UVB-Stimulated TNFα Release from Human Melanocyte and Melanoma Cells Is Mediated by p38 MAPK


Abstract
:1. Introduction
2. Results
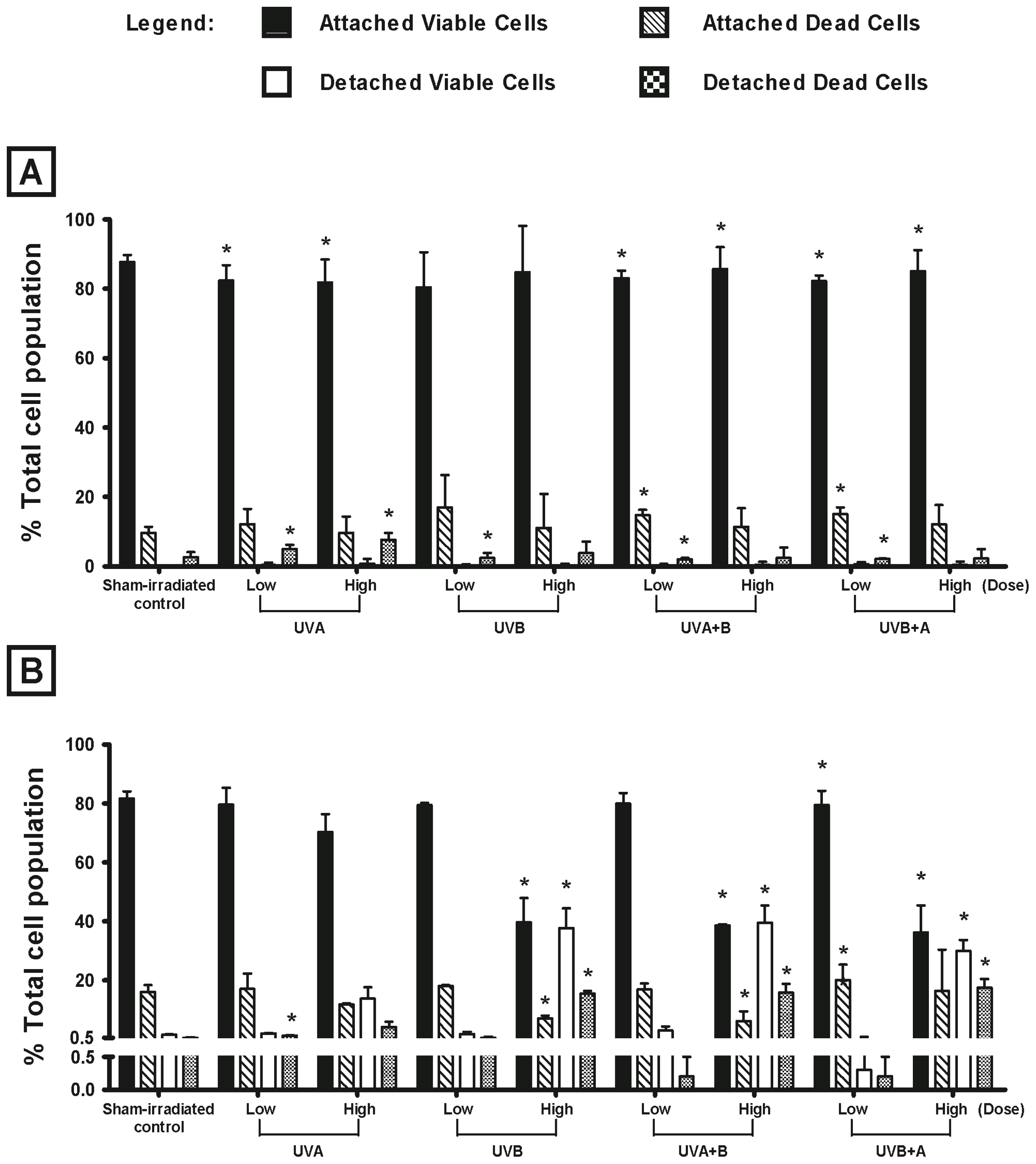
2.1. Effect of UV Radiation on the Viability of Melanocyte-Derived Cells
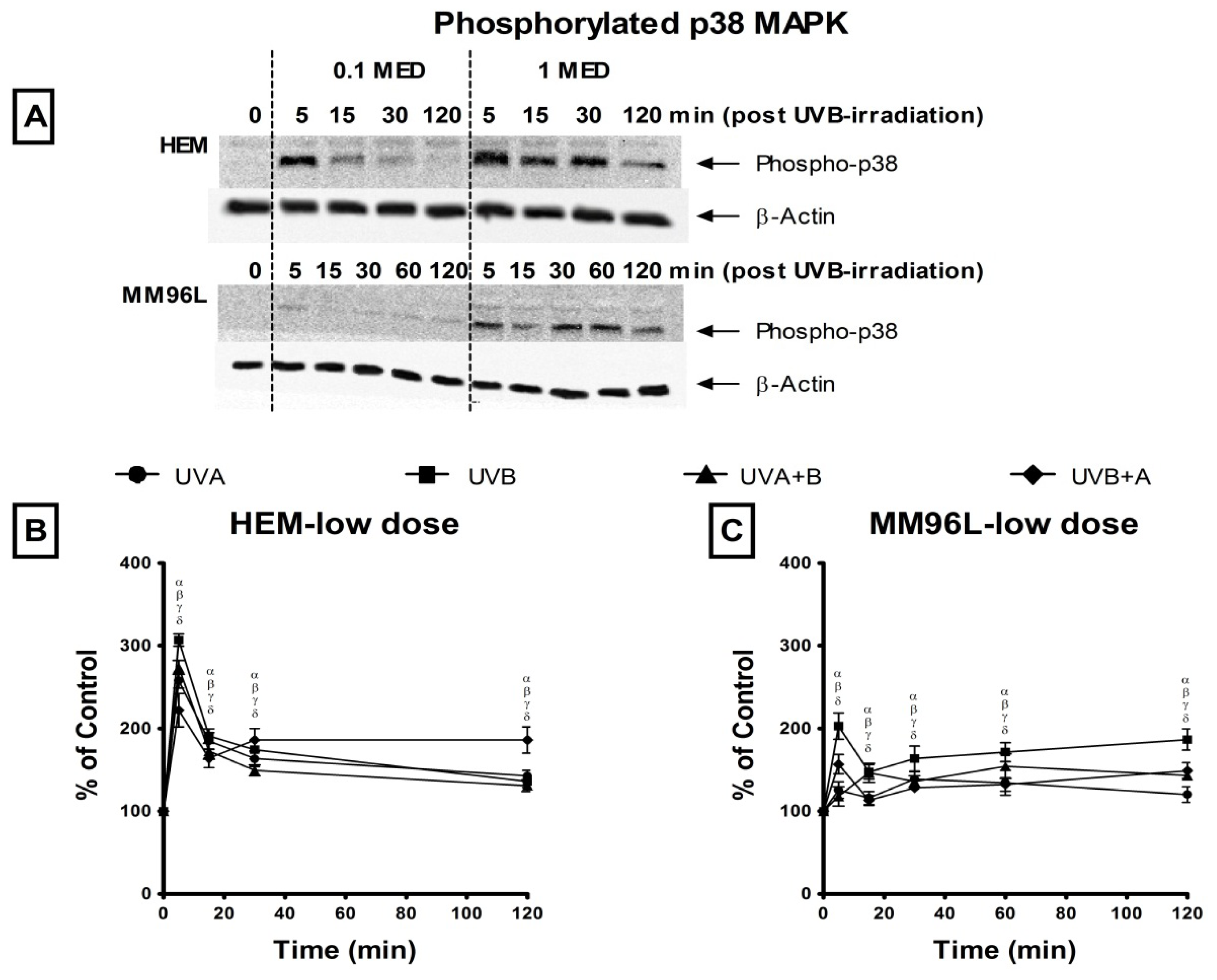
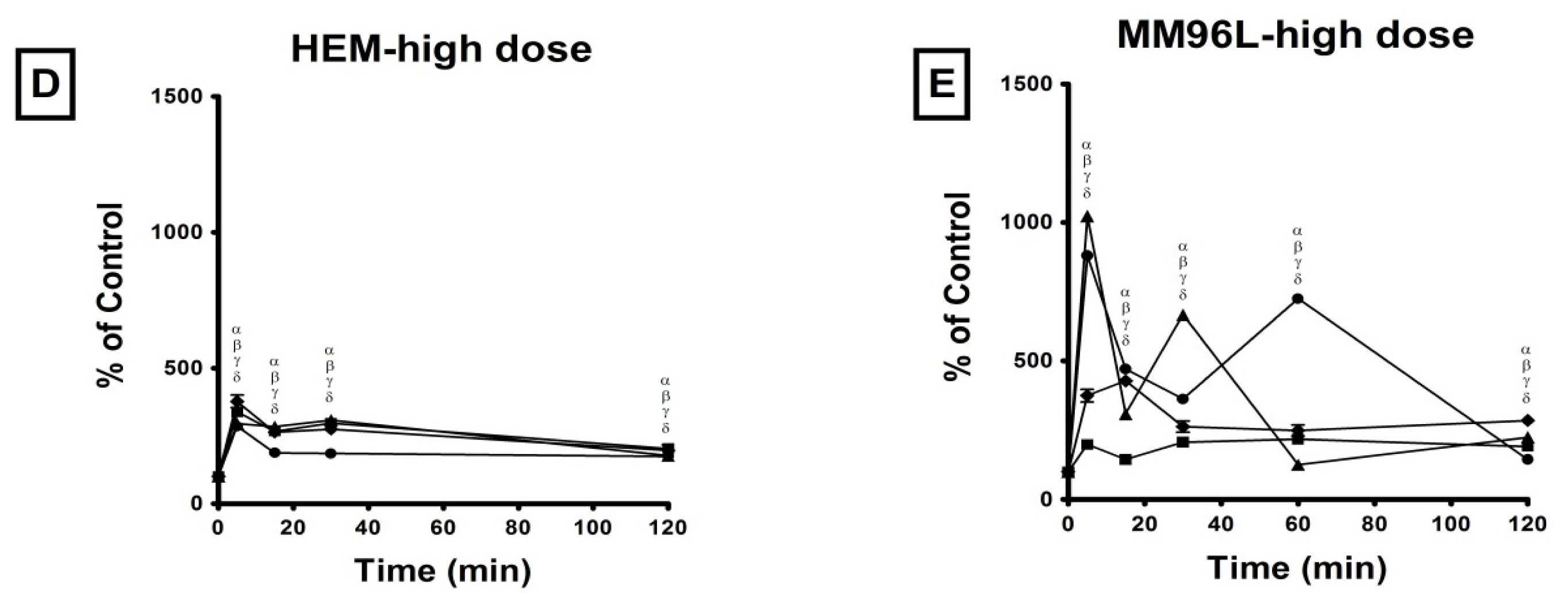
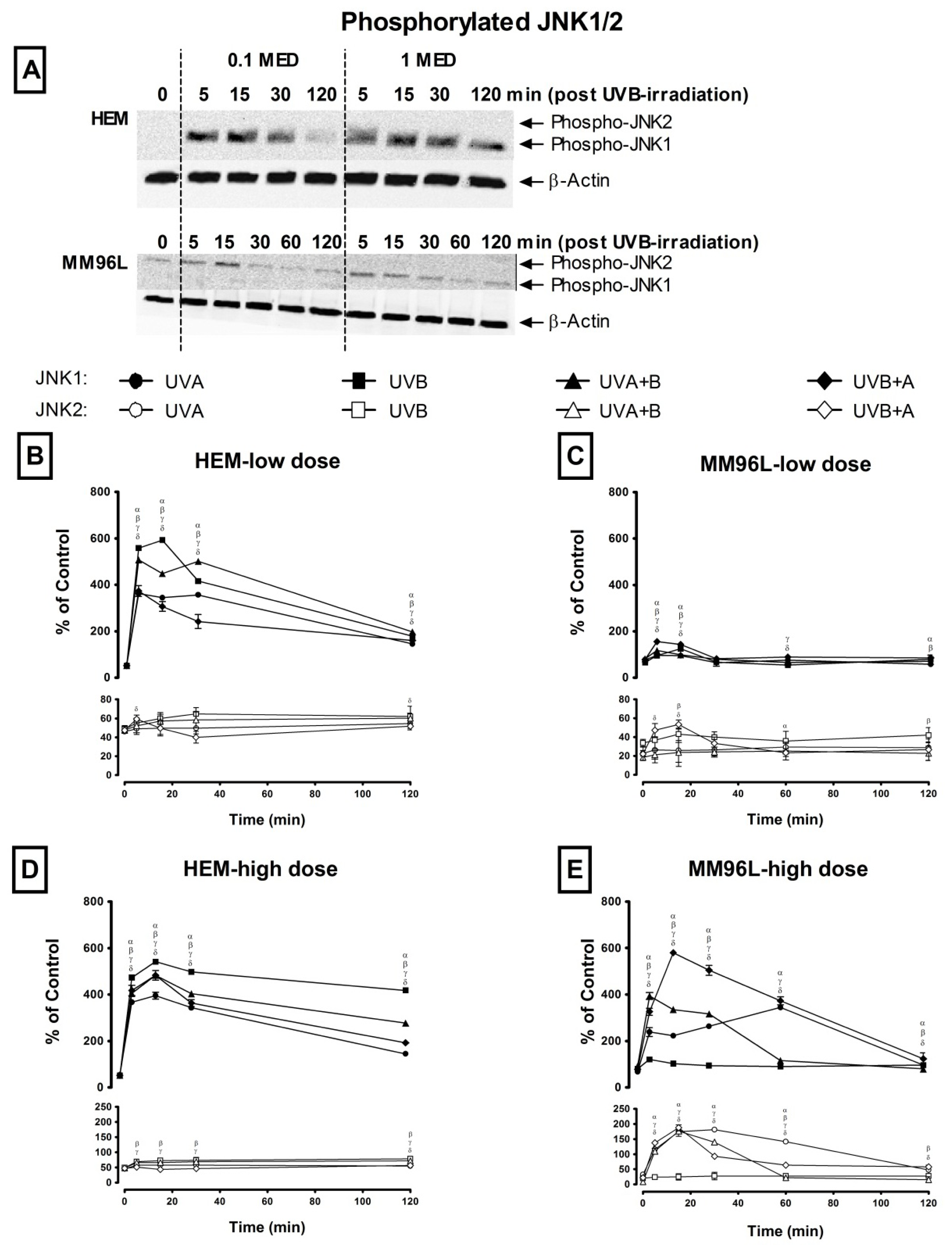
2.2. Effect of UV Radiation on the Activation of p38 MAPK, JNK and NFκB Pathways in Melanocyte-Derived Cells
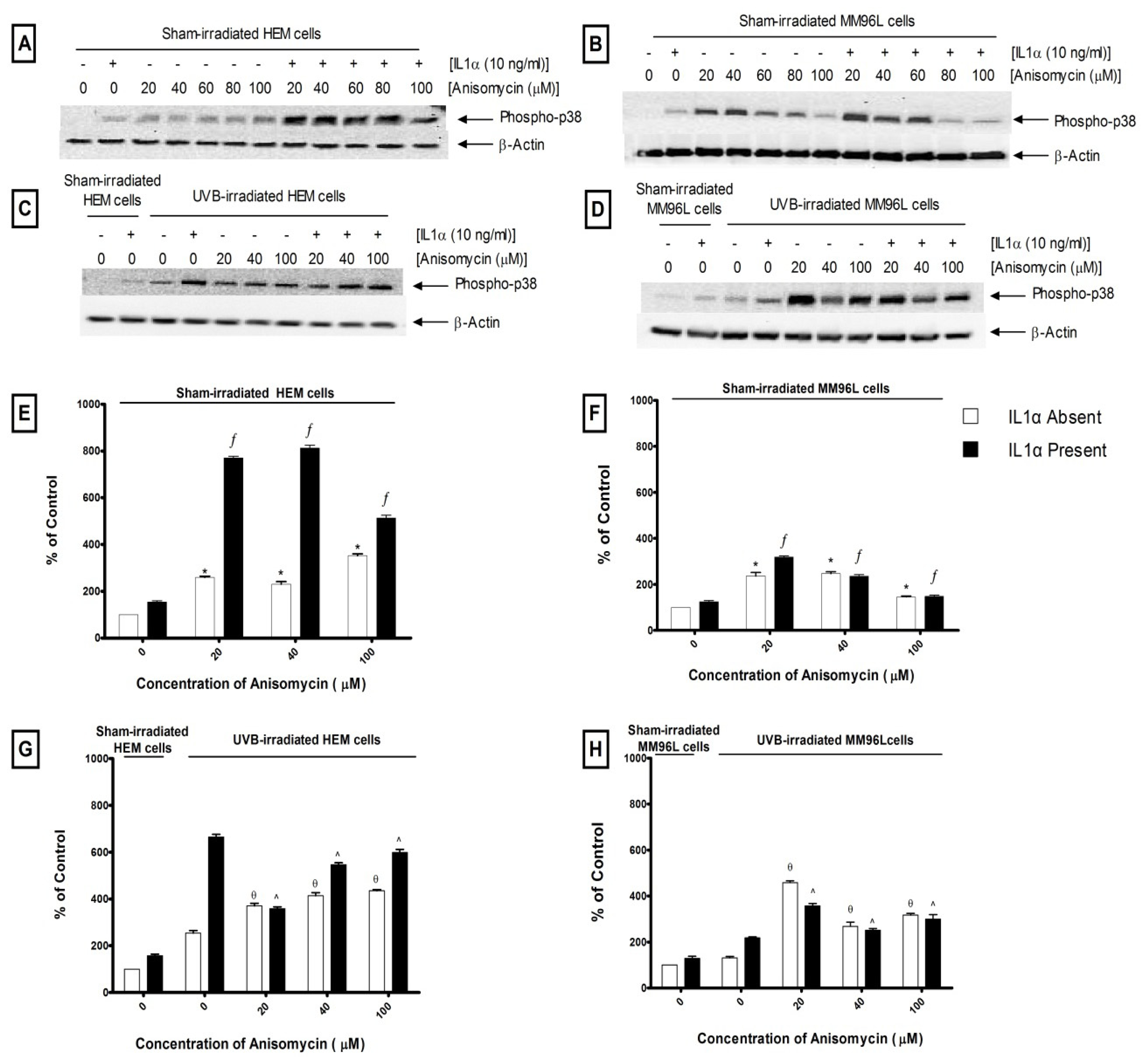
2.2.1. p38 MAPK Pathway
2.2.2. JNK Pathway
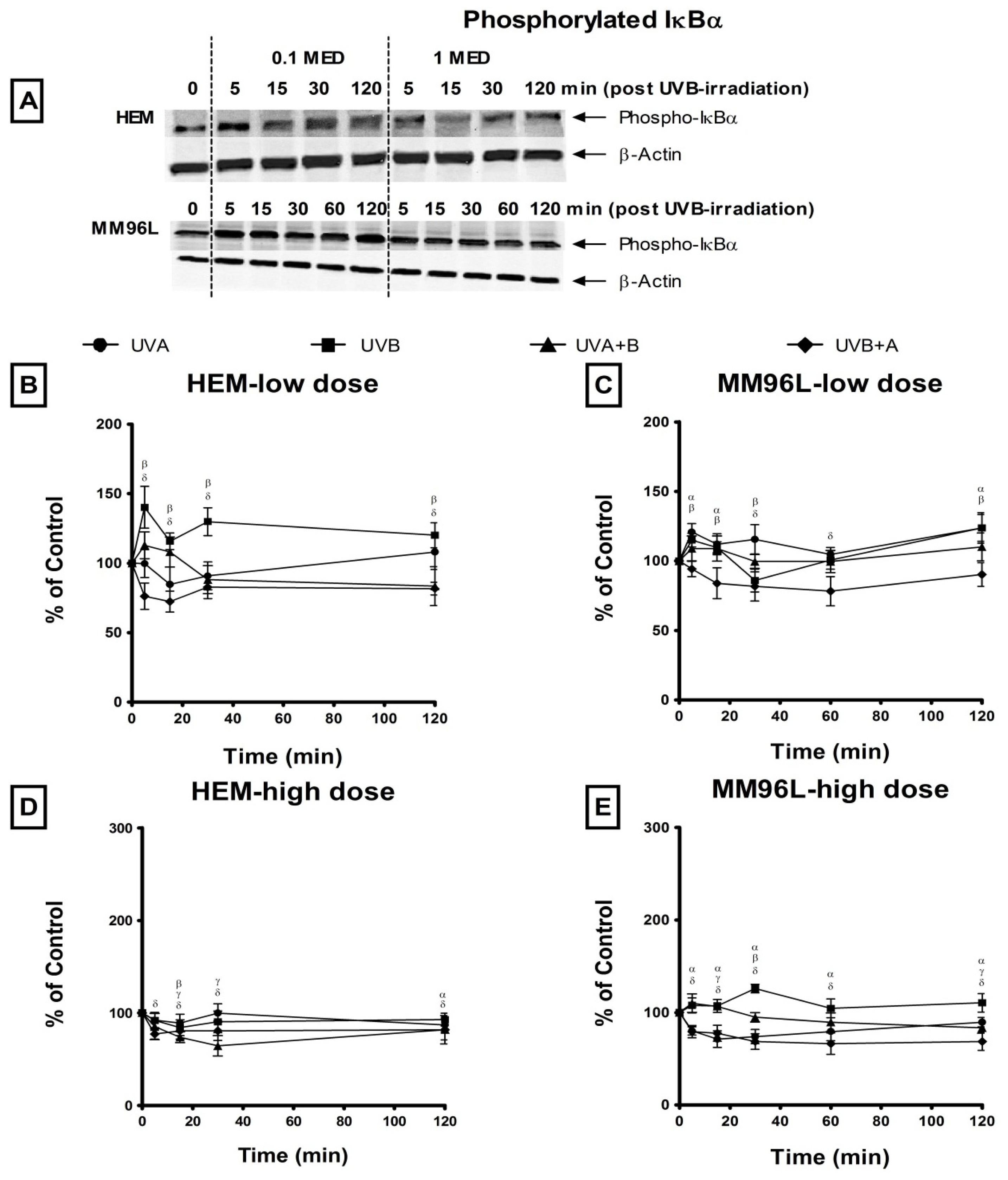
2.2.3. NFκB Pathway
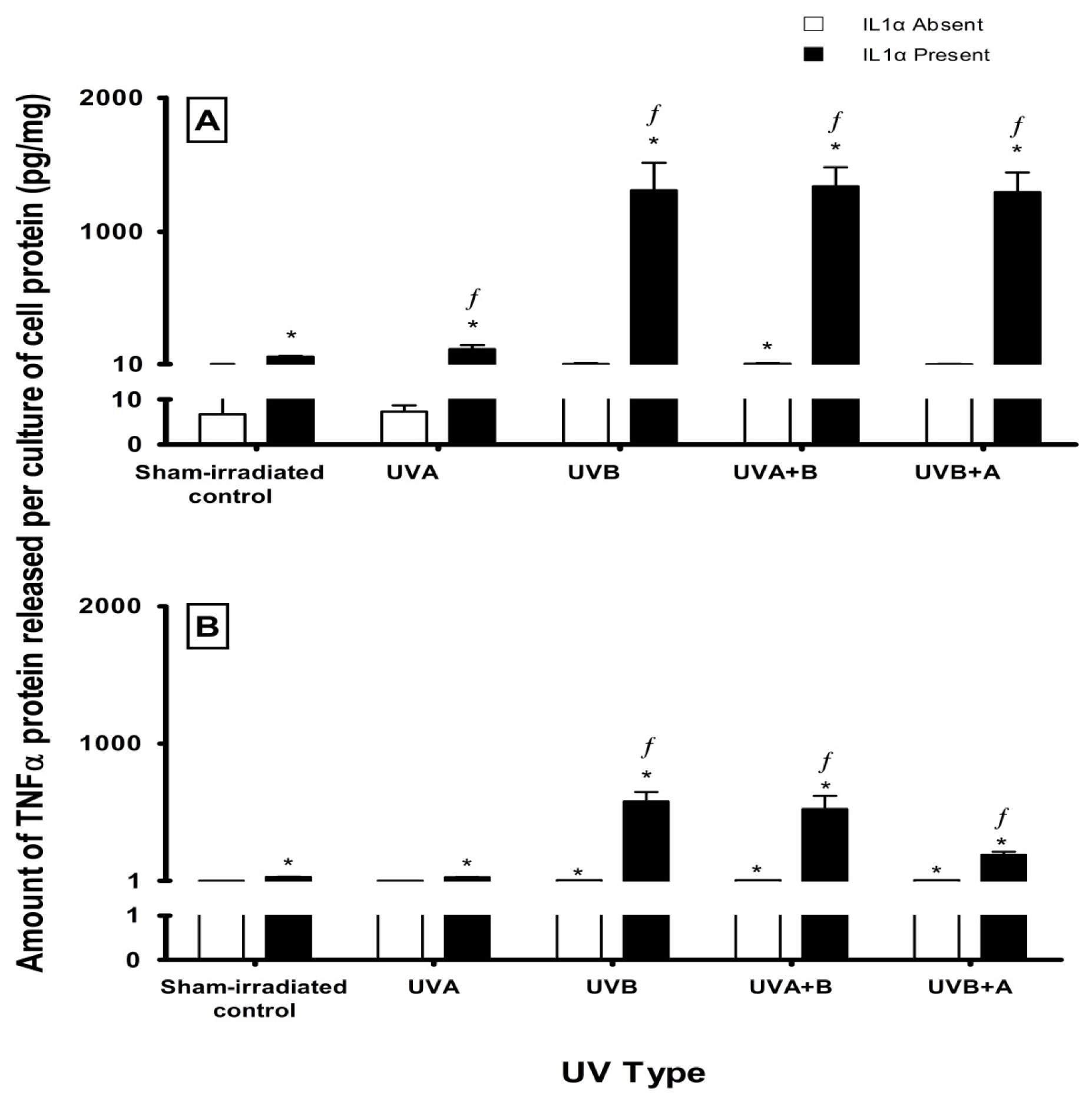
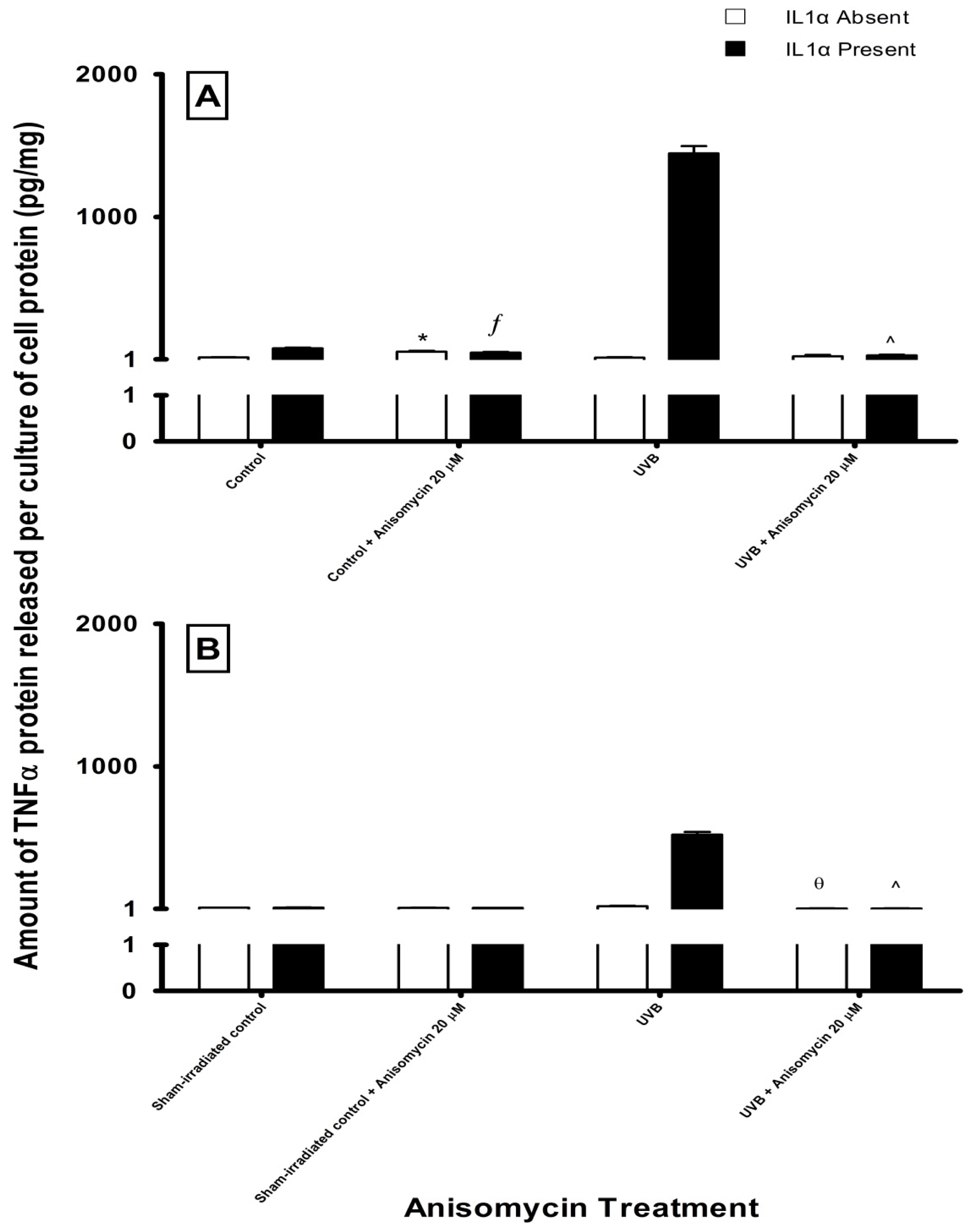
2.3. Effect of UV Radiation and IL1α on TNFα Release in Melanocyte-Derived Cells
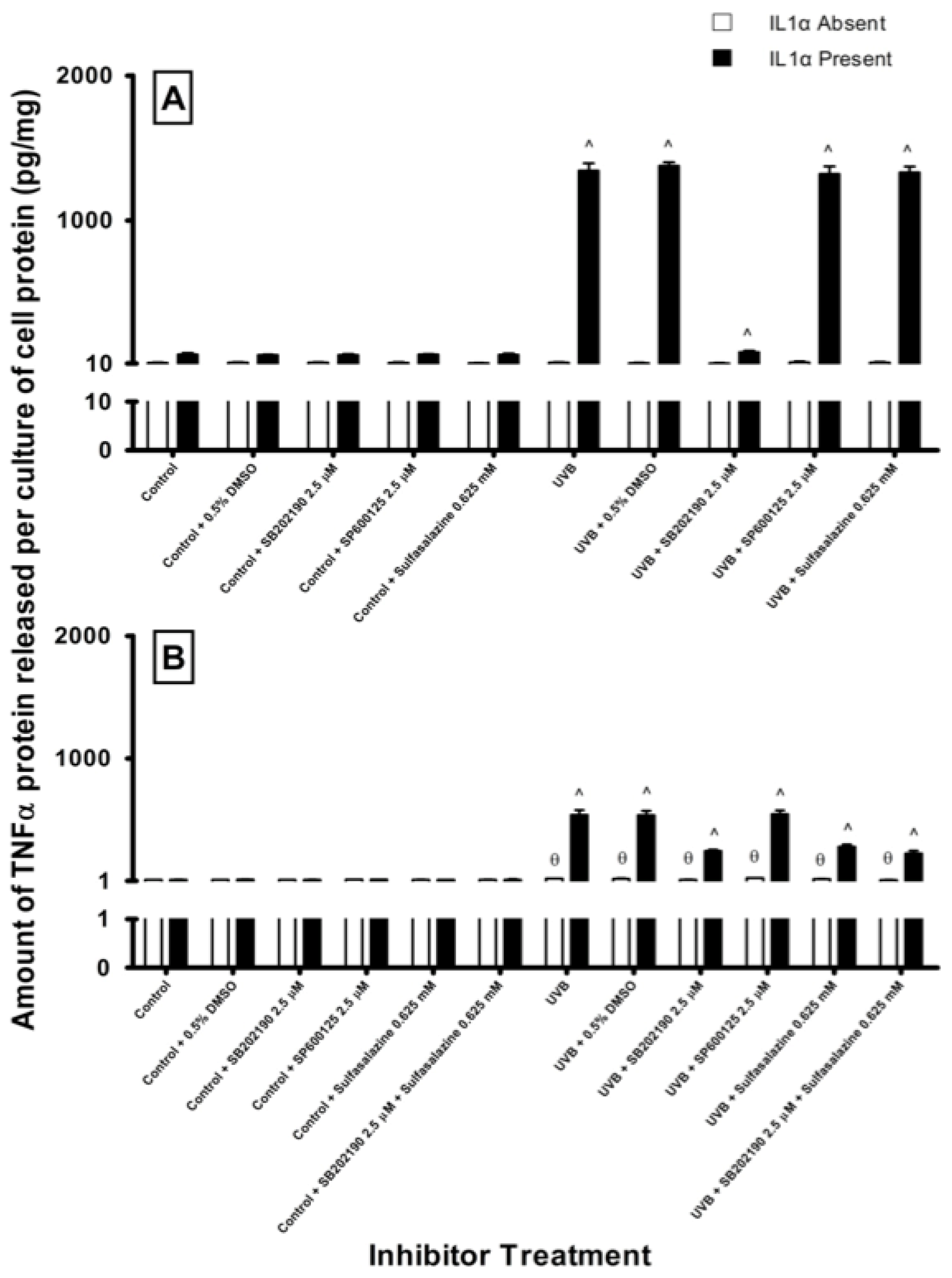
2.4. Effect of Pathway Specific Inhibitors on UV-Induced TNFα Release in Melanocyte-Derived Cells
3. Discussion
3.1. Choice of Cell Types and UV Radiation
3.2. Effect of UV Radiation on the Activation of the p38 MAPK, JNK and NFκB Pathways in Melanocyte-Derived Cells
3.3. Effect of UV Radiation and IL1α on TNFα Release in Melanocyte-Derived Cells
4. Experimental Section
4.1. Materials
4.2. Cell Types
Subculture
4.3. UV-Irradiation
4.4. Cell Viability
4.5. Inhibitor and Anisomycin Studies
4.6. Western Blotting
4.7. ELISA
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Muthusamy, V.; Piva, T.J. Melanoma Cell Signaling: Looking Beyond RAS-RAF-MEK. In Skin Cancers–Risk Factors, Prevention and Therapy; Porta, C.L., Ed.; InTech: Rijeka, Croatia, 2011; pp. 87–108. [Google Scholar]
- Sullivan, R.J.; Flaherty, K. MAP kinase signaling and inhibition in melanoma. Oncogene 2013, 32, 2373–2379. [Google Scholar]
- Liu, F.; Singh, A.; Yang, Z.; Garcia, A.; Kong, Y.; Meyskens, F.L., Jr. MiTF links Erk1/2 kinase and p21CIP1/WAF1 activation after UVC radiation in normal human melanocytes and melanoma cells. Mol. Cancer 2010, 9, 214. [Google Scholar]
