Misfolding and Amyloid Aggregation of Apomyoglobin

Abstract
:1. Introduction
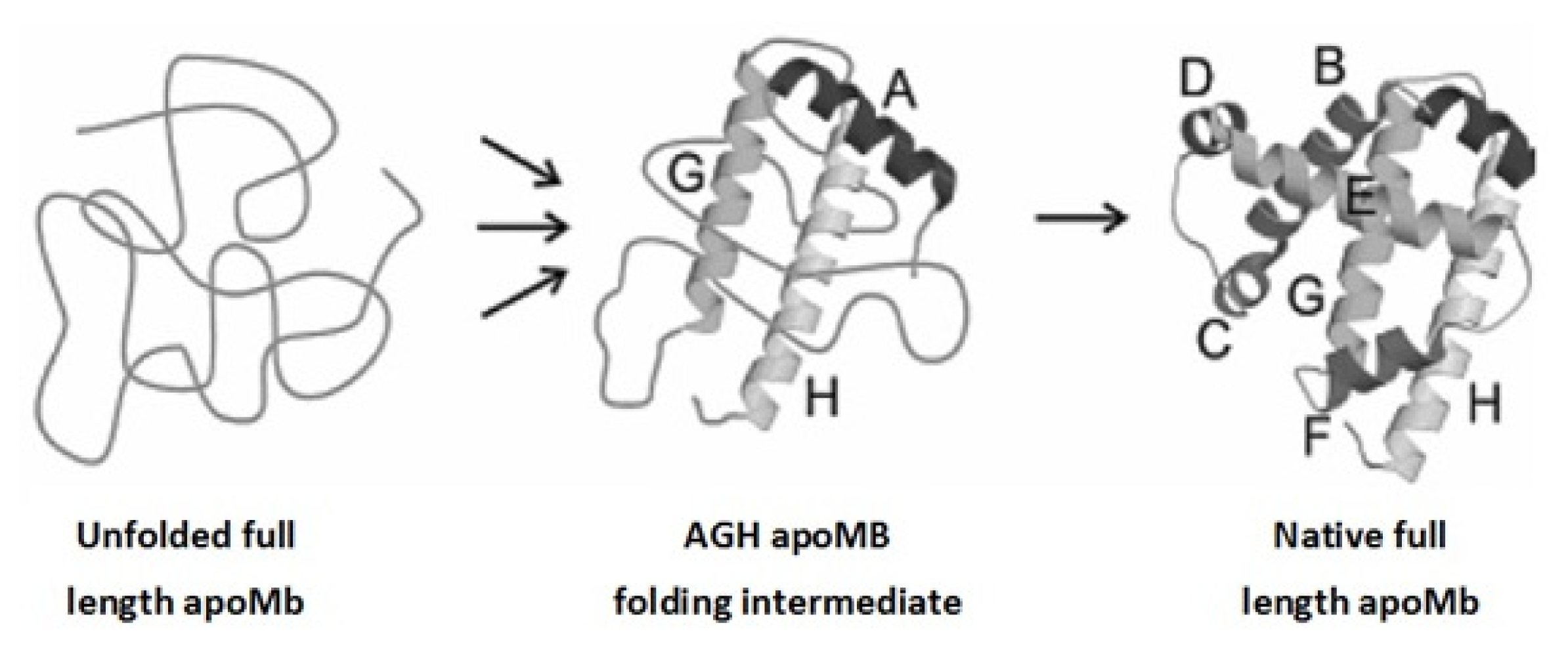
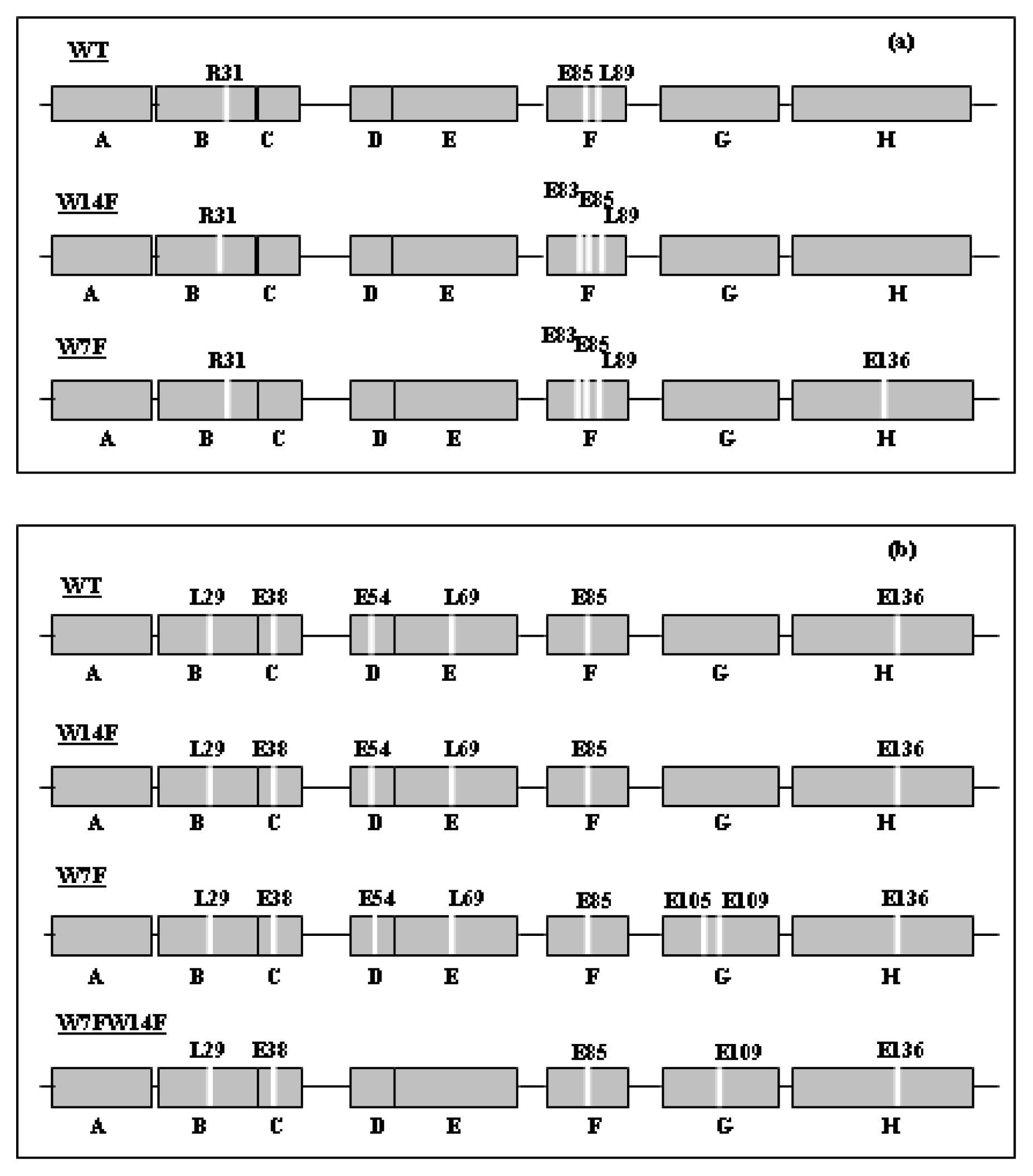
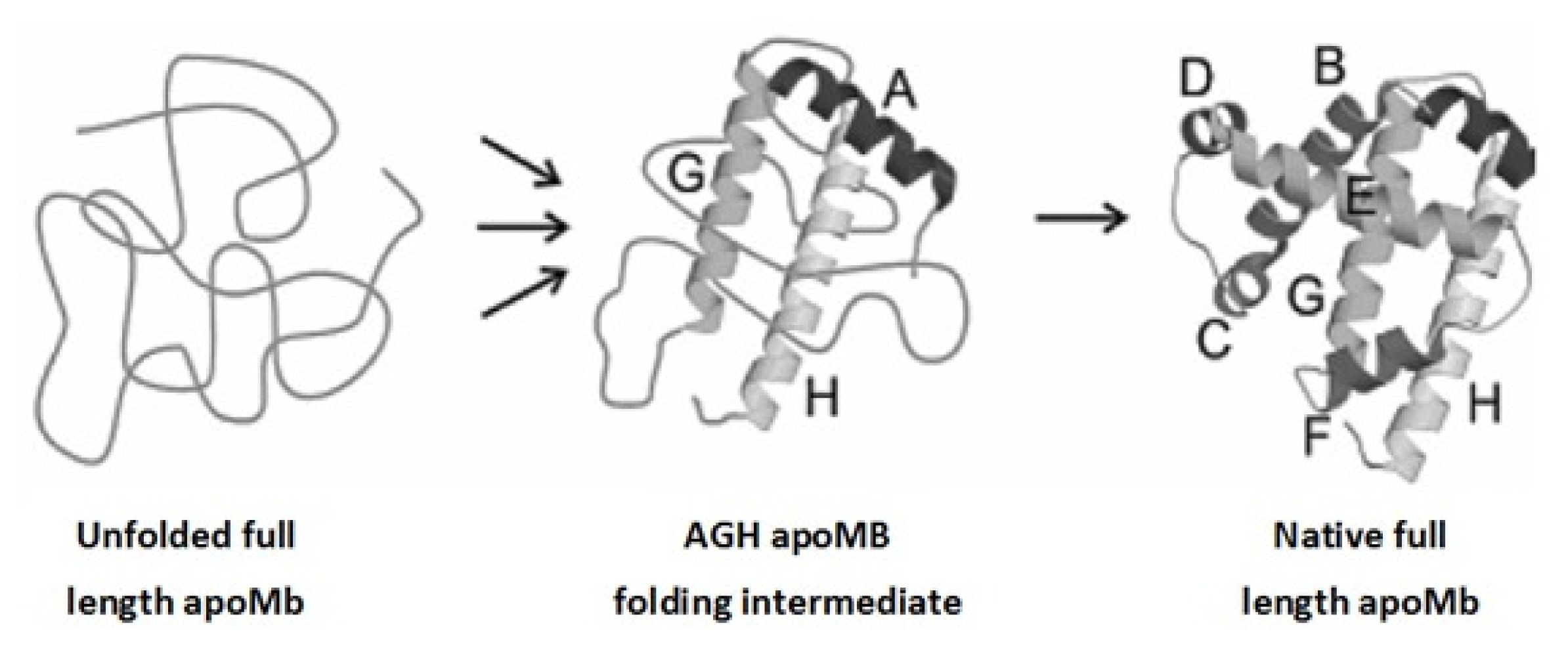
2. Apomyoglobin Folding
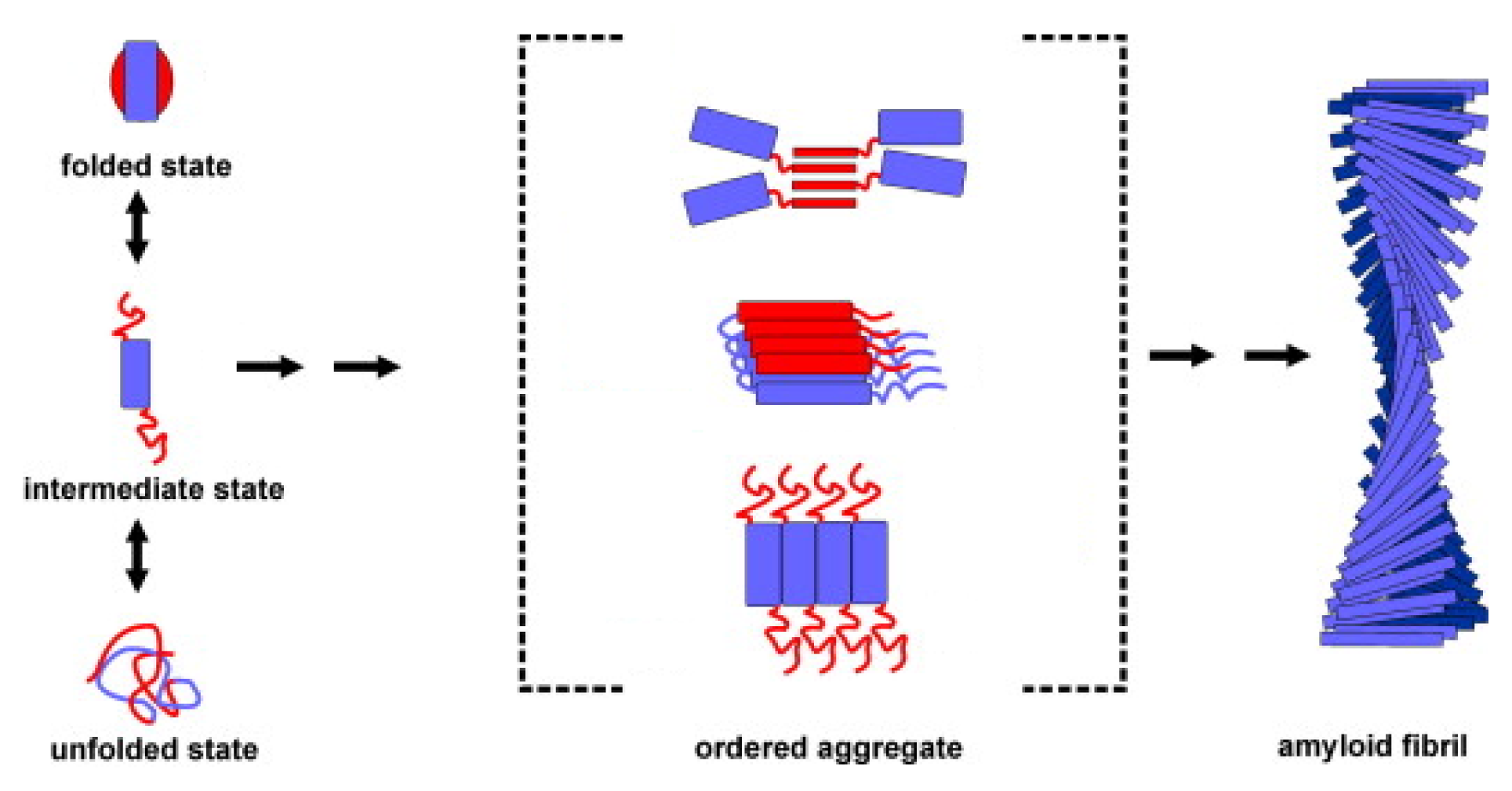
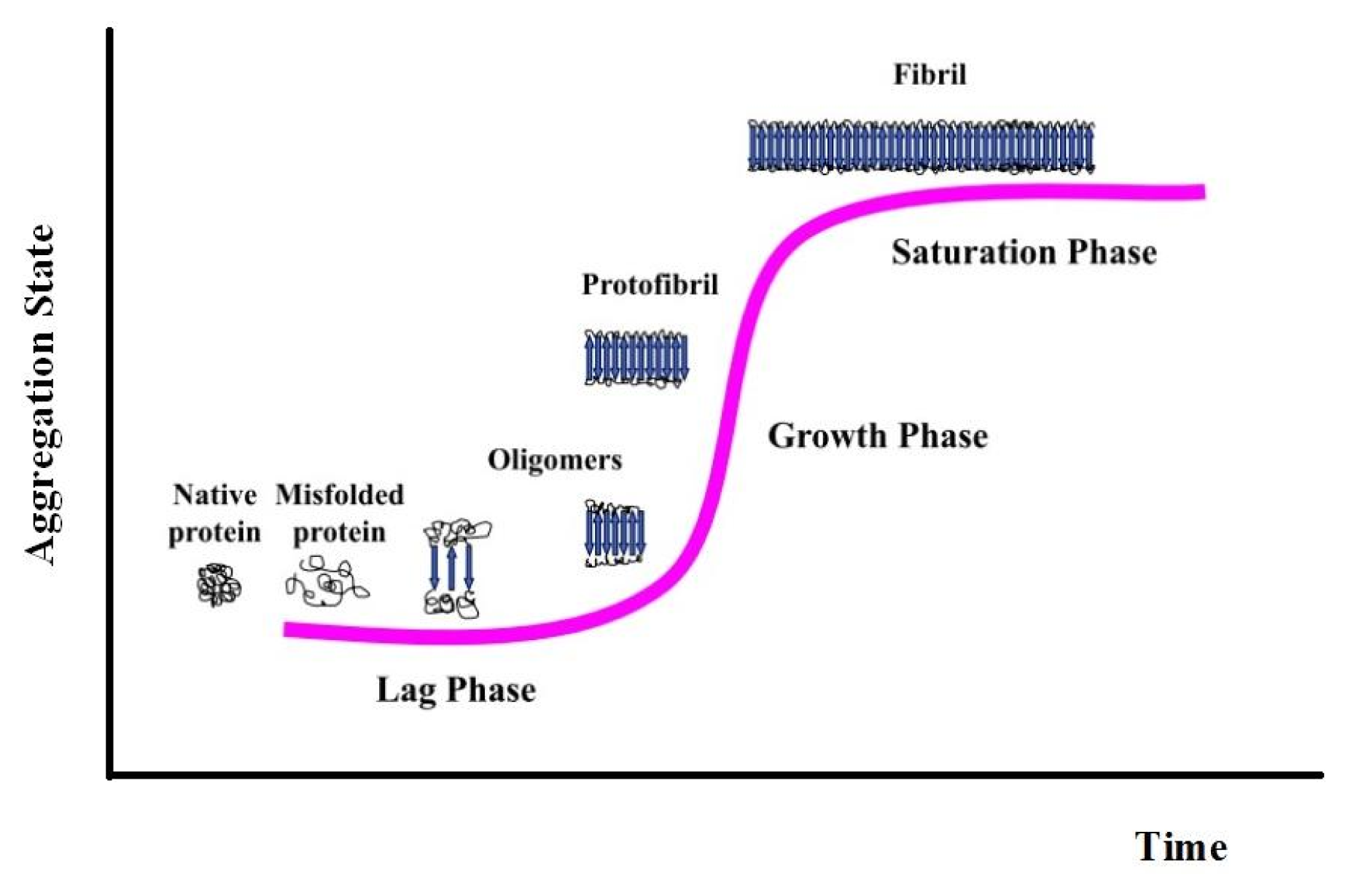
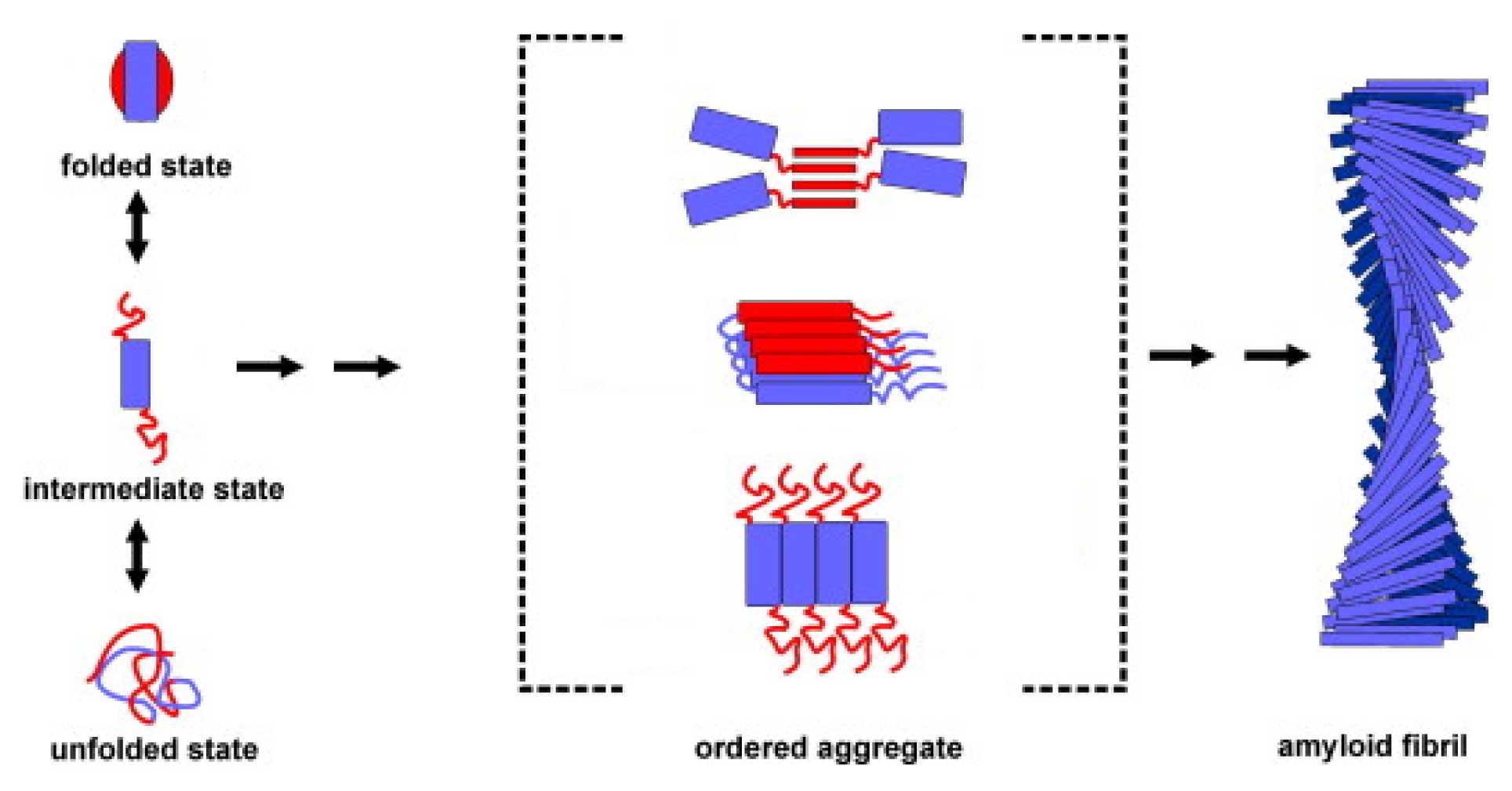
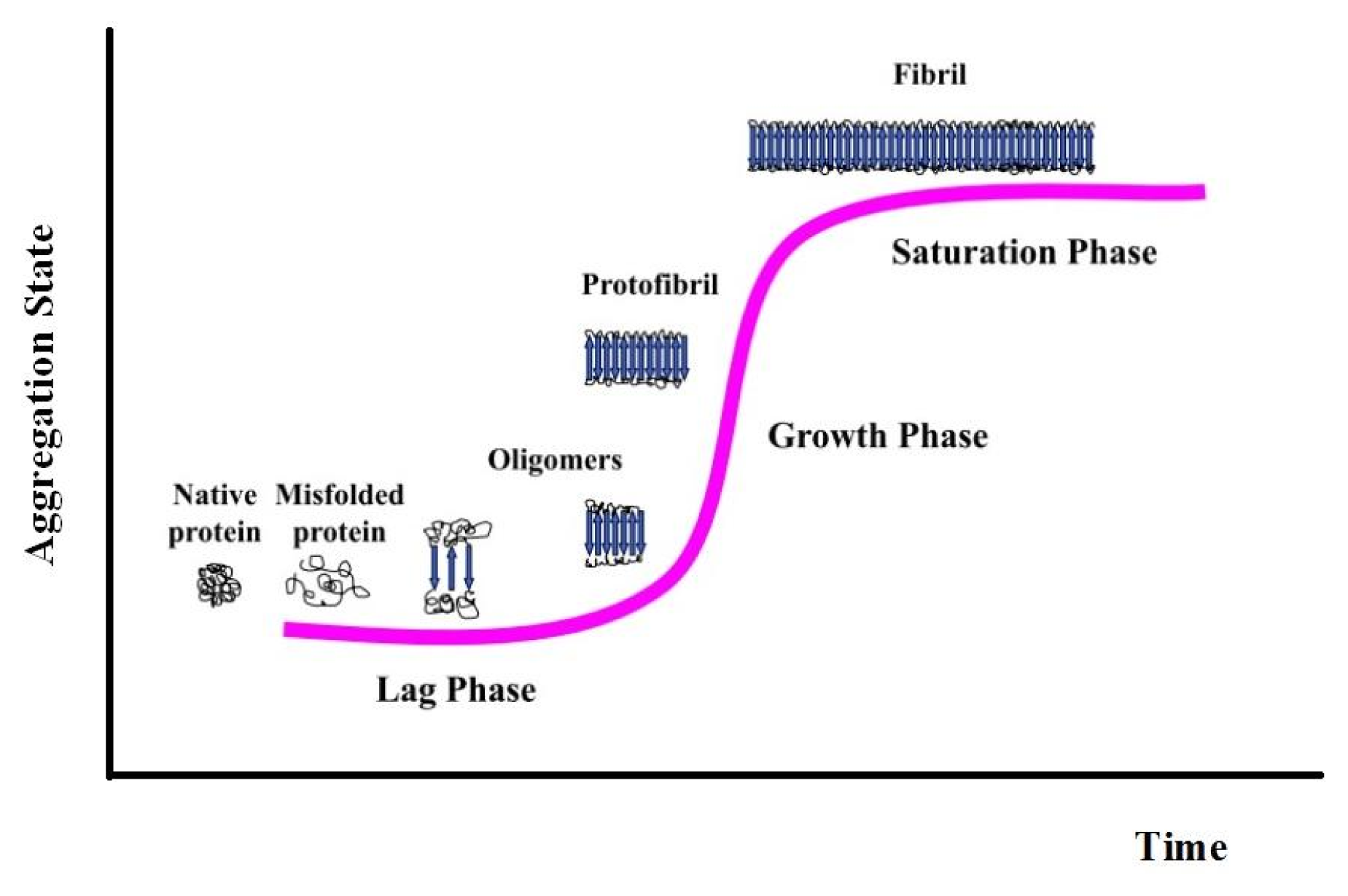
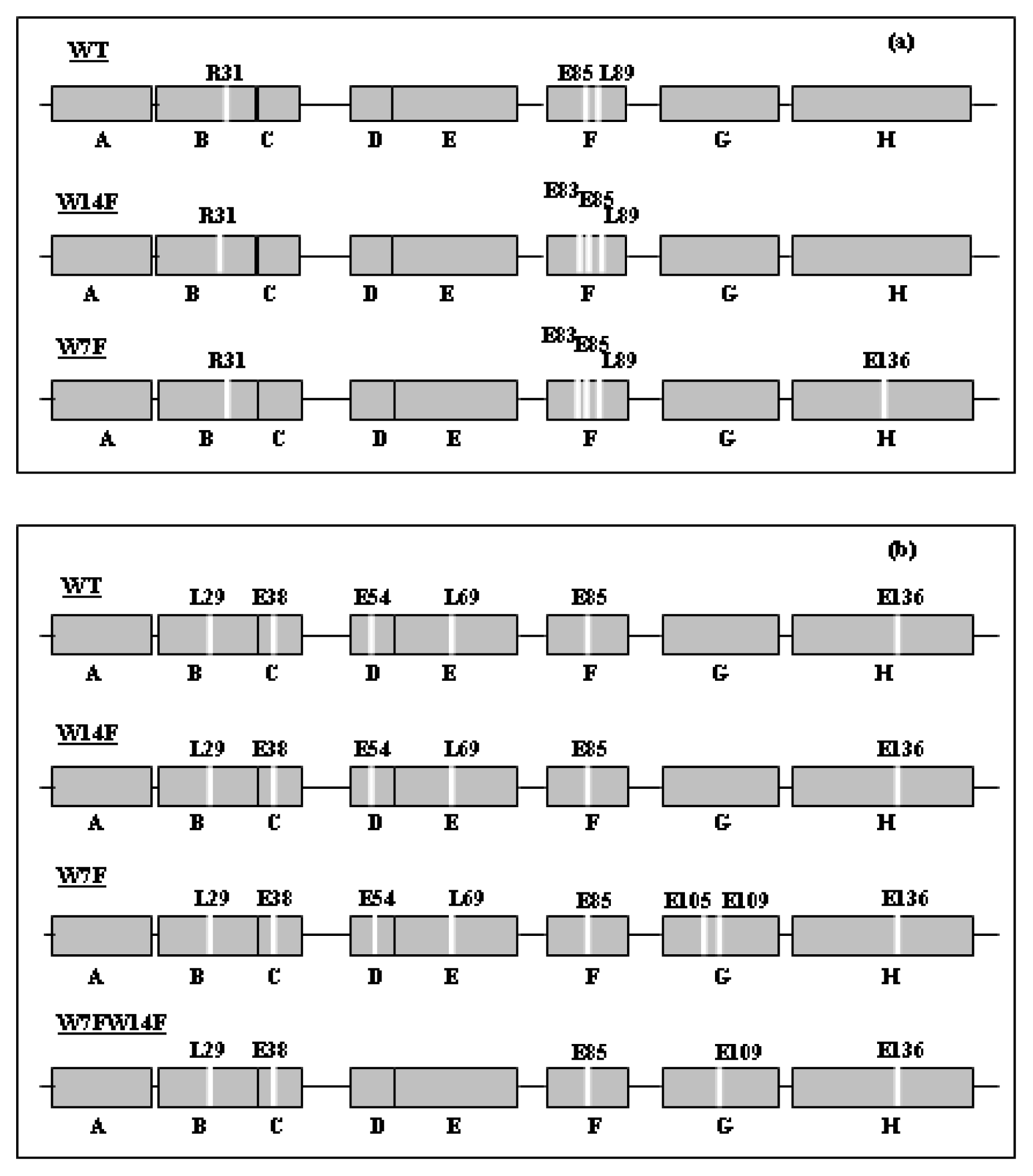
3. Apomyoglobin Misfolding and Amyloid Formation
4. Conclusions and Perspectives
Conflict of Interest
References
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med 2003, 81, 678–699. [Google Scholar]
- Stefani, M. Protein misfolding and aggregation: Newexamples in medicine and biology of the dark side of the protein world. Biochim. Biophys. Acta 2004, 1739, 5–25. [Google Scholar]
- Sipe, J.D.; Benson, M.D.; Buxbaum, J.N.; Ikeda, S.; Merlini, G.; Saraiva, M.J.; Westermark, P. Amyloid fibril protein nomenclature: 2010 recommendations of the nomenclature committee of the International Society of Amyloidosis. Amyloid 2010, 17, 101–104. [Google Scholar]
- Lansbury, P.T. Evolution of amyloid: What normal protein folding may tell us about fibrillogenesis and disease. Proc. Natl. Acad. Sci. USA 1999, 96, 3342–3344. [Google Scholar]
- Merlini, G.; Bellotti, V. Molecular mechanisms of amyloidosis. N. Engl. J. Med 2003, 349, 583–596. [Google Scholar]
- Sunde, M.; Blake, C.C. From the globular to the fibrous state: Protein structure and structural conversion in amyloid formation. Quart. Rev. Biophys 1998, 31, 1–39. [Google Scholar]
- Makin, O.S.; Atkins, E.; Sikorski, P.; Johansson, J.; Serpell, L.C. Molecular basis for amyloid fibril formation and stability. Proc. Natl. Acad. Sci. USA 2005, 102, 315–320. [Google Scholar]
- Makin, O.S.; Serpell, L.C. Structures for amyloid fibrils. FEBS J 2005, 272, 5950–5961. [Google Scholar]
- Jahn, T.R.; Makin, O.S.; Morris, K.L.; Marshall, K.E.; Tian, P.; Sikorski, P.; Serpell, L.C. The common architecture of cross-beta amyloid. J. Mol. Biol 2010, 395, 717–727. [Google Scholar]
- Sorrentino, A.; Giosafatto, C.V.; Sirangelo, I.; de Simone, C.; di Pierro, P.; Porta, R.; Mariniello, L. Higher susceptibility to amyloid fibril formation of the recombinant ovine prion protein modified by transglutaminase. Biochim. Biophys. Acta 2012, 1822, 1509–1515. [Google Scholar]
