Abstract
Accumulation of misfolded proteins has been implicated in a variety of neurodegenerative diseases including prion diseases, Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD). In the past decade, single-chain fragment variable (scFv) -based immunotherapies have been developed to target abnormal proteins or various forms of protein aggregates including Aβ, SNCA, Htt, and PrP proteins. The scFvs are produced by fusing the variable regions of the antibody heavy and light chains, creating a much smaller protein with unaltered specificity. Because of its small size and relative ease of production, scFvs are promising diagnostic and therapeutic reagents for protein misfolded diseases. Studies have demonstrated the efficacy and safety of scFvs in preventing amyloid protein aggregation in preclinical models. Herein, we discuss recent developments of these immunotherapeutics. We review efforts of our group and others using scFv in neurodegenerative disease models. We illustrate the advantages of scFvs, including engineering to enhance misfolded conformer specificity and subcellular targeting to optimize therapeutic action.
Keywords:
scFv; immunotherapy; Prion disease; Alzheimer’s disease; Parkinson’s disease; Huntington’s disease; PrP; Aβ; SNCA; Htt 1. Introduction
Correct folding of proteins is essential for their functions [1]. Abnormal protein folding could lead to inaction, degradation, incorrect trafficking, and aggregation of proteins. In the latter cases, aggregation of certain proteins, such as Prion protein (PrP), Huntingtin (Htt), amyloid β (Aβ), α-Synuclein (SNCA) and Cu/Zn superoxide dismutase (SOD1), are mechanistically linked to neurological disorders such as prion diseases, Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD) and amyotrophic lateral sclerosis (ALS) [2–8].
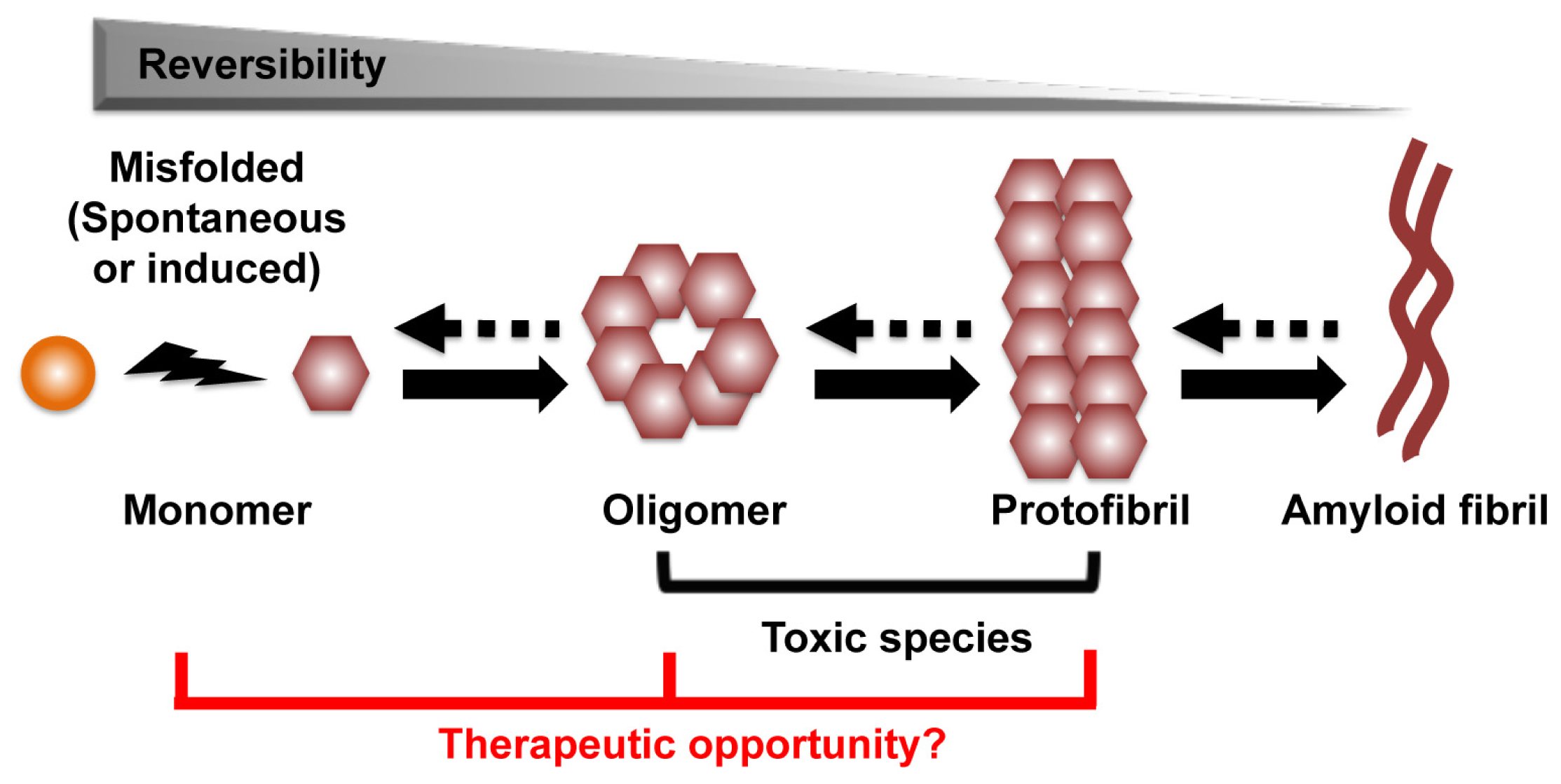
The process that leads to the formation of protein aggregates is a reversible heterogeneous multistep reaction. Amyloid formation can start from conversion of the native α-helix rich soluble cellular protein into the putatively pathogenic β-sheet rich isoforms, which are able to self-assemble through nucleation and monomer additions to form oligomers, protofibrils, and eventually amyloid fibril aggregates (Figure 1) [9,10]. Other aggregation pathways are also possible. Structural analysis and computer modeling suggested that amyloid aggregate can initiate directly from misfolded protein fragments, and that fibrils can act as seeds for protein aggregation [11,12]. An accumulated body of evidence indicates that the amyloid aggregates might not be responsible for the primary neurotoxic effects of these disorders. Rather, oligomers and protofibrils are presumptively the toxic species that drive neuronal dysfunction and death [12–15]. Thus, strategies to prevent oligomer/protofibril formation, promote their elimination or inhibit their toxic activity may be therapeutic. As an increase in monomer concentration enhances oligomer formation, approaches to reduce monomers also appear to be a viable therapeutic approach (Figure 1) [14].
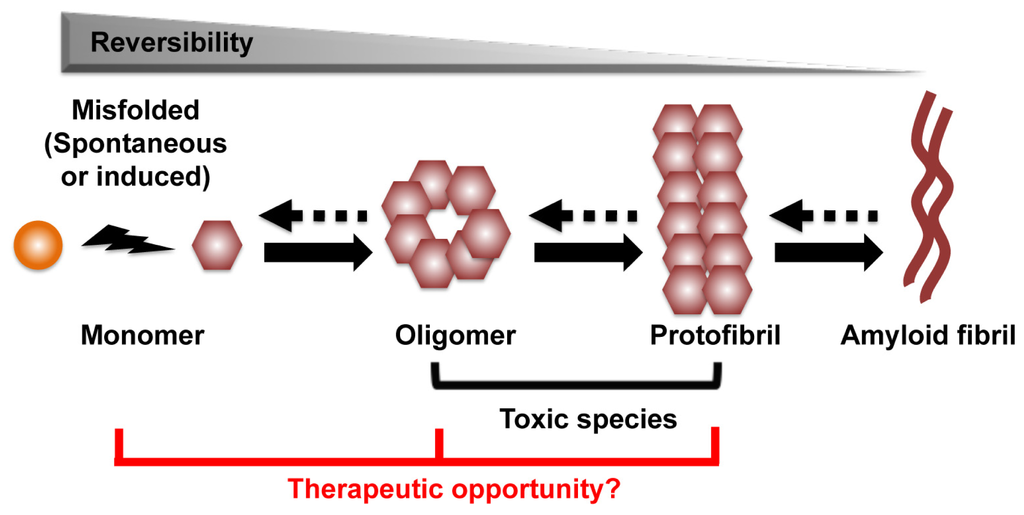
Figure 1.
