NADPH Oxidase Biology and the Regulation of Tyrosine Kinase Receptor Signaling and Cancer Drug Cytotoxicity

Abstract
:1. Introduction
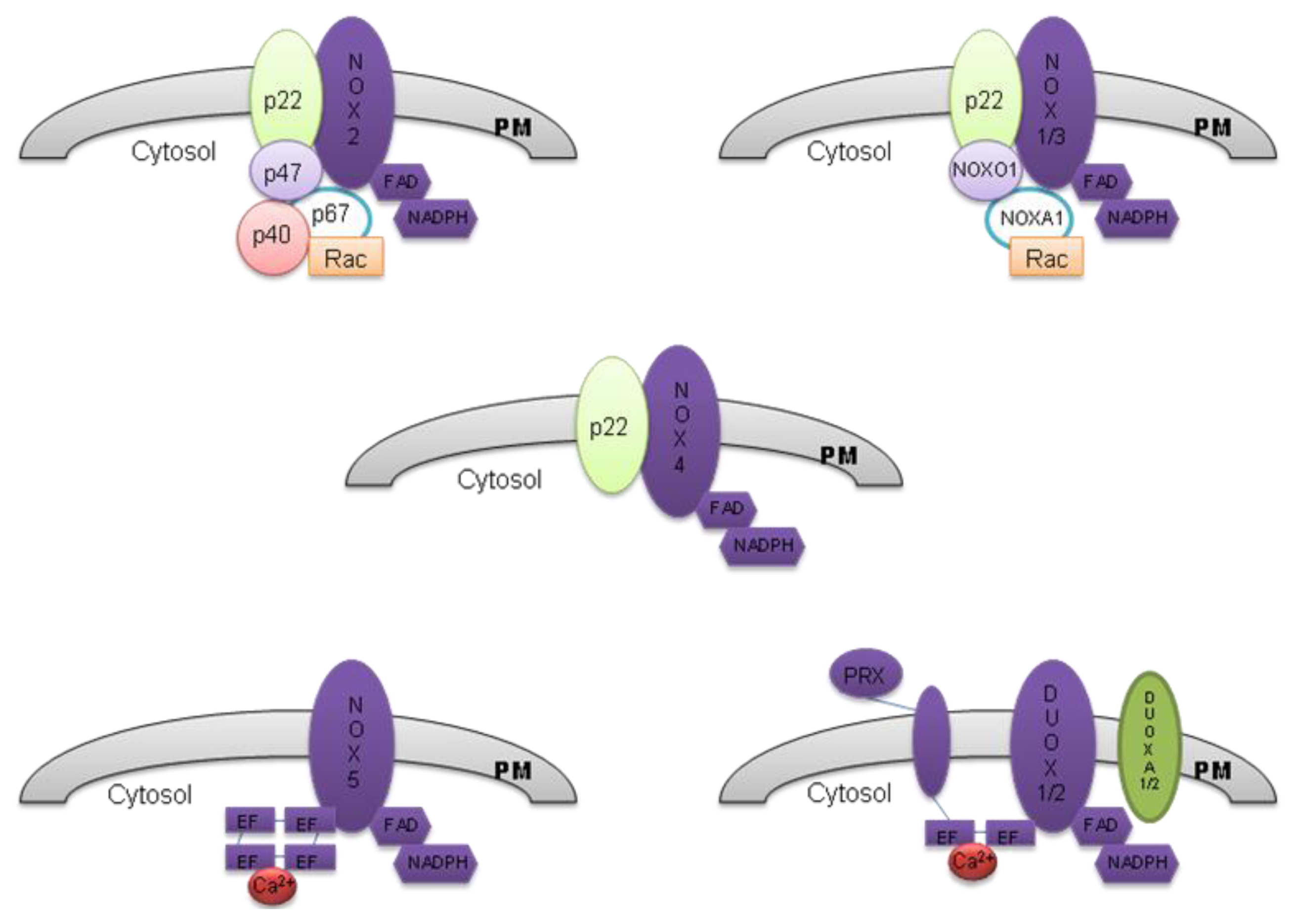
2. The NOX Family
2.1. NOX 2
2.2. NOX 1
2.3. NOX 3
2.4. NOX 4
2.5. NOX 5
2.6. DUOX 1 and DUOX 2
3. Reactive Oxygen Species
3.1. NOX-Derived ROS
4. Regulation of Cancer Cell Biology by NADPH Oxidase Activity: Implications in Hallmarks of Cancer
5. Redox-Sensitive Regulation of Signaling Pathways: NADPH-Derived ROS as Mediators
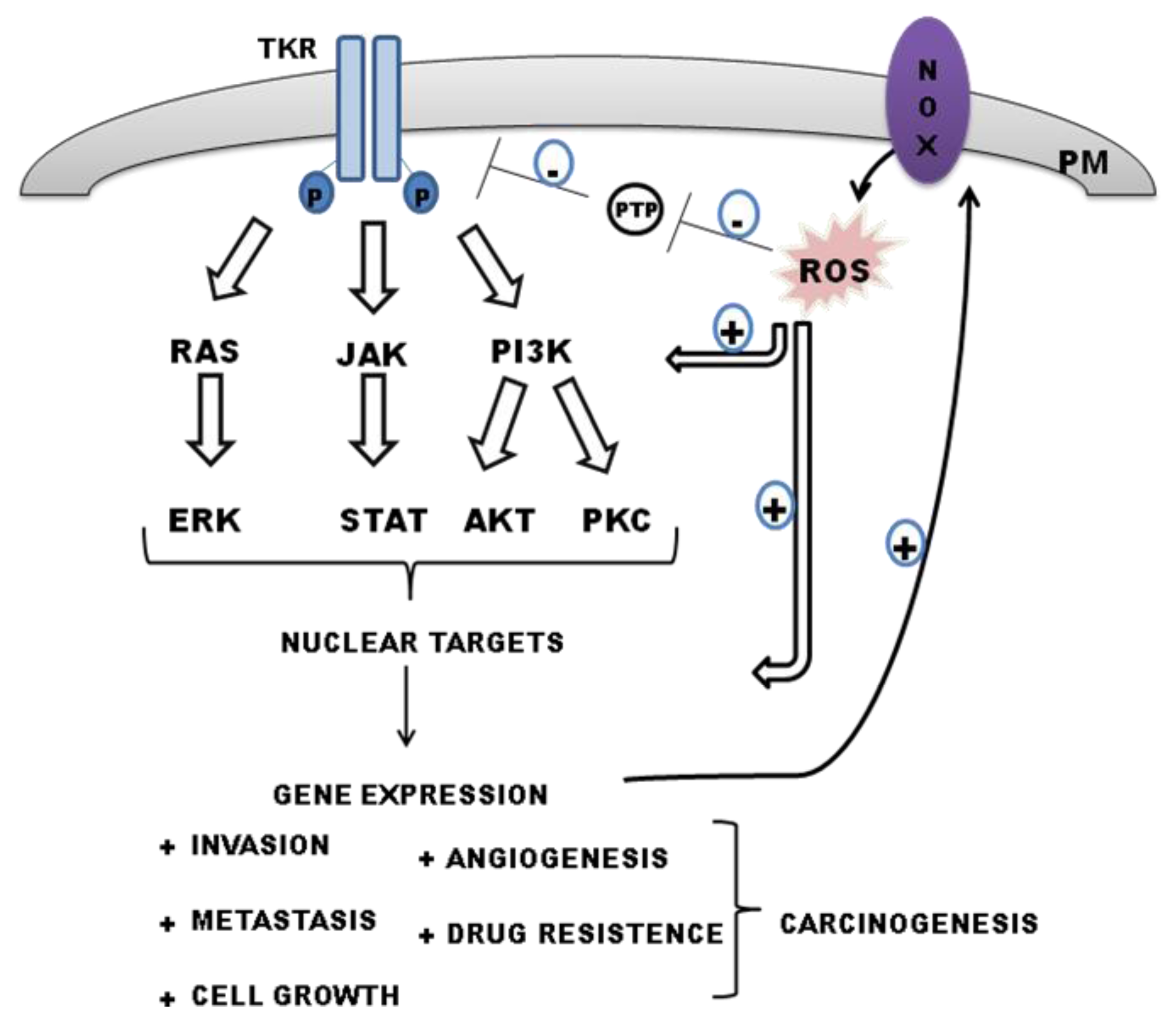
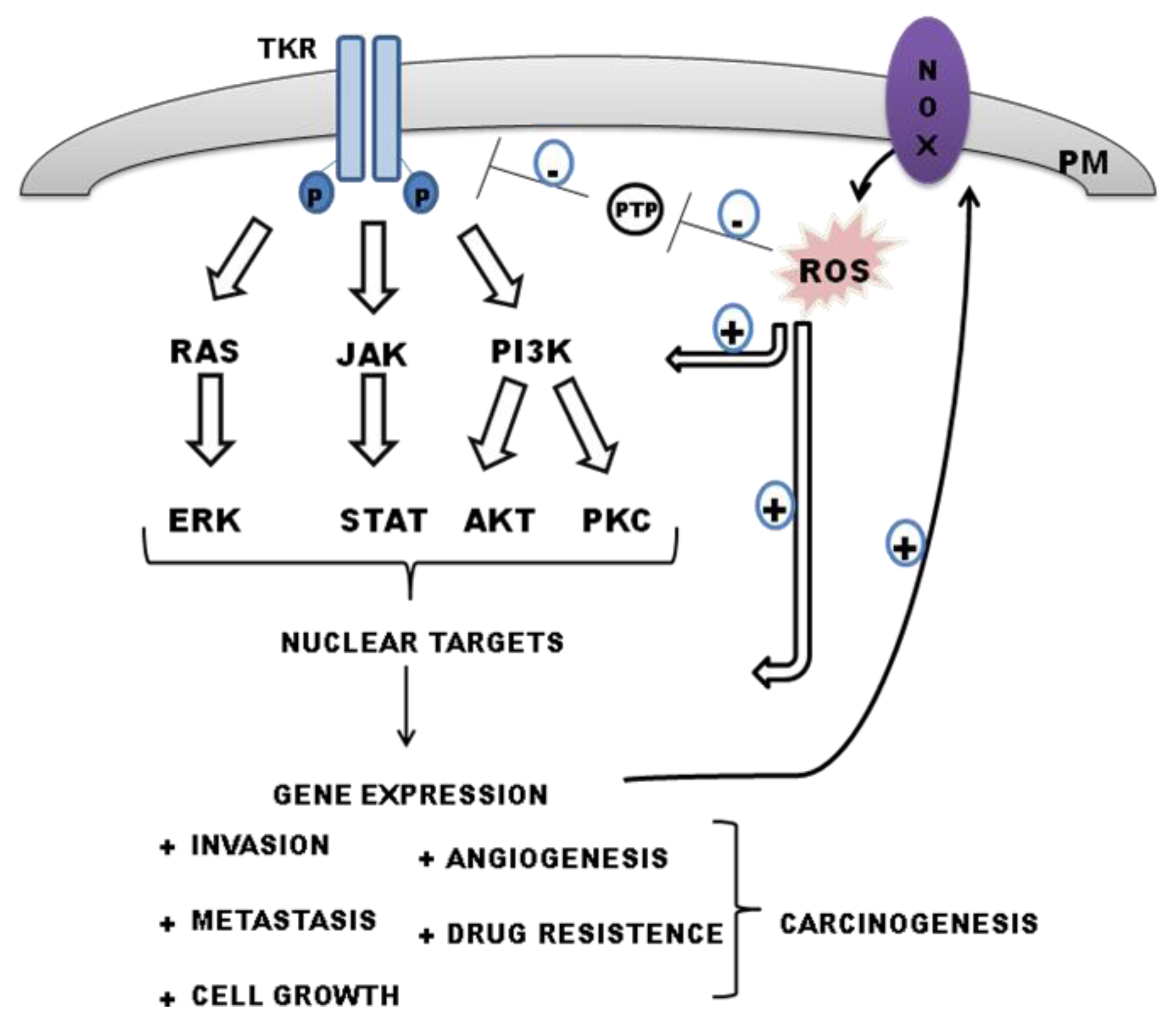
5.1. NOX Modulates PTP Activities: Maintaining TKR Signaling by ROS
6. Interplay between Tyrosine Kinase Receptor Signalling and ROS-Derived NADPH in Regulating Carcinogenesis
6.1. Epidermal Growth Factor Receptor (EGFR)
6.2. Vascular Endothelial Growth Factor Receptor (VEGFR)
7. NOX and Cancer Drug Therapy: Implications of ROS-Mediated Cytotoxicity and Resistance
7.1. ROS-Derived NADPH Oxidase Mediates Drug Cancer Cytotoxicity
7.2. Overcaming Drug Resistance in View of ROS-Derived NADPH Oxidase
8. Conclusions
Acknowledgments
Conflict of Interest
References
- Suzuki, Y.J.; Forman, H.J.; Sevanian, A. Oxidants as stimulators of signal transduction. Free Radic. Biol. Med 1997, 22, 269–285. [Google Scholar]
- Cosentino-Gomes, D.; Russo-Abrahão, T.; Fonseca-de-Souza, A.L.; Ferreira, C.R.; Galina, A.; Meyer-Fernandes, J.R. Modulation of Trypanosoma rangeliecto-phosphataseactivitybyhydrogen peroxide. Free Radic. Biol. Med 2009, 47, 152–158. [Google Scholar]
- Knock, G.A.; Ward, J.P.T. Redox regulation of protein kinases as a modulator of vascular function. Antioxid. Redox Signal 2011, 15, 1531–1547. [Google Scholar]
- Chiu, J.; Dawes, I.W. Redox control of cell proliferation. Trends Cell Biol 2012, 22, 592–601. [Google Scholar]
- Vernon, P.J.; Tang, D. Eat-me: Autophagy, phagocytosis, and reactive oxygen species signaling. Antioxid. Redox Signal 2013, 18, 677691. [Google Scholar]
- Altenhöfer, S.; Kleikers, P.W.; Radermacher, K.A.; Scheurer, P.; Rob Hermans, J.J.; Schiffers, P.; Ho, H.; Wingler, K.; Schmidt, H.H. The NOX toolbox: Validating the role of NADPH oxidases in physiology and disease. Cell. Mol. Life Sci 2012, 69, 2327–2343. [Google Scholar]
- Katsuyama, M.; Matsuno, K.; Yabe-Nishimura, C. Physiological roles of NOX/NADPH oxidase, the superoxide-generating enzyme. J. Clin. Biochem. Nutr 2012, 50, 9–22. [Google Scholar]
- Kleniewska, P.; Piechota, A.; Skibska, B.; Gorąca, A. The NADPH oxidase family and its inhibitors. Arch. Immunol. Ther. Exp 2012, 60, 277–294. [Google Scholar]
- Nisimoto, Y.; Tsubouchi, R.; Diebold, B.A.; Qiao, S.; Ogawa, H.; Ohara, T.; Tamura, M. Activation of NADPH oxidase 1 in tumour colon epithelial cells. Biochem. J 2008, 415, 57–65. [Google Scholar]
- Babior, B.M. NADPH oxidase: An update. Blood 1999, 93, 1464–1476. [Google Scholar]
- Raad, H.; Paclet, M.H.; Boussetta, T.; Kroviarski, Y.; Morel, F.; Quinn, M.T.; Gougerot-Pocidalo, M.A.; Dang, P.M.; El-Benna, J. Regulation of the phagocyte NADPH oxidase activity: Phosphorylation of gp91phox/NOX2 by protein kinase C enhances its diaphorase activity and binding to Rac2, p67phox, and p47phox. FASEB J 2009, 23, 1011–1022. [Google Scholar]
- Fontayne, A.; Dang, P.M.; Gougerot-Pocidalo, M.A.; El-Benna, J. Phosphorylation of p47phox sites by PKC alpha, beta II, delta, and zeta: Effect on binding to p22phox and on NADPH oxidase activation. Biochemistry 2002, 41, 7743–7750. [Google Scholar]
- Dewas, C.; Dang, P.M.; Gougerot-Pocidalo, M.A.; El-Benna, J. TNF-alpha induces phosphorylation of p47(phox) in human neutrophils: Partial phosphorylation of p47phox is a common event of priming of human neutrophils by TNF-alpha and granulocyte-macrophage colony-stimulating factor. J. Immunol 2003, 171, 4392–4398. [Google Scholar]