- Enzler, T.; Sano, Y.; Choo, M.K.; Cottam, H.B.; Karin, M.; Tsao, H.; Park, J.M. Cell-selective inhibition of NFκB signaling improves therapeutic index in a melanoma chemotherapy model. Cancer Discov 2011, 1, 496–507. [Google Scholar]
- Keuling, A.M.; Andrew, S.E.; Tron, V.A. Inhibition of p38 MAPK enhances ABT-737-induced cell death in melanoma cell lines: Novel regulation of PUMA. Pigment Cell Melanoma Res 2010, 23, 430–440. [Google Scholar]
- Selimovic, D.; Hassan, M.; Haikel, Y.; Hengge, U.R. Taxol-induced mitochondrial stress in melanoma cells is mediated by activation of c-Jun N-terminal kinase (JNK) and p38 pathways via uncoupling protein 2. Cell Signal 2008, 20, 311–322. [Google Scholar]
- Shieh, J.M.; Huang, T.F.; Hung, C.F.; Chou, K.H.; Tsai, Y.J.; Wu, W.B. Activation of c-jun N-terminal kinase is essential for mitochondrial membrane potential change and apoptosis induced by doxycycline in melanoma cells. Br. J. Pharmacol 2010, 160, 1171–1184. [Google Scholar]
- Alexaki, V.I.; Javelaud, D.; Mauviel, A. JNK supports survival in melanoma cells by controlling cell cycle arrest and apoptosis. Pigment Cell Melanoma Res 2008, 21, 429–438. [Google Scholar]
- Denkert, C.; Siegert, A.; Leclere, A.; Turzynski, A.; Hauptmann, S. An inhibitor of stress-activated MAP-kinases reduces invasion and MMP-2 expression of malignant melanoma cells. Clin. Exp. Metastasis 2002, 19, 79–85. [Google Scholar]
- Estrada, Y.; Dong, J.; Ossowski, L. Positive crosstalk between ERK and p38 in melanoma stimulates migration and in vivo proliferation. Pigment Cell Melanoma Res 2009, 22, 66–76. [Google Scholar]
- Ivanov, V.N.; Fodstad, O.; Ronai, Z. Expression of ring finger-deleted TRAF2 sensitizes metastatic melanoma cells to apoptosis via up-regulation of p38, TNFα and suppression of NFκB activities. Oncogene 2001, 20, 2243–2253. [Google Scholar]