The process of amyloid formation. The first step of the process can be a conversion from the native α-helix rich soluble cellular protein into the pathogenic β-sheet rich isoforms, in which they are able to self-assemble through a variety of subsequent nucleation and growth steps to form oligomer, protofibril, and eventually amyloid fibril aggregates. Oligomer and protofibril are putative toxic species that drive neuronal dysfunction. We hypothesize the therapeutic targets are monomer, oligomer and protofibril.
Active or passive immunotherapies directed against the disease-associated proteins have been prosecuted in preclinical and clinical studies [16–22]. Advancements in antibody technologies offer the scientific and clinical communities with better tools for diagnosis and treatment of neurological disease related to misfolded protein targets.
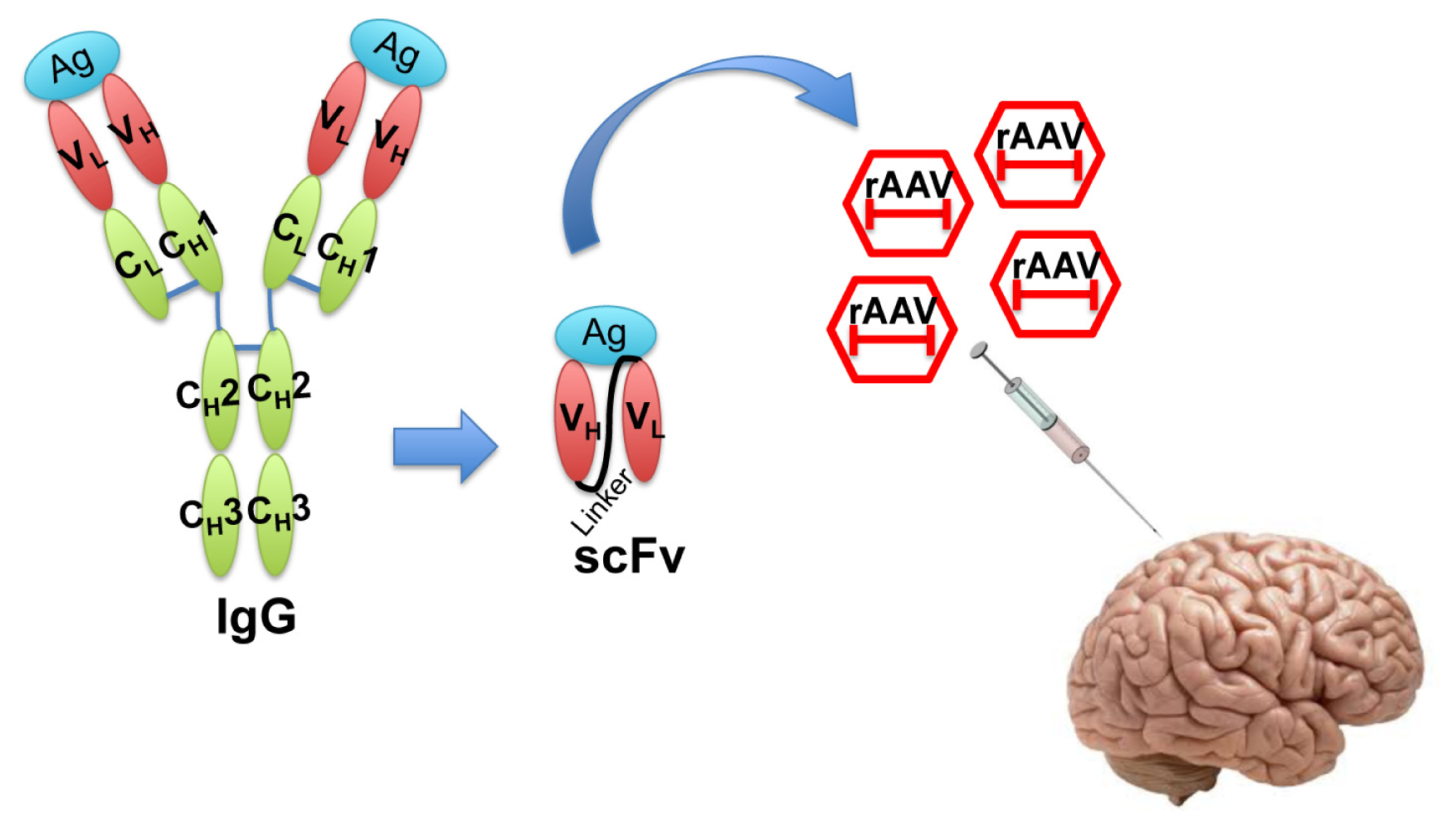
In the recent decade, single-chain fragment variable (scFv) antibodies have emerged as an option for passive immunotherapy [19]. The scFvs are generated by fusing the VH and VL fragments of an IgG with a suitable linker peptide [23]. The resulting molecule is relatively small and lacks the constant regions of the antibody, yet retains antigen-binding specificity. scFvs can be packaged in small viral vectors such as recombinant adeno-associated virus (rAAV) for direct injection into the central nervous system (Figure 2).
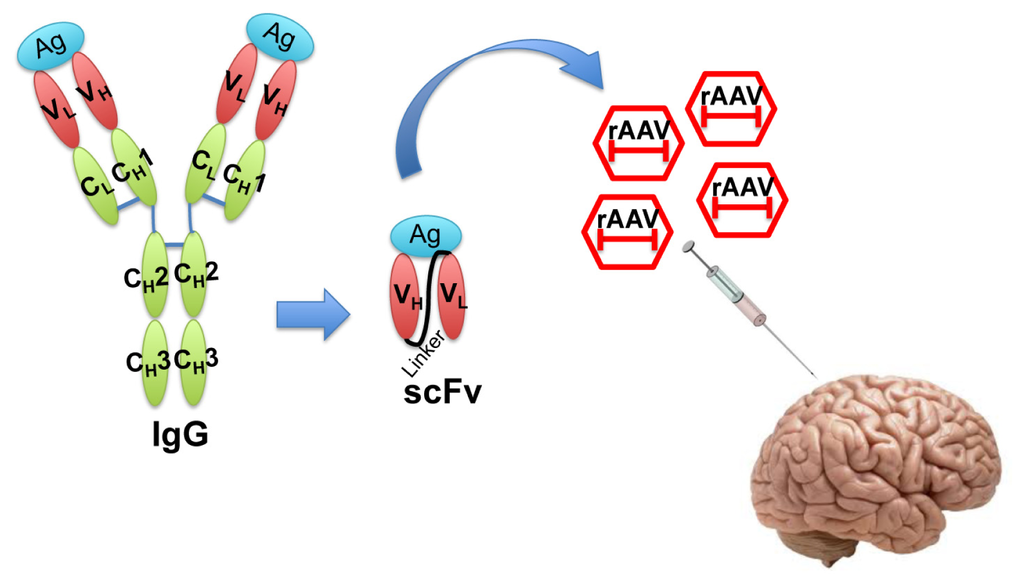
Figure 2.
