 2004, 84, 431–488. [Google Scholar]
- Vozzi, C.; Dupont, E.; Coppen, S.R.; Yeh, H.I.; Severs, N.J. Chamber-related differences in connexin expression in the human heart. J. Mol. Cell. Cardiol 1999, 31, 991–1003. [Google Scholar]
- Das Evcimen, N.; King, G.L. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol. Res 2007, 55, 498–510. [Google Scholar]
- Yu, L.; Zhao, Y.; Fan, Y.; Wang, M.; Xu, S.; Fu, G. Epigallocatechin-3 gallate, a green tea catechin, attenuated the downregulation of the cardiac gap junction induced by high glucose in neonatal rat cardiomyocytes. Cell. Physiol. Biochem 2010, 26, 403–412. [Google Scholar]
- Lin, H.; Ogawa, K.; Imanaga, I.; Tribulova, N. Remodeling of connexin 43 in the diabetic rat heart. Mol. Cell. Biochem 2006, 290, 69–78. [Google Scholar]
- Howarth, F.C.; Chandler, N.J.; Kharche, S.; Tellez, J.O.; Greener, I.D.; Yamanushi, T.T.; Billeter, R.; Boyett, M.R.; Zhang, H.; Dobrzynski, H. Effects of streptozotocin-induced diabetes on connexin43 mRNA and protein expression in ventricular muscle. Mol. Cell. Biochem 2008, 319, 105–114. [Google Scholar]
- Stilli, D.; Lagrasta, C.; Berni, R.; Bocchi, L.; Savi, M.; Delucchi, F.; Graiani, G.; Monica, M.; Maestri, R.; Baruffi, S.; et al. Preservation of ventricular performance at early stages of diabetic cardiomyopathy involves changes in myocyte size, number and intercellular coupling. Basic Res. Cardiol 2007, 102, 488–499. [Google Scholar]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar]
- Candido, R.; Forbes, J.M.; Thomas, M.C.; Thallas, V.; Dean, R.G.; Burns, W.C.; Tikellis, C.; Ritchie, R.H.; Twigg, S.M.; Cooper, M.E.; et al. A breaker of advanced glycation end products attenuates diabetes-induced myocardial structural changes. Circ. Res 2003, 92, 785–792. [Google Scholar]
- Shang, L.; Ananthakrishnan, R.; Li, Q.; Quadri, N.; Abdillahi, M.; Zhu, Z.; Qu, W.; Rosario, R.; Toure, F.; Yan, S.F.; et al. RAGE modulates hypoxia/reoxygenation injury in adult murine cardiomyocytes via JNK and GSK-3beta signaling pathways. PLoS One 2010, 5, e10092. [Google Scholar]
- Petrova, R.; Yamamoto, Y.; Muraki, K.; Yonekura, H.; Sakurai, S.; Watanabe, T.; Li, H.; Takeuchi, M.; Makita, Z.; Kato, I.; et al. Advanced glycation endproduct-induced calcium handling impairment in mouse cardiac myocytes. J. Mol. Cell. Cardiol 2002, 34, 1425–1431. [Google Scholar]
- Ma, H.; Li, S.Y.; Xu, P.; Babcock, S.A.; Dolence, E.K.; Brownlee, M.; Li, J.; Ren, J. Advanced glycation endproduct (AGE) accumulation and AGE receptor (RAGE) up-regulation contribute to the onset of diabetic cardiomyopathy. J. Cell. Mol. Med 2009, 13, 1751–1764. [Google Scholar]
- Ripplinger, C.M.; Li, W.; Hadley, J.; Chen, J.; Rothenberg, F.; Lombardi, R.; Wickline, S.A.; Marian, A.J.; Efimov, I.R. Enhanced transmural fiber rotation and connexin 43 heterogeneity are associated with an increased upper limit of vulnerability in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circ. Res 2007, 101, 1049–1057. [Google Scholar]
- Rucker-Martin, C.; Milliez, P.; Tan, S.; Decrouy, X.; Recouvreur, M.; Vranckx, R.; Delcayre, C.; Renaud, J.F.; Dunia, I.; Segretain, D.; et al. Chronic hemodynamic overload of the atria is an important factor for gap junction remodeling in human and rat hearts. Cardiovasc. Res 2006, 72, 69–79. [Google Scholar]
- Nygren, A.; Olson, M.L.; Chen, K.Y.; Emmett, T.; Kargacin, G.; Shimoni, Y. Propagation of the cardiac impulse in the diabetic rat heart: Reduced conduction reserve. J. Physiol 2007, 580, 543–560. [Google Scholar]
- De Mello, W.C. Impaired cell communication in the diabetic heart. The role of the renin angiotensin system. Mol. Cell. Biochem 2007, 296, 53–58. [Google Scholar]
- Zhang, M.; Kho, A.L.; Anilkumar, N.; Chibber, R.; Pagano, P.J.; Shah, A.M.; Cave, A.C. Glycated proteins stimulate reactive oxygen species production in cardiac myocytes: Involvement of Nox2 (gp91phox)-containing NADPH oxidase. Circulation 2006, 113, 1235–1243. [Google Scholar]