- Lewis, E.M.; Sergeant, S.; Ledford, B.; Stull, N.; Dinauer, M.C.; McPhail, L.C. Phosphorylation of p22phox on threonine 147 enhances NADPH oxidase activity by promoting p47phox binding. J. Biol. Chem 2010, 285, 2959–2967. [Google Scholar]
- Dang, P.M.; Morel, F.; Gougerot-Pocidalo, M.A.; El Benna, J. Phosphorylation of the NADPH oxidase component p67(PHOX) by ERK2 and P38MAPK: Selectivity of phosphorylated sites and existence of an intramolecular regulatory domain in the tetratricopeptide-rich region. Biochemistry 2003, 42, 4520–4526. [Google Scholar]
- Bromberg, Y.; Pick, E. Activation of NADPH-dependent superoxide production in a cell-free system by sodium dodecyl sulfate. J. Biol. Chem 1985, 260, 13539–13545. [Google Scholar]
- Brown, D.I.; Griendling, K.K. Nox proteins in signal transduction. Free Radic. Biol. Med 2009, 47, 1239–1253. [Google Scholar]
- Bánfi, B.; Malgrange, B.; Knisz, J.; Steger, K.; Dubois-Dauphin, M.; Krause, K.H. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J. Biol. Chem. 2004, 279, 46065–46072. [Google Scholar]
- Cheng, G.; Cao, Z.; Xu, X.; van Meir, E.G.; Lambeth, J.D. Homologs of gp91phox: Cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 2001, 269, 131–140. [Google Scholar]
- Martyn, K.D.; Frederick, L.M.; von Loehneysen, K.; Dinauer, M.C.; Knaus, U.G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal 2006, 18, 69–82. [Google Scholar]
- Lyle, A.N.; Deshpande, N.N.; Taniyama, Y.; Seidel-Rogol, B.; Pounkova, L.; Du, P.; Papaharalambus, C.; Lassègue, B.; Griendling, K.K. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ. Res 2009, 105, 249–259. [Google Scholar]
- Kawahara, T.; Lambeth, J.D. Phosphatidylinositol (4,5)-bisphosphate modulates Nox5 localization via an N-terminal polybasic region. Mol. Biol. Cell 2008, 19, 4020–4031. [Google Scholar]
- Meitzler, J.L.; de Montellano, P.R.O. Structural stability and heme binding potential of the truncated human dual oxidase 2 (DUOX2) peroxidase domain. Arch. Biochem. Biophys 2011, 512, 197–203. [Google Scholar]
- Hoste, C.; Dumont, J.E.; Miot, F.; de deken, X. The type of DUOX-dependent ROS production is dictated by defined sequences in DUOXA. Exp. Cell Res 2012, 318, 2353–2364. [Google Scholar]
- Rigutto, S.; Hoste, C.; Grasberger, H.; Milenkovic, M.; Communi, D.; Dumont, J.E.; Corvilain, B.; Miot, F.; de deken, X. Activation of dual oxidases Duox1 and Duox2: Differential regulation mediated by camp-dependent protein kinase and protein kinase C-dependent phosphorylation. J. Biol. Chem 2009, 284, 6725–6734. [Google Scholar]
- Rossi, F.; Zatti, M. Biochemical aspects of phagocytosis in polymorphonuclear leucocytes. NADH and NADPH oxidation by the granules of resting and phagocytizing cells. Experientia 1964, 20, 21–23. [Google Scholar]
- Vignais, P.V. The superoxide-generating NADPH oxidase: Structural aspects and activation mechanism. Cell Mol. Life Sci 2002, 59, 1428–1459. [Google Scholar]
- Rada, B.; Hably, C.; Meczner, A.; Timár, C.; Lakatos, G.; Enyedi, P.; Ligeti, E. Role of Nox2 in elimination of microorganisms. Semin Immunopathol 2008, 30, 237–253. [Google Scholar]
- Pendyala, S.; Natarajan, V. Redox regulation of Nox proteins. Respir. Physiol. Neurobiol 2010, 174, 265–271. [Google Scholar]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev 2007, 87, 245–313. [Google Scholar]
- Jung, O.; Schreiber, J.G.; Geiger, H.; Pedrazzini, T.; Busse, R.; Brandes, R.P. gp91phox-containing NADPH oxidase mediates endothelial dysfunction in renovascular hypertension. Circulation 2004, 109, 1795–1801. [Google Scholar]
- Suh, Y.A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell transformation by the superoxide-generating oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar]