- Kannaiyan, R.; Manu, K.A.; Chen, L.; Li, F.; Rajendran, P.; Subramaniam, A.; Lam, P.; Kumar, A.P.; Sethi, G. Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c-Jun N-terminal kinase and suppression of PI3K/Akt signaling pathways. Apoptosis 2011, 16, 1028–1041. [Google Scholar]
- Ke, H.; Augustine, C.K.; Gandham, V.D.; Jin, J.Y.; Tyler, D.S.; Akiyama, S.K.; Hall, R.P.; Zhang, J.Y. Cyld inhibits melanoma growth and progression through suppression of the JNK/AP-1 and β1-integrin signaling pathways. J. Investig. Dermatol 2013, 133, 221–229. [Google Scholar]
- Hayden, M.S.; Ghosh, S. Signaling to NFκB. Genes Dev 2004, 18, 2195–2224. [Google Scholar]
- McNulty, S.E.; del Rosario, R.; Cen, D.; Meyskens, F.L., Jr; Yang, S. Comparative expression of NFκB proteins in melanocytes of normal skin vs. benign intradermal naevus and human metastatic melanoma biopsies. Pigment Cell Res. 2004, 17, 173–180. [Google Scholar]
- McNulty, S.E.; Tohidian, N.B.; Meyskens, F.L., Jr. RelA, p50 and inhibitor of κBα are elevated in human metastatic melanoma cells and respond aberrantly to ultraviolet light B. Pigment Cell Res. 2001, 14, 456–465. [Google Scholar]
- Muthusamy, V.; Hodges, L.D.; Macrides, T.A.; Boyle, G.M.; Piva, T.J. Effect of novel marine nutraceuticals on IL-1α-mediated TNFα release from UVB-irradiated human melanocyte-derived cells. Oxid. Med. Cell Longev 2011, 2011, 728645. [Google Scholar]
- Ivanov, V.N.; Ronai, Z. Down-regulation of tumor necrosis factor α expression by activating transcription factor 2 increases UVC-induced apoptosis of late-stage melanoma cells. J. Biol. Chem 1999, 274, 14079–14089. [Google Scholar]
- Gray-Schopfer, V.C.; Karasarides, M.; Hayward, R.; Marais, R. Tumor necrosis factor-α blocks apoptosis in melanoma cells when BRAF signaling is inhibited. Cancer Res 2007, 67, 122–129. [Google Scholar]
- Tandle, A.; Hanna, E.; Lorang, D.; Hajitou, A.; Moya, C.A.; Pasqualini, R.; Arap, W.; Adem, A.; Starker, E.; Hewitt, S.; et al. Tumor vasculature-targeted delivery of tumor necrosis factor-α. Cancer 2009, 115, 128–139. [Google Scholar]