- Rochet, J.C.; Lansbury, P.J. Amyloid fibrillogenesis: Themes and variations. Curr. Opin. Struct. Biol 2000, 10, 60–68. [Google Scholar]
- Hurle, M.R.; Helms, L.R.; Li, L.; Chan, W.; Wetzel, R. A role for destabilizing amino acid replacements in light-chain amyloidosis. Proc. Natl. Acad. Sci. USA 1994, 91, 5446–5450. [Google Scholar]
- Goedert, M.; Ghetti, B.; Spillantini, M.G. Tau gene mutations in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Their relevance for understanding the neurogenerative process. Ann. N. Y. Acad. Sci 2000, 920, 74–83. [Google Scholar]
- Quintas, A.; Vaz, D.C.; Cardoso, I.; Saraiva, M.J.; Brito, R.M. Tetramer dissociation and monomer partial unfolding precedes protofibril formation in amyloidogenic transthyretin variants. J. Biol. Chem 2001, 276, 27202–27213. [Google Scholar]
- Canet, D.; Last, A.M.; Tito, P.; Sunde, M.; Spencer, A.; Archer, D.B.; Redfield, C.; Robinson, C.V.; Dobson, C.M. Local cooperativity in the unfolding of an amyloidogenic variant of human lysozyme. Nat. Struct. Biol 2002, 9, 308–315. [Google Scholar]
- Niraula, T.N.; Haraoka, K.; Ando, Y.; Li, H.; Yamada, H.; Akasaka, K. Decreased thermodynamic stability as a crucial factor for familial amyloidotic polyneuropathy. J. Mol. Biol 2002, 320, 333–342. [Google Scholar]
- Konno, T. Amyloid-induced aggregation and precipitation of soluble proteins: An electrostatic contribution of the Alzheimer’s beta (25–35) amyloid fibril. Biochemistry 2001, 40, 2148–2154. [Google Scholar]
- Chiti, F.; Calamai, M.; Taddei, N.; Stefani, M.; Ramponi, G.; Dobson, C.M. Studies of the aggregation of mutant proteins in vitro provide insights into the genetics of amyloid diseases. Proc. Natl. Acad. Sci. USA 2002, 99, 16419–16426. [Google Scholar]
- Chiti, F.; Stefani, M.; Taddei, N.; Ramponi, G.; Dobson, C.M. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature 2003, 424, 805–808. [Google Scholar]
- Tjernberg, L.; Hosia, W.; Bark, N.; Thyberg, J.; Johansson, J. Charge attraction and beta propensity are necessary for amyloid fibril formation from tetrapeptides. J. Biol. Chem 2002, 277, 43243–43246. [Google Scholar]