- Bánfi, B.; Maturana, A.; Jaconi, S.; Arnaudeau, S.; Laforge, T.; Sinha, B.; Ligeti, E.; Demaurex, N.; Krause, K.H. A mammalian H+ channel generated through alternative splicing of the NADPH oxidase homolog NOH-1. Science 2000, 287, 138–142. [Google Scholar]
- Rokutan, K.; Kawahara, T.; Kuwano, Y.; Tominaga, K.; Sekiyama, A.; Teshima-Kondo, S. NADPH oxidases in the gastrointestinal tract: A potential role of Nox1 in innate immune response and carcinogenesis. Antioxid. Redox Signal 2006, 8, 1573–1582. [Google Scholar]
- Rokutan, K.; Kawahara, T.; Kuwano, Y.; Tominaga, K.; Nishida, K.; Teshima-Kondo, S. Nox enzymes and oxidative stress in the immunopathology of the gastrointestinal tract. Semin. Immunopathol 2008, 30, 315–327. [Google Scholar]
- Kikuchi, H.; Hikage, M.; Miyashita, H.; Fukumoto, M. NADPH oxidase subunit, gp91(phox) homologue, preferentially expressed in human colon epithelial cells. Gene 2000, 254, 237–243. [Google Scholar]
- Geiszt, M.; Kopp, J.B.; Várnai, P.; Leto, T.L. Identification of renox, an NAD(P)H oxidase in kidney. Proc. Natl. Acad. Sci. USA 2000, 97, 8010–8014. [Google Scholar]
- Yang, S.; Madyastha, P.; Bingel, S.; Ries, W.; Key, L. A new superoxide-generating oxidase in murine osteoclasts. J. Biol. Chem 2001, 276, 5452–5458. [Google Scholar]
- Kawahara, T.; Ritsick, D.; Cheng, G.; Lambeth, J.D. Point mutations in the proline-rich region of p22phox are dominant inhibitors of Nox1- and Nox2-dependent reactive oxygen generation. J. Biol. Chem 2005, 280, 31859–31869. [Google Scholar]
- Serrander, L.; Jaquet, V.; Bedard, K.; Plastre, O.; Hartley, O.; Arnaudeau, S.; Demaurex, N.; Schlegel, W.; Krause, K.H. NOX5 is expressed at the plasma membrane and generates superoxide in response to protein kinase C activation. Biochimie 2007, 89, 1159–1167. [Google Scholar]
- Kamiguti, A.S.; Serrander, L.; Lin, K.; Harris, R.J.; Cawley, J.C.; Allsup, D.J.; Slupsky, J.R.; Krause, K.H.; Zuzel, M. Expression and activity of NOX5 in the circulating malignant B cells of hairy cell leukemia. J. Immunol 2005, 175, 8424–8430. [Google Scholar]
- Brar, S.S.; Kennedy, T.P.; Sturrock, A.B.; Huecksteadt, T.P.; Quinn, M.T.; Whorton, A.R.; Hoidal, J.R. An NAD(P)H oxidase regulates growth and transcription in melanoma cells. Am. J. Physiol. Cell Physiol 2002, 282, 1212–1224. [Google Scholar]
- Brar, S.S.; Corbin, Z.; Kennedy, T.P.; Hemendinger, R.; Thornton, L.; Bommarius, B.; Arnold, R.S.; Whorton, A.R.; Sturrock, A.B.; Huecksteadt, T.P.; et al. NOX5 NAD(P)H oxidase regulates growth and apoptosis in DU 145 prostate cancer cells. Am. J. Physiol. Cell Physiol 2003, 285, 353–369. [Google Scholar]
- Fu, X.; Beer, D.G.; Behar, J.; Wands, J.; Lambeth, D.; Cao, W. cAMP-response element-binding protein mediates acid-induced NADPH oxidase NOX5-S expression in Barrett esophageal adenocarcinoma cells. J. Biol. Chem 2006, 281, 20368–20382. [Google Scholar]
- Dupuy, C.; Ohayon, R.; Valent, A.; Noël-Hudson, M.S.; Dème, D.; Virion, A. Purification of a novel flavoprotein involved in the thyroid NADPH oxidase. Cloning of the porcine and human cdnas. J. Biol. Chem 1999, 274, 37265–37269. [Google Scholar]
- De Deken, X.; Wang, D.; Many, M.C.; Costagliola, S.; Libert, F.; Vassart, G.; Dumont, J.E.; Miot, F. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J. Biol. Chem 2000, 275, 23227–23233. [Google Scholar]
- Meitzler, J.L.; de Montellano, P.R.O. Caenorhabditis elegans and human dual oxidase 1 (DUOX1) “peroxidase” domains: Insights into heme binding and catalytic activity. J. Biol. Chem 2009, 284, 18634–18643. [Google Scholar]
- Harper, R.W.; Xu, C.; Eiserich, J.P.; Chen, Y.; Kao, C.Y.; Thai, P.; Setiadi, H.; Wu, R. Differential regulation of dual NADPH oxidases/peroxidases, Duox1 and Duox2, by Th1 and Th2 cytokines in respiratory tract epithelium. FEBS Lett. 2005, 579, 4911–4917. [Google Scholar]