- Ivanov, V.N.; Ronai, Z. p38 protects human melanoma cells from UV-induced apoptosis through down-regulation of NFκB activity and Fas expression. Oncogene 2000, 19, 3003–3012. [Google Scholar]
- Halliday, G.M.; Lyons, J.G. Inflammatory doses of UV may not be necessary for skin carcinogenesis. Photochem. Photobiol 2008, 84, 272–283. [Google Scholar]
- Muthusamy, V.; Piva, T.J. The UV response of the skin: A review of the MAPK, NFκB and TNFα signal transduction pathways. Arch. Dermatol. Res 2010, 302, 5–17. [Google Scholar]
- Johnson, G.L.; Nakamura, K. The c-Jun kinase/stress-activated pathway: Regulation, function and role in human disease. Biochim. Biophys. Acta 2007, 1773, 1341–1348. [Google Scholar]
- Hayden, M.S.; Ghosh, S. Shared principles in NFκB signaling. Cell 2008, 132, 344–362. [Google Scholar]
- Devalaraja, M.N.; Wang, D.Z.; Ballard, D.W.; Richmond, A. Elevated constitutive IκB kinase activity and IκBα phosphorylation in Hs294T melanoma cells lead to increased basal MGSA/GROα transcription. Cancer Res 1999, 59, 1372–1377. [Google Scholar]
- Clydesdale, G.J.; Dandie, G.W.; Muller, H.K. Ultraviolet light induced injury: Immunological and inflammatory effects. Immunol. Cell Biol 2001, 79, 547–568. [Google Scholar]
- Duthie, M.S.; Kimber, I.; Norval, M. The effects of ultraviolet radiation on the human immune system. Br. J. Dermatol 1999, 140, 995–1009. [Google Scholar]
- Ullrich, S.E.; Byrne, S.N. The immunologic revolution: Photoimmunology. J. Investig. Dermatol 2012, 132, 896–905. [Google Scholar]
- Bashir, M.M.; Sharma, M.R.; Werth, V.P. UVB and proinflammatory cytokines synergistically activate TNFα production in keratinocytes through enhanced gene transcription. J. Investig. Dermatol 2009, 129, 994–1001. [Google Scholar]
- Hazzalin, C.A.; Le Panse, R.; Cano, E.; Mahadevan, L.C. Anisomycin selectively desensitizes signaling components involved in stress kinase activation and fos and jun induction. Mol. Cell Biol 1998, 18, 1844–1854. [Google Scholar]