- Ciani, B.; Hutchinson, E.G.; Sessions, R.B.; Woolfson, D.N. A designed system for assessing how sequence affects alpha to beta conformational transitions in proteins. J. Biol. Chem 2002, 277, 10150–10155. [Google Scholar]
- Monsellier, E.; Ramazzotti, M.; de Laureto, P.P.; Tartaglia, G.G.; Taddei, N.; Fontana, A.; Vendruscolo, M.; Chiti, F. The distribution of residues in a polypeptide sequence is a determinant of aggregation optimized by evolution. Biophys. J 2007, 93, 4382–4391. [Google Scholar]
- Tartaglia, G.G.; Pawar, A.P.; Campioni, S.; Dobson, C.M.; Chiti, F.; Vendruscolo, M. Prediction of aggregation-prone regions in structured proteins. J. Mol. Biol 2008, 380, 425–443. [Google Scholar]
- Uversky, V.N.; Fink, A.L. Conformational constraints for amyloid fibrillation: The importance of being unfolded. Biochim. Biophys. Acta 2004, 1698, 131–153. [Google Scholar]
- Bousset, L.; Thomson, N.H.; Radford, S.E.; Melki, R. The yeast prion Ure2p retains its native alpha-helical conformation upon assembly into protein fibrils in vitro. EMBO J 2002, 21, 2903–2911. [Google Scholar]
- Chow, M.K.M.; Ellisdon, A.M.; Cabrita, L.D.; Bottomley, S.P. Polyglutamine expansion in ataxin-3 does not affect protein stability: Implications for misfolding and disease. J. Biol. Chem 2004, 279, 47643–47651. [Google Scholar]
- Pedersen, J.S.; Christensen, G.; Otze, D.E. Modulation of S6 fibrillation by unfolding rates and gatekeeper residues. J. Mol. Biol 2004, 341, 575–588. [Google Scholar]
- Plakoutsi, G.; Taddei, N.; Stefani, M.; Chiti, F. Aggregation of the Acylphosphatase from Sulfolobus solfataricus: The folded and partially unfolded states can both be precursors for amyloid formation. J. Biol. Chem 2004, 279, 14111–14119. [Google Scholar]
- Chiti, F.; Webster, P.; Taddei, N.; Clark, A.; Stefani, M.; Ramponi, G.; Dobson, C.M. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci USA 1999, 96, 3590–3594. [Google Scholar]
- Dobson, C.M. The structural basis of protein folding and its links with human disease. Philos. Trans. R. Soc. Lond. B 2001, 356, 133–145. [Google Scholar]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar]
- Stefani, M. Structural features and cytotoxicity of amyloid oligomers: Implications in Alzheimer’s disease and other diseases with amyloid deposits. Prog. Neurobiol 2012, 99, 226–245. [Google Scholar]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem 2006, 75, 333–366. [Google Scholar]