- Moreno, J.C.; Bikker, H.; Kempers, M.J.; van Trotsenburg, A.S.; Baas, F.; de Vijlder, J.J.; Vulsma, T.; Ris-Stalpers, C. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N. Engl. J. Med 2002, 347, 95–102. [Google Scholar]
- Biswas, S.; Chida, A.S.; Rahman, I. Redox modifications of protein-thiols: Emerging roles in cell signaling. Biochem. Pharmacol 2006, 71, 551–564. [Google Scholar]
- Bartosz, G. Reactive oxygen species: Destroyers or messengers? Biochem. Pharmacol 2009, 77, 1303–1315. [Google Scholar]
- Cosentino-Gomes, D.; Rocco-Machado, N.; Meyer-Fernandes, J.R. Cell signaling through protein kinase C oxidation and activation. Int. J. Mol. Sci 2012, 13, 10697–10721. [Google Scholar]
- Vásquez-Vivar, J.; Kalyanaraman, B.; Martásek, P.; Hogg, N.; Masters, B.S.; Karoui, H.; Tordo, P.; Pritchard, K.A., Jr. Superoxide generation by endothelial nitric oxide synthase: The influence of cofactors. Proc. Natl. Acad. Sci. USA 1998, 95, 9220–9225. [Google Scholar]
- McNally, J.S.; Davis, M.E.; Giddens, D.P.; Saha, A.; Hwang, J.; Dikalov, S.; Jo, H.; Harrison, D.G. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am. J. Physiol. Heart Circ. Physiol 2003, 285, 2290–2297. [Google Scholar]
- Morand, S.; Ueyama, T.; Tsujibe, S.; Saito, N.; Korzeniowska, A.; Leto, T.L. Duox maturation factors form cell surface complexes with Duox affecting the specificity of reactive oxygen species generation. FASEB J 2009, 23, 1205–1218. [Google Scholar]
- Takac, I.; Schröder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem 2011, 286, 13304–13313. [Google Scholar]
- Dikalov, S.I.; Dikalova, A.E.; Bikineyeva, A.T.; Schmidt, H.H.; Harrison, D.G.; Griendling, K.K. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic. Biol. Med 2008, 45, 1340–1351. [Google Scholar]
- Dupuy, C.; Kaniewski, J.; Dème, D.; Pommier, J.; Virion, A. NADPH-dependent H2O2 generation catalyzed by thyroid plasma membranes. Studies with electron scavengers. Eur. J. Biochem 1989, 185, 597–603. [Google Scholar]
- Block, K.; Gorin, Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat Rev Cancer 2012, 12, 627–637. [Google Scholar]
- Weinberg, F.; Chandel, N.S. Reactive oxygen species-dependent signaling regulates cancer. Cell Mol. Life Sci 2009, 66, 3663–3673. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar]
- Benhar, M.; Dalyot, I.; Engelberg, D.; Levitzki, A. Enhanced ROS production in oncogenically transformed cells potentiates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase activation and sensitization to genotoxic stress. Mol. Cell Biol 2001, 21, 6913–6926. [Google Scholar]
- Hayes, P.; Knaus, U.G. Balancing reactive oxygen species in the epigenome: NADPH oxidases as target and perpetrator. Antioxid. Redox Signal 2013. [Google Scholar] [CrossRef]
- Cross, J.V.; Templeton, D.J. Regulation of signal transduction through protein cysteine oxidation. Antioxid. Redox Signal 2006, 8, 1819–1827. [Google Scholar]
- Salmeen, A.; Barford, D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid. Redox Signal 2005, 7, 560–577. [Google Scholar]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumoursuppressor. Nat. Rev. Mol. Cell Biol 2012, 13, 283–296. [Google Scholar]
- Lee, S.R.; Yang, K.S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem 2002, 277, 20336–20342. [Google Scholar]
- Kwon, J.; Lee, S.R.; Yang, K.S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA 2004, 10, 16419–16424. [Google Scholar]
- Chetram, M.A.; Don-Salu-Hewage, A.S.; Hinton, C.V. ROS enhances CXCR4-mediated functions through inactivation of PTEN in prostate cancer cells. Biochem. Biophys. Res. Commun 2011, 410, 195–200. [Google Scholar]
- Lee, J.K.; Edderkaoui, M.; Truong, P.; Ohno, I.; Jang, K.T.; Berti, A.; Pandol, S.J.; Gukovskaya, A.S. NADPH oxidase promotes pancreatic cancer cell survival via inhibiting JAK2 dephosphorylation by tyrosine phosphatases. Gastroenterology 2007, 133, 1637–1648. [Google Scholar]