- Johansson, P.; Pavey, S.; Hayward, N. Confirmation of a BRAF mutation-associated gene expression signature in melanoma. Pigment Cell Res 2007, 20, 216–221. [Google Scholar]
- Castellano, M.; Pollock, P.M.; Walters, M.K.; Sparrow, L.E.; Down, L.M.; Gabrielli, B.G.; Parsons, P.G.; Hayward, N.K. CDKN2A/p16 is inactivated in most melanoma cell lines. Cancer Res 1997, 57, 4868–4875. [Google Scholar]
- Pope, J.H.; Morrison, L.; Moss, D.J.; Parsons, P.G.; Sister, Regius Mary. Human malignant melanoma cell lines. Pathology 1979, 11, 191–195. [Google Scholar]
- Kadekaro, A.L.; Kavanagh, R.J.; Wakamatsu, K.; Ito, S.; Pipitone, M.A.; Abdel-Malek, Z.A. Cutaneous photobiology. The melanocyte vs. the sun: Who will win the final round? Pigment Cell Res 2003, 16, 434–447. [Google Scholar]
- Kaidbey, K.H.; Agin, P.P.; Sayre, R.M.; Kligman, A.M. Photoprotection by melanin—A comparison of black and caucasian skin. J. Am. Acad. Dermatol 1979, 1, 249–260. [Google Scholar]
- Kobayashi, N.; Nakagawa, A.; Muramatsu, T.; Yamashina, Y.; Shirai, T.; Hashimoto, M.W.; Ishigaki, Y.; Ohnishi, T.; Mori, T. Supranuclear melanin caps reduce ultraviolet induced DNA photoproducts in human epidermis. J. Investig. Dermatol 1998, 110, 806–810. [Google Scholar]
- Yohn, J.J.; Norris, D.A.; Yrastorza, D.G.; Buno, I.J.; Leff, J.A.; Hake, S.S.; Repine, J.E. Disparate antioxidant enzyme activities in cultured human cutaneous fibroblasts, keratinocytes, and melanocytes. J. Investig. Dermatol 1991, 97, 405–409. [Google Scholar]
- Koch-Paiz, C.A.; Amundson, S.A.; Bittner, M.L.; Meltzer, P.S.; Fornace, A.J., Jr. Functional genomics of UV radiation responses in human cells. Mutat. Res. 2004, 549, 65–78. [Google Scholar]
- Besaratinia, A.; Kim, S.I.; Pfeifer, G.P. Rapid repair of UVA-induced oxidized purines and persistence of UVB-induced dipyrimidine lesions determine the mutagenicity of sunlight in mouse cells. FASEB J 2008, 22, 2379–2392. [Google Scholar]