- Wiseman, R.L.; Powers, E.T.; Kelly, J.W. Partitioning conformational intermediates between competing refolding and aggregation pathways: Insights into transthyretin amyloid disease. Biochemistry 2005, 44, 16612–16623. [Google Scholar]
- Lashuel, H.A.; Hartley, D.M.; Petre, B.M.; Wall, J.S.; Simon, M.N.; Walz, T.; Lansbury, P.T., Jr. Mixtures of wild-type and a pathogenic (E22G) form of Abeta40 in vitro accumulate protofibrils, including amyloid pores. J. Mol. Biol. 2003, 332, 795–808. [Google Scholar]
- Poirier, M.A.; Li, H.; Macosko, J.; Cail, S.; Amzel, M.; Ross, C.A. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrillization. J. Biol. Chem 2002, 277, 41032–41037. [Google Scholar]
- Serpell, L.C.; Sunde, M.; Benson, M.D.; Tennent, G.A.; Pepys, M.B.; Fraser, P.E. The protofilament substructure of amyloid fibrils. J. Mol. Biol 2000, 300, 1033–1039. [Google Scholar]
- Maliskauskas, M.; Zamotin, V.; Jass, J.; Noppe, W.; Dobson, C.M.; Morozova-Roche, L.A. Amyloid protofilaments from the calcium-binding protein equine lysozyme: Formation of ring and linear structures depends on pH and metal ion concentration. J. Mol. Biol 2003, 330, 879–890. [Google Scholar]
- Hatters, D.M.; MacPhee, C.E.; Lawrence, L.J.; Sawyer, W.H.; Howlett, G.J. Human apolipoprotein C-II forms twisted amyloid ribbons and closed loops. Biochemistry 2000, 39, 8276–8283. [Google Scholar]
- Porat, Y.; Kolusheva, S.; Jelinek, R.; Gazil, E. The human islet amyloid polypeptide forms transient membrane-active prefibrillar assemblies. Biochemistry 2003, 42, 10971–10977. [Google Scholar]
- Ding, T.T.; Lee, S.J.; Rochet, J.C.; Lansbury, P.T., Jr. Annular alpha-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry 2002, 41, 10209–10217. [Google Scholar]
- Relini, A.; Torrasa, S.; Rolandi, R.; Gliozzi, A.; Rosano, C.; Canale, C.; Bolognesi, M.; Plakoutsi, G.; Bucciantini, M.; Chiti, F.; et al. Monitoring the process of HypF fibrillization and liposome permeabilization by protofibrils. J. Mol. Biol 2004, 338, 943–957. [Google Scholar]
- Lee, C.C.; Nayak, A.; Sethuraman, A.; Belfort, G.; McRae, G.J. A three-stage kinetic model of amyloid fibrillation. Biophys. J 2007, 92, 3448–3458. [Google Scholar]