Illustration of single-chain fragment variable (scFv) structure and recombinant adeno-associated virus (rAAV) delivery of scFv. Engineered scFv lacking the constant regions of IgG is a much smaller molecule that still retains the antigen binding affinity. scFvs can be packaged into rAAV and efficiently delivered in vivo for therapeutic purposes. Ag: Antigen; V: variable region; C: constant region; L: light chain; H: heavy chain.
Compared with chimeric or humanized antibodies, scFvs have several advantages. First of all, they are among the smallest antibodies that still retain antigen-binding specificity although affinity can be reduced. Through intracerebral injection, intranasal administration and viral transduction, scFvs can be delivered and spread throughout the brain [17,24–26]. scFvs have characteristics that may improve their function compared to conventional murine monoclonal antibody (mAb), Fab or (Fab’)2 fragments. scFvs have little immunogenicity and because of the absence of a constant region, they do not fix complement. The small size of the scFvs yield better tissue penetration but they tend to have shorter half-lives. Furthermore, since scFvs do not require glycosylation, they can be produced in a bacterial expression system thus allowing production at a substantial scale [27].
To develop effective scFv immunotherapy for the neurodegenerative diseases associated with protein misfolding, two questions should be addressed: (1) Is there a specific conformation of misfolded protein target that is causally linked to disease mechanism? (2) Does the specific misfolded and pathogenic target produce injury in an extracellular, intracellular or subcellular compartment? Herein, we review studies on scFv mediated immunotherapeutic approaches for several neurodegenerative diseases caused by protein misfolding.
2. scFv Therapy in Prion Diseases
Prion diseases or transmissible spongiform encephalopathies (TSE) are a group of diverse transmissible, fatal diseases that feature the conversion of normal cellular expressed prion proteins (PrPC) into a pathological conformation (PrPSc). The human forms of the disease include fatal familial insomnia, kuru, Creutzfeldt-Jakob disease, and Gerstmann-Straussler-Scheinker disease [9,28]. Antibodies against PrPc could, indirectly, promote the clearance of the PrPsc, prevent the conversion from PrPc into PrPsc [26], inhibit the transport of PrPc to the cell surface [29], or enhance the degradation of cellular PrPc [26]. Generally, PrPc is considered as a monomeric isoform of the prion protein rich in α-helical structure and thus sensitive to digestion by proteinase K; while the PrPsc is a higher order form rich in β-pleated sheet structure and is resistant to proteinase K digestion [30,31].
There are at least 20 studies that used scFv to target prion proteins [24–26,29,32–48]. Many of the scFvs are capable of inhibiting PrP aggregation and reducing PrPsc-related cellular toxicity. Earlier attempts were made by Leclerc et al. and Flego et al. using phage display libraries expressing human scFvs to isolate anti-PrP antibody fragments [32,35]. scFvs can also be engineered based on the coding sequences of the variable regions of established murine monoclonal antibodies against prion proteins [25,33,49,50].