- Schieke, S.M.; Ruwiedel, K.; Gers-Barlag, H.; Grether-Beck, S.; Krutmann, J. Molecular crosstalk of the ultraviolet A and ultraviolet B signaling responses at the level of mitogen-activated protein kinases. J. Investig. Dermatol 2005, 124, 857–859. [Google Scholar]
- Sabapathy, K.; Wagner, E.F. Jnk2: A negative regulator of cellular proliferation. Cell Cycle 2004, 3, 1520–1523. [Google Scholar]
- Chen, N.; Nomura, M.; She, Q.B.; Ma, W.Y.; Bode, A.M.; Wang, L.; Flavell, R.A.; Dong, Z. Suppression of skin tumorigenesis in c-Jun NH2-terminal kinase-2-deficient mice. Cancer Res 2001, 61, 3908–3912. [Google Scholar]
- Tao, J.; Gao, Y.; Li, M.O.; He, W.; Chen, L.; Harvev, B.; Davis, R.J.; Flavell, R.A.; Yin, Z. JNK2 negatively regulates CD8+ T cell effector function and anti-tumor immune response. Eur. J. Immunol 2007, 37, 818–829. [Google Scholar]
- Alsina, J.; Gorsk, D.H.; Germino, F.J.; Shih, W.; Lu, S.E.; Zhang, Z.G.; Yang, J.M.; Hait, W.N.; Goydos, J.S. Detection of mutations in the mitogen-activated protein kinase pathway in human melanoma. Clin. Cancer Res 2003, 9, 6419–6425. [Google Scholar]
- Hoshino, R.; Chatani, Y.; Yamori, T.; Tsuruo, T.; Oka, H.; Yoshida, O.; Shimada, Y.; Ari-i, S.; Wada, H.; Fujimoto, J.; et al. Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene 1999, 18, 813–822. [Google Scholar]
- Collisson, E.A.; de, A.; Suzuki, H.; Gambhir, S.S.; Kolodney, M.S. Treatment of metastatic melanoma with an orally available inhibitor of the Ras-Raf-MAPK cascade. Cancer Res 2003, 63, 5669–5673. [Google Scholar]
- Pavey, S.; Johansson, P.; Packer, L.; Taylor, J.; Stark, M.; Pollock, P.M.; Walker, G.J.; Boyle, G.M.; Harper, U.; Cozzi, S.J.; et al. Microarray expression profiling in melanoma reveals a BRAF mutation signature. Oncogene 2004, 23, 4060–4067. [Google Scholar]