- Harper, J.D.; Lansbury, P.T., Jr. Models of amyloid seeding in Alzheimer’s disease and scrapie: Mechanistic truths and physiological consequences of the time dependent solubility of amyloid proteins. Annu. Rev. Biochem. 1997, 66, 385–407. [Google Scholar]
- Serpell, L.C.; Sunde, M.; Blake, C.C. The molecular basis of amyloidosis. Cell. Mol. Life Sci 1997, 53, 871–887. [Google Scholar]
- Bhak, G.; Choe, Y.J.; Paik, S.R. Mechanism of amyloidogenesis: Nucleation-dependent fibrillation versus double-concerted fibrillation. BMB Rep 2009, 42, 541–551. [Google Scholar]
- Nguyen, H.D.; Hall, H.K. Molecular dynamics simulations of spontaneous fibril formation by random-coil peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 16180–16185. [Google Scholar]
- Harper, J.D.; Lieber, C.M.; Lansbury, P.T., Jr. Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer's disease amyloid-beta protein. Chem. Biol. 1997, 4, 951–959. [Google Scholar]
- Stefani, M. Protein folding and misfolding on surfaces. Int. J. Mol. Sci 2008, 9, 2515–2542. [Google Scholar]
- Necula, M.; Kayed, R.; Milton, S.; Glabe, C. Small molecules inhibitors distinguish between amyloid beta oligomerization and fibrillization pathways. J. Biol. Chem 2007, 28, 10311–10324. [Google Scholar]
- Uversky, V.N. Mysterious oligomerization of the amyloidogenic proteins. FEBS J 2010, 277, 2940–2953. [Google Scholar]
- Baldwin, R.L.; Rose, G.D. Is protein folding hierarchic? II. Folding intermediates and transition states. Trends Biochem. Sci 1999, 24, 77–83. [Google Scholar]
- Aurora, R.; Creamer, T.P.; Srinivasan, R.; Rose, G.D. Local interactions in protein folding: Lessons from the alpha-helix. J. Biol. Chem 1997, 272, 1413–1416. [Google Scholar]
- Baldwin, R.L. In search of the energetic role of peptide hydrogen bonds. J. Biol. Chem 2003, 278, 17581–17588. [Google Scholar]
- Kauzmann, W. Some factors in the interpretation of protein denaturation. Adv. Protein Chem 1959, 14, 1–63. [Google Scholar]
- Tanford, C. The Hydrophobic Effect; Wiley: New York, NY, USA, 1973; pp. 120–125. [Google Scholar]
- Dill, K.A. Theory for the folding and stability of globular proteins. Biochemistry 1985, 24, 1501–1509. [Google Scholar]
- Kim, P.S.; Baldwin, R.L. Intermediates in the folding reactions of small proteins. Annu. Rev. Biochem 1990, 59, 631–660. [Google Scholar]