- Sun, C.; Liang, C.; Ren, Y.; Zhen, Y.; He, Z.; Wang, H.; Tan, H.; Pan, X.; Wu, Z. Advanced glycation end products depress function of endothelial progenitor cells via p38 and ERK 1/2 mitogen-activated protein kinase pathways. Basic Res. Cardiol 2009, 104, 42–49. [Google Scholar]
- Giles, T.D.; Ouyang, J.; Kerut, E.K.; Given, M.B.; Allen, G.E.; McIlwain, E.F.; Greenberg, S.S. Changes in protein kinase C in early cardiomyopathy and in gracilis muscle in the BB/Wor diabetic rat. Am. J. Physiol 1998, 274, H295–H307. [Google Scholar]
- Fernandes, R.; Girao, H.; Pereira, P. High glucose down-regulates intercellular communication in retinal endothelial cells by enhancing degradation of connexin 43 by a proteasome-dependent mechanism. J. Biol. Chem 2004, 279, 27219–27224. [Google Scholar]
- Trudeau, K.; Muto, T.; Roy, S. Downregulation of mitochondrial connexin 43 by high glucose triggers mitochondrial shape change and cytochrome C release in retinal endothelial cells. Invest. Ophthalmol. Vis. Sci 2012, 53, 6675–6681. [Google Scholar]
- Liu, L.; Hu, X.; Cai, G.Y.; Lv, Y.; Zhuo, L.; Gao, J.J.; Cui, S.Y.; Feng, Z.; Fu, B.; Chen, X.M. High glucose-induced hypertrophy of mesangial cells is reversed by connexin43 overexpression via PTEN/Akt/mTOR signaling. Nephrol. Dial. Transplant 2012, 27, 90–100. [Google Scholar]
- Li, A.F.; Roy, S. High glucose-induced downregulation of connexin 43 expression promotes apoptosis in microvascular endothelial cells. Invest. Ophthalmol. Vis. Sci 2009, 50, 1400–1407. [Google Scholar]
- Bidasee, K.R.; Zhang, Y.; Shao, C.H.; Wang, M.; Patel, K.P.; Dincer, U.D.; Besch, H.R., Jr. Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes 2004, 53, 463–473. [Google Scholar]
- Lin, F.L.; Chang, C.I.; Chuang, K.P.; Wang, C.Y.; Liu, H.J. Advanced glycation end products down-regulate gap junctions in human hepatoma SKHep 1 cells via the activation of Src-dependent ERK1/2 and JNK/SAPK/AP1 signaling pathways. J. Agric. Food Chem 2010, 58, 8636–8642. [Google Scholar]
- Wang, C.Y.; Liu, H.J.; Chen, H.J.; Lin, Y.C.; Wang, H.H.; Hung, T.C.; Yeh, H.I. AGE-BSA down-regulates endothelial connexin43 gap junctions. BMC Cell Biol 2011, 12, 19. [Google Scholar]
- Rubenstein, D.A.; Maria, Z.; Yin, W. Glycated albumin modulates endothelial cell thrombogenic and inflammatory responses. J. Diabetes Sci. Technol 2011, 5, 703–713. [Google Scholar]
- Sakata, N.; Meng, J.; Takebayashi, S. Effects of advanced glycation end products on the proliferation and fibronectin production of smooth muscle cells. J. Atheroscler. Thromb 2000, 7, 169–176. [Google Scholar]
- Chen, J.; Song, M.; Yu, S.; Gao, P.; Yu, Y.; Wang, H.; Huang, L. Advanced glycation endproducts alter functions and promote apoptosis in endothelial progenitor cells through receptor for advanced glycation endproducts mediate overpression of cell oxidant stress. Mol. Cell. Biochem 2010, 335, 137–146. [Google Scholar]
- Li, J.; Liu, N.F.; Wei, Q. Effect of rosiglitazone on cardiac fibroblast proliferation, nitric oxide production and connective tissue growth factor expression induced by advanced glycation end-products. J. Int. Med. Res 2008, 36, 329–335. [Google Scholar]
- Kato, T.; Yamashita, T.; Sekiguchi, A.; Tsuneda, T.; Sagara, K.; Takamura, M.; Kaneko, S.; Aizawa, T.; Fu, L.T. AGEs-RAGE system mediates atrial structural remodeling in the diabetic rat. J. Cardiovasc. Electrophysiol 2008, 19, 415–420. [Google Scholar]
- Kang, R.; Tang, D.; Livesey, K.M.; Schapiro, N.E.; Lotze, M.T.; Zeh, H.J., III. The Receptor for Advanced Glycation End-products (RAGE) protects pancreatic tumor cells against oxidative injury. Antioxid. Redox. Signal. 2011, 15, 2175–2184. [Google Scholar]
- Li, J.; Schmidt, A.M. Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J. Biol. Chem 1997, 272, 16498–16506. [Google Scholar]
- Duffy, H.S.; Ashton, A.W.; O’Donnell, P.; Coombs, W.; Taffet, S.M.; Delmar, M.; Spray, D.C. Regulation of connexin43 protein complexes by intracellular acidification. Circ. Res 2004, 94, 215–222. [Google Scholar]