- Lopez-Bergami, P.; Huang, C.; Goydos, J.S.; Yip, D.; Bar-Eli, M.; Herlyn, M.; Smalley, K.S.; Mahale, A.; Eroshkin, A.; Aaronson, S.; et al. Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell 2007, 11, 447–460. [Google Scholar]
- Shang, J.; Schwarz, C.; Sanchez Ruderisch, H.; Hertting, T.; Orfanos, C.E.; Tebbe, B. Effects of UVA and l-ascorbic acid on nuclear factor-κB in melanocytes and in HaCaT keratinocytes. Skin Pharmacol. Appl. Skin Physiol 2002, 15, 353–359. [Google Scholar]
- Djavaheri-Mergny, M.; Gras, M.P.; Mergny, J.L.; Dubertret, L. UVA-induced decrease in nuclear factor-κB activity in human keratinocytes. Biochem. J 1999, 338, 607–613. [Google Scholar]
- Kondo, S.; Jimbow, K. Dose-dependent induction of IL-12 but not IL-10 from human keratinocytes after exposure to ultraviolet light a. J. Cell Physiol 1998, 177, 493–498. [Google Scholar]
- Basile, J.R.; Eichten, A.; Zacny, V.; Munger, K. NF-κB-mediated induction of p21(cip1/waf1) by tumor necrosis factor α induces growth arrest and cytoprotection in normal human keratinocytes. Mol. Cancer Res 2003, 1, 262–270. [Google Scholar]
- Szoltysek, K.; Pietranek, K.; Kalinowska-Herok, M.; Pietrowska, M.; Kimmel, M.; Widlak, P. TNFα-induced activation of NFκB protects against UV-induced apoptosis specifically in p53-proficient cells. Acta Biochim. Pol 2008, 55, 741–748. [Google Scholar]
- Ravi, D.; Muniyappa, H.; Das, K.C. Caffeine inhibits UV-mediated NFκB activation in A2058 melanoma cells: An ATM-PKCδ-p38 MAPK-dependent mechanism. Mol. Cell Biochem 2008, 308, 193–200. [Google Scholar]
- Jinlian, L.; Yingbin, Z.; Chunbo, W. p38 MAPK in regulating cellular responses to ultraviolet radiation. J. Biomed. Sci 2007, 14, 303–312. [Google Scholar]
- Weston, C.R.; Davis, R.J. The JNK signal transduction pathway. Curr. Opin. Cell Biol 2007, 19, 142–149. [Google Scholar]
- Krohne, T.U.; Hunt, S.; Holz, F.G. Effect of 308 nm excimer laser irradiation on retinal pigment epithelium cell viability in vitro. Br. J. Ophthalmol 2009, 93, 91–95. [Google Scholar]