- Hughson, F.M.; Wright, P.E.; Baldwin, R.L. Structural characterization of a partly folded apomyoglobin intermediate. Science 1990, 249, 1544–1548. [Google Scholar]
- Kay, M.S.; Baldwin, R.L. Packing interactions in the apomyglobin folding intermediate. Nat. Struct. Biol 1996, 3, 439–445. [Google Scholar]
- Barrick, D.; Baldwin, R.L. Stein and Moore award address the molten globule state intermediate of apomyoglobin and the process of protein folding. Protein Sci 1993, 2, 869–876. [Google Scholar]
- Sirangelo, I.; Bismuto, E.; Tavassi, S.; Irace, G. Apomyoglobin folding intermediates characterized by the hydrophobic fluorescent probe 8-anilino-1-naphthalene sulfonate. Biochem. Biophys. Acta 1998, 1385, 69–77. [Google Scholar]
- Sirangelo, I.; Dal Piaz, F.; Malmo, C.; Casillo, M.; Birolo, L.; Pucci, P.; Marino, G.; Irace, G. Hexafluoroisopropanol and acid destabilized forms of apomyoglobin exhibit structural differences. Biochemistry 2003, 42, 312–319. [Google Scholar]
- Sirangelo, I.; Iannuzzi, C.; Malmo, C.; Irace, G. Tryptophanyl substitutions in apomyoglobin affect conformation and dynamic properties of AGH subdomain. Biopolymers 2003, 70, 649–654. [Google Scholar]
- 6Uzawa, T.; Akiyama, S.; Kimura, T.; Takahashi, S.; Ishimori, K.; Morishima, I.; Fujisawa, T. Collapse and search dynamics of apomyoglobin folding revealed by submillisecond observations of alphahelical content and compactness. Proc. Natl. Acad. Sci. USA 2004, 101, 1171–1176. [Google Scholar]
- Jennings, P.A.; Wright, P.E. Formation of a molten globule intermediate early in the kinetic folding pathway of apomyoglobin. Science 1993, 262, 892–896. [Google Scholar]
- Sirangelo, I.; Malmo, C.; Casillo, M.; Mezzogiorno, A.; Papa, M.; Irace, G. Tryptophanyl substitutions in apomyoglobin determine protein aggregation and amyloid-like fibril formation at physiological pH. J. Biol. Chem 2002, 277, 45887–45891. [Google Scholar]
- Sirangelo, I.; Malmo, C.; Iannuzzi, C.; Mezzogiorno, A.; Bianco, M.R.; Papa, M.; Irace, G. Fibrillogenesis and cytotoxic activity of the amyloid-forming apomyoglobin mutant W7FW14F. J. Biol. Chem 2004, 279, 13183–13189. [Google Scholar]
- Sirangelo, I.; Tavassi, S.; Martelli, P.L.; Casadio, R.; Irace, G. The effect of tryptophanyl substitution on folding and structure of myoglobin. Eur. J. Biochem 2000, 267, 3937–3945. [Google Scholar]