- Leithe, E.; Rivedal, E. Epidermal growth factor regulates ubiquitination, internalization and proteasome-dependent degradation of connexin43. J. Cell. Sci 2004, 117, 1211–1220. [Google Scholar]
- Jia, G.; Cheng, G.; Gangahar, D.M.; Agrawal, D.K. Involvement of connexin 43 in angiotensin II-induced migration and proliferation of saphenous vein smooth muscle cells via the MAPK-AP-1 signaling pathway. J. Mol. Cell. Cardiol 2008, 44, 882–890. [Google Scholar]
- Liang, J.Y.; Wang, S.M.; Chung, T.H.; Yang, S.H.; Wu, J.C. Effects of 18-glycyrrhetinic acid on serine 368 phosphorylation of connexin43 in rat neonatal cardiomyocytes. Cell. Biol. Int 2008, 32, 1371–1379. [Google Scholar]
- Musil, L.S.; Cunningham, B.A.; Edelman, G.M.; Goodenough, D.A. Differential phosphorylation of the gap junction protein connexin43 in junctional communication-competent and -deficient cell lines. J. Cell. Biol 1990, 111, 2077–2088. [Google Scholar]
- Kwak, B.R.; Jongsma, H.J. Regulation of cardiac gap junction channel permeability and conductance by several phosphorylating conditions. Mol. Cell. Biochem 1996, 157, 93–99. [Google Scholar]
- Beardslee, M.A.; Lerner, D.L.; Tadros, P.N.; Laing, J.G.; Beyer, E.C.; Yamada, K.A.; Kleber, A.G.; Schuessler, R.B.; Saffitz, J.E. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ. Res 2000, 87, 656–662. [Google Scholar]
- Polontchouk, L.; Ebelt, B.; Jackels, M.; Dhein, S. Chronic effects of endothelin 1 and angiotensin II on gap junctions and intercellular communication in cardiac cells. FASEB J 2002, 16, 87–89. [Google Scholar]
- Huang, Y.S.; Tseng, Y.Z.; Wu, J.C.; Wang, S.M. Mechanism of oleic acid-induced gap junctional disassembly in rat cardiomyocytes. J. Mol. Cell. Cardiol 2004, 37, 755–766. [Google Scholar]
- Zhang, F.L.; Gao, H.Q.; Shen, L. Inhibitory effect of GSPE on RAGE expression induced by advanced glycation end products in endothelial cells. J. Cardiovasc. Pharmacol 2007, 50, 434–440. [Google Scholar]
- Zheng, F.; Cai, W.; Mitsuhashi, T.; Vlassara, H. Lysozyme enhances renal excretion of advanced glycation endproducts in vivo and suppresses adverse age-mediated cellular effects in vitro: A potential AGE sequestration therapy for diabetic nephropathy? Mol. Med 2001, 7, 737–747. [Google Scholar]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Kloting, I.; et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 2001, 50, 2792–2808. [Google Scholar]
- Blondel, B.; Roijen, I.; Cheneval, J.P. Heart cells in culture: A simple method for increasing the proportion of myoblasts. Experientia 1971, 27, 356–358. [Google Scholar]







© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yu, L.; Zhao, Y.; Xu, S.; Ding, F.; Jin, C.; Fu, G.; Weng, S. Advanced Glycation End Product (AGE)-AGE Receptor (RAGE) System Upregulated Connexin43 Expression in Rat Cardiomyocytes via PKC and Erk MAPK Pathways. Int. J. Mol. Sci. 2013, 14, 2242-2257. https://doi.org/10.3390/ijms14022242
Yu L, Zhao Y, Xu S, Ding F, Jin C, Fu G, Weng S. Advanced Glycation End Product (AGE)-AGE Receptor (RAGE) System Upregulated Connexin43 Expression in Rat Cardiomyocytes via PKC and Erk MAPK Pathways. International Journal of Molecular Sciences. 2013; 14(2):2242-2257. https://doi.org/10.3390/ijms14022242
Chicago/Turabian StyleYu, Lu, Yanbo Zhao, Shengjie Xu, Fang Ding, Chongying Jin, Guosheng Fu, and Shaoxiang Weng. 2013. "Advanced Glycation End Product (AGE)-AGE Receptor (RAGE) System Upregulated Connexin43 Expression in Rat Cardiomyocytes via PKC and Erk MAPK Pathways" International Journal of Molecular Sciences 14, no. 2: 2242-2257. https://doi.org/10.3390/ijms14022242
APA StyleYu, L., Zhao, Y., Xu, S., Ding, F., Jin, C., Fu, G., & Weng, S. (2013). Advanced Glycation End Product (AGE)-AGE Receptor (RAGE) System Upregulated Connexin43 Expression in Rat Cardiomyocytes via PKC and Erk MAPK Pathways. International Journal of Molecular Sciences, 14(2), 2242-2257. https://doi.org/10.3390/ijms14022242