- Mikita, N.; Kanazawa, N.; Yoshimasu, T.; Ikeda, T.; Li, H.J.; Yamamoto, Y.; Furukawa, F. The protective effects of ultraviolet A1 irradiation on spontaneous lupus erythematosus-like skin lesions in MRL/lpr mice. Clin. Dev. Immunol 2009, 2009, 673952. [Google Scholar]
- Kuchel, J.M.; Barnetson, R.S.; Halliday, G.M. Ultraviolet A augments solar-simulated ultraviolet radiation-induced local suppression of recall responses in humans. J. Investig. Dermatol 2002, 118, 1032–1037. [Google Scholar]
- Huynh, T.T.; Chan, K.S.; Piva, T.J. Effect of ultraviolet radiation on the expression of pp38MAPK and furin in human keratinocyte-derived cell lines. Photodermatol. Photoimmunol. Photomed 2009, 25, 20–29. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Sham | UVA | UVB | UVA + B | UVB + A |
|---|---|---|---|---|---|
| HEM | 10 ± 4 | 17 ± 6 | 109 ± 40 | 103 ± 26 | 130 ± 24 |
| MM96L | 15 ± 1 | 14 ± 4 | 97 ± 22 | 88 ± 23 | 32 ± 4 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Muthusamy, V.; Piva, T.J. UVB-Stimulated TNFα Release from Human Melanocyte and Melanoma Cells Is Mediated by p38 MAPK. Int. J. Mol. Sci. 2013, 14, 17029-17054. https://doi.org/10.3390/ijms140817029
Muthusamy V, Piva TJ. UVB-Stimulated TNFα Release from Human Melanocyte and Melanoma Cells Is Mediated by p38 MAPK. International Journal of Molecular Sciences. 2013; 14(8):17029-17054. https://doi.org/10.3390/ijms140817029
Chicago/Turabian StyleMuthusamy, Visalini, and Terrence J. Piva. 2013. "UVB-Stimulated TNFα Release from Human Melanocyte and Melanoma Cells Is Mediated by p38 MAPK" International Journal of Molecular Sciences 14, no. 8: 17029-17054. https://doi.org/10.3390/ijms140817029
APA StyleMuthusamy, V., & Piva, T. J. (2013). UVB-Stimulated TNFα Release from Human Melanocyte and Melanoma Cells Is Mediated by p38 MAPK. International Journal of Molecular Sciences, 14(8), 17029-17054. https://doi.org/10.3390/ijms140817029