- Fändrich, M.; Forge, V.; Buder, K.; Kittler, M.; Dobson, C.M.; Diekmann, S. Myoglobin forms amyloid fibrils by association of unfolded polypeptide segments. Proc. Natl. Acad. Sci. USA 2003, 100, 15463–15468. [Google Scholar]
- Vilasi, S.; Sarcina, R.; Maritato, R.; de Simone, A.; Irace, G.; Sirangelo, I. Heparin induces harmless fibril formation in amyloidogenic W7FW14F apomyoglobin and amyloid aggregation in wild-type protein in vitro. PLoS One 2011, 6, e22076. [Google Scholar]
- Infusini, G.; Iannuzzi, C.; Vilasi, S.; Birolo, L.; Pagnozzi, D.; Pucci, P.; Irace, G.; Sirangelo, I. Resolution of the effects induced by W→F substitutions on the conformation and dynamics of the amyloid-forming apomyoglobin mutant W7FW14F. Eur. Biophys. J 2012, 41, 615–627. [Google Scholar]
- Iannuzzi, C.; Vilasi, S.; Portaccio, M.; Irace, G.; Sirangelo, I. Heme binding inhibits the fibrillization of amyloidogenic apomyoglobin and determines lack of aggregate cytotoxicity. Protein Sci 2007, 16, 507–516. [Google Scholar]
- Chow, C.C.; Chow, C.; Raghunathan, V.; Huppert, T.J.; Kimball, E.B.; Cavagnero, S. Chain length dependence of apomyoglobin folding: Structural evolution from misfolded sheets to native helices. Biochemistry 2003, 42, 7090–7099. [Google Scholar]
- Ribeiro, E.A., Jr; Ramos, C.H.I. Circular permutation and deletion of myoglobin indicate that the correct position of its N-terminus is required foe native stability and solubility but not for nativelike heme binding and folding. Biochemistry 2005, 44, 4699–4709. [Google Scholar]
- Nishimura, C.; Dyson, H.J.; Wright, P. Identification of native and non-native structure in kinetic folding intermediates of apomyoglobin. J. Mol. Biol 2006, 355, 139–156. [Google Scholar]
- Nishimura, C.; Dyson, H.J.; Wright, P.E. Energetic frustration of apomyoglobin folding: Role of the B helix. J. Mol. Biol 2010, 396, 1319–1328. [Google Scholar]
- Infusini, G.; Iannuzzi, C.; Vilasi, S.; Maritato, R.; Birolo, L.; Pagnozzi, D.; Pucci, P.; Irace, G.; Sirangelo, I. W-F substitutions in apomyoglobin increase the local flexibility of the N-terminal region causing amyloid aggregation: A H/D exchange study. Protein Pept. Lett. 2013, in press. [Google Scholar]