- Russell, J.S.; Colevas, A.D. The use of epidermal growth factor receptor monoclonal antibodies in squamous cell carcinoma of the head and neck. Chemother. Res. Pract 2012, 2012, 1–13. [Google Scholar]
- Martinelli, E.; de Palma, R.; Orditura, M.; de Vita, F.; Ciardiello, F. Anti-epidermal growth factor receptor monoclonal antibodies in câncer therapy. Clin. Exp. Immunol 2009, 158, 1–9. [Google Scholar]
- Rocha-Lima, C.M.; Soares, H.P.; Raez, L.E.; Singal, R. EGFR targeting of solid tumors. Cancer Control 2007, 14, 295–304. [Google Scholar]
- Sancho, P.; Fabregat, I. NADPH oxidase NOX1 controls autocrine growth of liver tumor cells through up-regulation of the epidermal growth factor receptor pathway. J. Biol. Chem 2010, 285, 24815–24824. [Google Scholar]
- Mesquita, F.S.; Dyer, S.N.; Heinrich, D.A.; Bulun, S.E.; Marsh, E.E.; Nowak, R.A. Reactive oxygen species mediate mitogenic growth factor signaling pathways in human leiomyoma smooth muscle cells. Biol. Reprod 2010, 82, 341–351. [Google Scholar]
- Caja, L.; Sancho, P.; Bertran, E.; Iglesias-Serret, D.; Gil, J.; Fabregat, I. Over activation of the MEK/ERK pathway in liver tumor cells confers resistance to TGF-β-induced cell death through impairing up-regulation of the NADPH oxidase NOX4. Cancer Res 2009, 69, 7595–7602. [Google Scholar]
- Sancho, P.; Bertrán, E.; Caja, L.; Carmona-Cuenca, I.; Murillo, M.M.; Fabregat, I. The inhibition of the epidermal growth factor (EGF) pathway enhances TGF-β-induced apoptosis in rat hepatoma cells through inducing oxidative stress coincident with a change in the expression pattern of the NADPH oxidases (NOX) isoforms. Biochim. Biophys. Acta 2009, 1793, 253–263. [Google Scholar]
- Carmona-Cuenca, I.; Roncero, C.; Sancho, P.; Caja, L.; Fausto, N.; Fernández, M.; Fabregat, I. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J. Hepatol 2008, 49, 965–976. [Google Scholar]
- Uemura, T.; Hibi, K.; Kaneko, T.; Takeda, S.; Inoue, S.; Okochi, O.; Nagasaka, T.; Nakao, A. Detection of K-ras mutations in the plasma DNA of pancreatic cancer patients. J. Gastroenterol 2004, 39, 56–60. [Google Scholar]
- Adachi, Y.; Shibai, Y.; Mitsushita, J.; Shang, W.H.; Hirose, K.; Kamata, T. Oncogenic Ras upregulates NADPH oxidase 1 gene expression through MEK-ERK-dependent phosphorylation of GATA-6. Oncogene 2008, 27, 4921–4932. [Google Scholar]
- Kissil, J.L.; Walmsley, M.J.; Hanlon, L.; Haigis, K.M.; Bender Kim, C.F.; Sweet-Cordero, A.; Eckman, M.S.; Tuveson, D.A.; Capobianco, A.J.; Tybulewicz, V.L.; et al. Requirement for Rac1 in a K-ras induced lung cancer in the mouse. Cancer Res 2007, 67, 8089–8094. [Google Scholar]
- Du, J.; Liu, J.; Smith, B.J.; Tsao, M.S.; Cullen, J.J. Role of Rac1-dependent NADPH oxidase in the growth of pancreatic cancer. Cancer Gene Ther 2011, 18, 135–143. [Google Scholar]
- Du, J.; Nelson, E.S.; Simons, A.L.; Olney, K.E.; Moser, J.C.; Schrock, H.E.; Wagner, B.A.; Buettner, G.R.; Smith, B.J.; Teoh, M.L.; et al. Regulation of pancreatic cancer growth by superoxide. Mol. Carcinog. 2013. [Google Scholar] [CrossRef]
- Ranjan, P.; Anathy, V.; Burch, P.M.; Weirather, K.; Lambeth, J.D.; Heintz, N.H. Redox-dependent expression of cyclin D1 and cell proliferation by Nox1 in mouse lung epithelial cells. Antioxid. Redox Signal 2006, 8, 1447–1459. [Google Scholar]
- Shinohara, M.; Shang, W.H.; Kubodera, M.; Harada, S.; Mitsushita, J.; Kato, M.; Miyazaki, H.; Sumimoto, H.; Kamata, T. Nox1 redox signaling mediates oncogenic Ras-induced disruption of stress fibers and focal adhesions by down-regulating Rho. J. Biol. Chem 2007, 282, 17640–17648. [Google Scholar]
- Shinohara, M.; Adachi, Y.; Mitsushita, J.; Kuwabara, M.; Nagasawa, A.; Harada, S.; Furuta, S.; Zhang, Y.; Seheli, K.; Miyazaki, H.; et al. Reactive oxygen generated by NADPH oxidase 1 (Nox1) contributes to cell invasion by regulating matrix metalloprotease-9 production and cell migration. J. Biol. Chem 2010, 285, 4481–4488. [Google Scholar]
- Liebmann, C. EGF receptor activation by GPCRs: An universal pathway reveals different versions. Mol. Cell Endocrinol 2011, 331, 222–231. [Google Scholar]
- Ritter, S.L.; Hall, R.A. Fine-tuning of GPCR activity by receptor-interacting proteins. Nat. Rev. Mol. Cell Biol 2009, 10, 819–830. [Google Scholar]
- Lemjabbar-Alaoui, H.; Sidhu, S.S.; Mengistab, A.; Gallup, M.; Basbaum, C. TACE/ADAM-17 phosphorylation by PKC-epsilon mediates premalignant changes in tobacco smoke-exposed lung cells. PLoS One 2011, 6, e17489. [Google Scholar]
- Gianni, D.; Bohl, B.; Courtneidge, S.A.; Bokoch, G.M. The involvement of the tyrosine kinase c-Src in the regulation of reactive oxygen species generation mediated by NADPH oxidase-1. Mol. Biol. Cell 2008, 19, 2984–2994. [Google Scholar]
- Lin, C.C.; Lee, I.T.; Wu, W.L.; Lin, W.N.; Yang, C.M. Adenosine triphosphate regulates NADPH oxidase activity leading to hydrogen peroxide production and COX-2/PGE2 expression in A549 cells. Am. J. Physiol. Lung Cell Mol. Physiol 2012, 303, L401–L412. [Google Scholar]
- Biscardi, J.S.; Maa, M.C.; Tice, D.A.; Cox, M.E.; Leu, T.H.; Parsons, S.J. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J. Biol. Chem 1999, 274, 8335–8343. [Google Scholar]
- Cattaneo, F.; Iaccio, A.; Guerra, G.; Montagnani, S.; Ammendola, R. NADPH-oxidase-dependent reactive oxygen species mediate EGFR transactivation by FPRL1 in WKYMVm-stimulated human lung cancer cells. Free Radic. Biol. Med 2011, 51, 1126–1136. [Google Scholar]
- Moody, T.W.; Sancho, V.; di Florio, A.; Nuche-Berenguer, B.; Mantey, S.; Jensen, R.T. Bombesin receptor subtype-3 agonists stimulate the growth of lung cancer cells and increase EGF receptor tyrosine phosphorylation. Peptides 2011, 32, 1677–1684. [Google Scholar]
- Lemos, C.; Sack, U.; Schmid, F.; Juneja, M.; Stein, U. Anti-metastatic treatment in colorectal cancer: Targeting signaling pathways. Curr. Pharm. Des 2013, in press. [Google Scholar]
- McMahon, B.J. Epidemiology and natural history of hepatitis B. Semin. Liver Dis 2005, 25, 3–8. [Google Scholar]
- Pallis, A.G.; Syrigos, K.N. Targeting tumor neovasculature in non-small-cell lung cancer. Crit. Rev. Oncol. Hematol 2013, in press. [Google Scholar]
- Guo, S.; Colbert, L.S.; Fuller, M.; Zhang, Y.; Gonzalez-Perez, R.R. Vascular endothelial growth factor receptor-2 in breast cancer. Biochim. Biophys. Acta 2010, 1806, 108–121. [Google Scholar]
- McMahon, G. VEGF receptor signaling in tumorangiogenesis. Oncologist 2000, 5, S3–S10. [Google Scholar]
- Arbiser, J.L.; Petros, J.; Klafter, R.; Govindajaran, B.; McLaughlin, E.R.; Brown, L.F.; Cohen, C.; Moses, M.; Kilroy, S.; Arnold, R.S.; et al. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc. Natl. Acad. Sci. USA 2002, 99, 715–720. [Google Scholar]
- Meng, D.; Mei, A.; Liu, J.; Kang, X.; Shi, X.; Qian, R.; Chen, S. NADPH oxidase 4 mediates insulin-stimulated HIF-1α and VEGF expression, and angiogenesis in vitro. PLoS One 2012, 7, e48393. [Google Scholar]
- Hsieh, C.H.; Shyu, W.C; Chiang, C.Y.; Kuo, J.W.; Shen, W.C.; Liu, R.S. NADPH oxidase subunit 4-mediated reactive oxygen species contribute to cycling hypoxia-promoted tumor progression in glioblastoma multiforme. PLoS One 2011, 6, e23945. [Google Scholar]
- Calvani, M.; Comito, G.; Giannoni, E.; Chiarugi, P. Time-dependent stabilization of hypoxia inducible factor-1α by different intracellular sources of reactive oxygen species. PLoS One 2012, 7, e38388. [Google Scholar]