- Fandrich, M.; Fletcher, M.A.; Dobson, C.M. Amyloid fibrils from muscle myoglobin. Nature 2001, 410, 165–166. [Google Scholar]
- Waltho, J.P.; Feher, V.A.; Merutka, G.; Dyson, H.J.; Wright, P.E. Peptide models of protein folding initiation sites. 1. Secondary structure formation by peptides corresponding to the G- and H-helices of myoglobin. Biochemistry 1993, 32, 6337–6347. [Google Scholar]
- Picotti, P.; de Franceschi, G.; Frare, E.; Spolaore, B.; Zambonin, M.; Chiti, F.; de Laureto, P.P.; Fontana, A. Amyloid fibril formation and disaggregation of fragment 1–29 of apomyoglobin: Insights into the effect of pH on protein fibrillogenesis. J. Mol. Biol. 2007, 367, 1237–1245. [Google Scholar]
- Fabiani, E.; Stadler, A.M.; Madern, D.; Koza, M.M.; Tehei, M.; Hirai, M.; Zaccai, G. Dynamics of apomyoglobin in the a-to-b transition and of partially unfolded aggregated protein. Eur. Biophys. J 2009, 38, 237–244. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH 7.0 | wt | W7F | W14 | W7FW14F |
|---|---|---|---|---|
| α | 0.65 | 0.52 | 0.56 | 0.56 |
| β | 0.04 | 0.09 | 0.07 | 0.09 |
| Turn | 0.09 | 0.14 | 0.12 | 0.14 |
| Unordered | 0.22 | 0.25 | 0.25 | 0.21 |
| pH 4.0 | wt | W7F | W14 | W7FW14F |
| α | 0.46 | 0.26 | 0.37 | 0.28 |
| β | 0.10 | 0.23 | 0.13 | 0.22 |
| Turn | 0.17 | 0.21 | 0.21 | 0.22 |
| Unordered | 0.27 | 0.30 | 0.29 | 0.28 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Iannuzzi, C.; Maritato, R.; Irace, G.; Sirangelo, I. Misfolding and Amyloid Aggregation of Apomyoglobin. Int. J. Mol. Sci. 2013, 14, 14287-14300. https://doi.org/10.3390/ijms140714287
Iannuzzi C, Maritato R, Irace G, Sirangelo I. Misfolding and Amyloid Aggregation of Apomyoglobin. International Journal of Molecular Sciences. 2013; 14(7):14287-14300. https://doi.org/10.3390/ijms140714287
Chicago/Turabian StyleIannuzzi, Clara, Rosa Maritato, Gaetano Irace, and Ivana Sirangelo. 2013. "Misfolding and Amyloid Aggregation of Apomyoglobin" International Journal of Molecular Sciences 14, no. 7: 14287-14300. https://doi.org/10.3390/ijms140714287
APA StyleIannuzzi, C., Maritato, R., Irace, G., & Sirangelo, I. (2013). Misfolding and Amyloid Aggregation of Apomyoglobin. International Journal of Molecular Sciences, 14(7), 14287-14300. https://doi.org/10.3390/ijms140714287