- Kim, J.; Koyanagi, T.; Mochly-Rosen, D. PKCδ activation mediates angiogenesis via NADPH oxidase activity in PC-3 prostate cancer cells. Prostate 2011, 71, 946–954. [Google Scholar]
- Nayak, B.K.; Feliers, D.; Sudarshan, S.; Friedrichs, W.E.; Day, R.T.; New, D.D.; Fitzgerald, J.P.; Eid, A.; Denapoli, T.; Parekh, D.J.; et al. Stabilization of HIF-2α through redox regulation of mTORC2 activation and initiation of mRNA translation. Oncogene 2013. [Google Scholar] [CrossRef]
- Komatsu, D.; Kato, M.; Nakayama, J.; Miyagawa, S.; Kamata, T. NADPH oxidase 1 plays a critical mediating role in oncogenic Ras-induced vascular endothelial growth factor expression. Oncogene 2008, 27, 4724–4732. [Google Scholar]
- Maraldi, T.; Prata, C.; Caliceti, C.; Vieceli Dalla Sega, F.; Zambonin, L.; Fiorentini, D.; Hakim, G. VEGF-induced ROS generation from NAD(P)H oxidases protects human leukemic cells from apoptosis. Int. J. Oncol 2010, 36, 1581–1589. [Google Scholar]
- Garrido-Urbani, S.; Jemelin, S.; Deffert, C.; Carnesecchi, S.; Basset, O.; Szyndralewiez, C.; Heitz, F.; Page, P.; Montet, X.; Michalik, L.; et al. Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARα mediated mechanism. PLoS One 2011, 6, e14665. [Google Scholar]
- Meissner, M.; Stein, M.; Urbich, C.; Reisinger, K.; Suske, G.; Staels, B.; Kaufmann, R.; Gille, J. PPARalpha activators inhibit vascular endothelial growth factor receptor-2 expression by repressing Sp1-dependent DNA binding and transactivation. Circ. Res 2004, 94, 324–332. [Google Scholar]
- Barth, B.M.; Gustafson, S.J.; Young, M.M.; Fox, T.E.; Shanmugavelandy, S.S.; Kaiser, J.M.; Cabot, M.C.; Kester, M.; Kuhn, T.B. Inhibition of NADPH oxidase by glucosylceramide confers chemoresistance. Cancer Biol. Ther 2010, 10, 1126–1136. [Google Scholar]
- Witte, A.B.; Anestål, K.; Jerremalm, E.; Ehrsson, H.; Arnér, E.S. Inhibition of thioredoxin reductase but not of glutathione reductase by the major classes of alkylating and platinum-containing anticancer compounds. Free Radic. Biol. Med 2005, 39, 696–703. [Google Scholar]
- Alexandre, J.; Hu, Y.; Lu, W.; Pelicano, H.; Huang, P. Novel action of paclitaxel against cancer cells: Bystander effect mediated by reactive oxygen species. Cancer Res 2007, 67, 3512–3517. [Google Scholar]
- Honeychurch, J.; Alduaij, W.; Azizyan, M.; Cheadle, E.J.; Pelicano, H.; Ivanov, A.; Huang, P.; Cragg, M.S.; Illidge, T.M. Antibody-induced nonapoptotic cell death in human lymphoma and leukemia cells is mediated through a novel reactive oxygen species-dependent pathway. Blood 2012, 119, 3523–3533. [Google Scholar]
- Orcutt, K.P.; Parsons, A.D.; Sibenaller, Z.A.; Scarbrough, P.M.; Zhu, Y.; Sobhakumari, A.; Wilke, W.W.; Kalen, A.L.; Goswami, P.; Miller, F.J., Jr; et al. Erlotinib-mediated inhibition of EGFR signaling induces metabolic oxidative stress through NOX4. Cancer Res 2011, 71, 3932–3940. [Google Scholar]
- Ortiz, C.; Caja, L.; Sancho, P.; Bertran, E.; Fabregat, I. Inhibition of the EGF receptor blocks autocrine growth and increases the cytotoxic effects of doxorubicin in rat hepatoma cells: Role of reactive oxygen species production and glutathione depletion. Biochem. Pharmacol 2008, 75, 1935–1945. [Google Scholar]
- Dahan, L.; Sadok, A.; Formento, J.L.; Seitz, J.F.; Kovacic, H. Modulation of cellular redox state underlies antagonism between oxaliplatin and cetuximab in human colorectal cancer cell lines. Br. J. Pharmacol 2009, 158, 610–620. [Google Scholar]
- Wartenber, M.; Hoffmann, E.; Schwindt, H.; Grünheck, F.; Petros, J.; Arnold, J.R.S.; Hescheler, J.; Sauer, H. Reactive oxygen species-linked regulation of the multidrug resistance transporter P-glycoprotein in Nox-1 overexpressing prostate tumor spheroids. FEBS Lett 2005, 579, 4541–4549. [Google Scholar]
- Giordano, C.R.; Mueller, K.L.; Terlecky, L.J.; Krentz, K.A.; Bollig-Fischer, A.; Terlecky, S.R.; Boerner, J.L. A targeted enzyme approach to sensitization of tyrosine kinase inhibitor-resistant breast cancer cells. Exp. Cell Res 2012, 318, 2014–2021. [Google Scholar]
- Huang, W.C.; Chen, Y.J.; Li, L.Y.; Wei, Y.L.; Hsu, S.C.; Tsai, S.L.; Chiu, P.C.; Huang, W.P.; Wang, Y.N.; Chen, C.H.; et al. Nuclear translocation of epidermal growth factor receptor by Akt-dependent phosphorylation enhances breast cancer-resistant protein expression in gefitinib-resistant cells. J. Biol. Chem 2011, 286, 20558–20568. [Google Scholar]
- Petruccelli, L.A.; Dupéré-Richer, D.; Pettersson, F.; Retrouvey, H.; Skoulikas, S.; Miller, W.H., Jr. Vorinostat induces reactive oxygen species and DNA damage in acute myeloid leukemia cells. PLoS One 2011, 6, e20987. [Google Scholar]
- Premkumar, D.R.; Jane, E.P.; Agostino, N.R.; Didomenico, J.D.; Pollack, I.F. Bortezomib-induced sensitization of malignant human glioma cells to vorinostat-induced apoptosis depends on reactive oxygen species production, mitochondrial dysfunction, Noxa upregulation, Mcl-1 cleavage, and DNA damage. Mol. Carcinog 2013, 52, 118–133. [Google Scholar]
- Hu, Y.; Lu, W.; Chen, G.; Zhang, H.; Jia, Y.; Wei, Y.; Yang, H.; Zhang, W.; Fiskus, W.; Bhalla, K.; et al. Overcoming resistance to histone deacetylase inhibitors in human leukemia with the redox modulating compound β-phenylethyl isothiocyanate. Blood 2010, 116, 2732–2741. [Google Scholar]
- Lu, J.P.; Monardo, L.; Bryskin, I.; Hou, Z.F.; Trachtenberg, J.; Wilson, B.C.; Pinthus, J.H. Androgens induce oxidative stress and radiation resistance in prostate cancer cells though NADPH oxidase. Prostate Cancer Prostatic Dis 2010, 13, 39–46. [Google Scholar]
- Sun, Y.; St. Clair, D.K.; Xu, Y.; Crooks, P.A.; St. Clair, W.H. A NADPH oxidase-dependent redox signaling pathway mediates the selective radiosensitization effect of parthenolide in prostate cancer cells. Cancer Res 2010, 70, 2880–2890. [Google Scholar]
- Schramm, A.; Matusik, P.; Osmenda, G.; Guzik, T.J. Targeting NADPH oxidases in vascular pharmacology. Vascul. Pharmacol 2012, 56, 216–231. [Google Scholar]


{kind=link}
{kind=link}
| NOX isoforms | Subunits | Regulators | References |
|---|---|---|---|
| NOX1 | p22phox, NOXA1, NOXO1 and RAC1 | ANG II, PDGF | [8,9] |
| NOX2 | gp91phox, p22phox, p40phox, p47phox, p67phox, RAC1 | PKC, (TNF)-α, phosphatidic acid | [7,8,10–16] |
| NOX3 | p22phox, NOXO1, NOXA1, RAC1 | Unknown | [8,17–19] |
| NOX4 | P22phox | Poldip2 | [20,21] |
| NOX5 | NONE | Ca2+, ptdlns(4,5)p2 | [7,8,22] |
| DUOX1 | DUOXA1, DUOXA2 | IL-4, IL-3,Camp, PKA | [7,23–25] |
| DUOX2 | DUOXA1, DUOXA2 | IFN-γ, PLC, PKC | [7,23–25] |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Paletta-Silva, R.; Rocco-Machado, N.; Meyer-Fernandes, J.R. NADPH Oxidase Biology and the Regulation of Tyrosine Kinase Receptor Signaling and Cancer Drug Cytotoxicity. Int. J. Mol. Sci. 2013, 14, 3683-3704. https://doi.org/10.3390/ijms14023683
Paletta-Silva R, Rocco-Machado N, Meyer-Fernandes JR. NADPH Oxidase Biology and the Regulation of Tyrosine Kinase Receptor Signaling and Cancer Drug Cytotoxicity. International Journal of Molecular Sciences. 2013; 14(2):3683-3704. https://doi.org/10.3390/ijms14023683
Chicago/Turabian StylePaletta-Silva, Rafael, Nathália Rocco-Machado, and José Roberto Meyer-Fernandes. 2013. "NADPH Oxidase Biology and the Regulation of Tyrosine Kinase Receptor Signaling and Cancer Drug Cytotoxicity" International Journal of Molecular Sciences 14, no. 2: 3683-3704. https://doi.org/10.3390/ijms14023683
APA StylePaletta-Silva, R., Rocco-Machado, N., & Meyer-Fernandes, J. R. (2013). NADPH Oxidase Biology and the Regulation of Tyrosine Kinase Receptor Signaling and Cancer Drug Cytotoxicity. International Journal of Molecular Sciences, 14(2), 3683-3704. https://doi.org/10.3390/ijms14023683