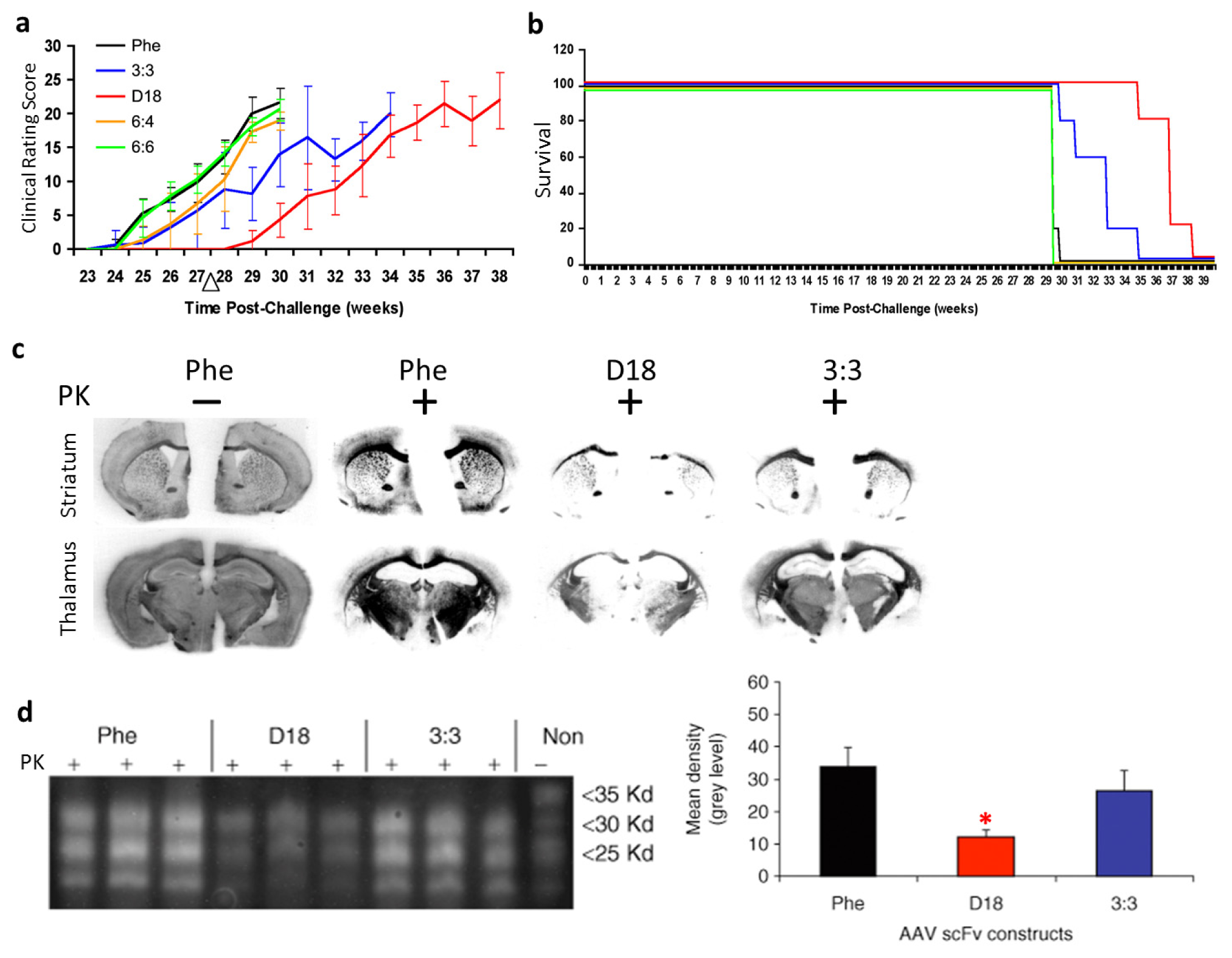
Our group developed passive immunomodulation with rAAV2 delivered anti-PrPc scFv in a mouse model of prion disease [39]. We first identified three novel anti-PrPc scFvs (3:3, 6:4, and 6:6) by screening the pAPII6 phagemid library. The scFv version of D18, a previously characterized anti-PrP Fab [13], was also generated. The four rAAV vectors expressing the anti-PrP scFvs, respectively, and a control scFv against an irrelevant antigen, phenobarbital (Phe), were administered intracerebrally into mouse prion model. All scFvs were engineered to be secreted efficiently with the aid of a murine immunoglobulin κ-secretory signal. Our results showed that treatment with anti-PrPc rAAVscFv delayed the onset of the prion disease in a mouse model. Clinical signs of prion disease in mice treated with scFvD18 appeared about 1 month later than all other groups, as evaluated by clinical rating scores (Figure 3a). The incubation period of the disease was significantly delayed in mice treated with scFv3:3 and scFvD18 (Figure 3b). The secreted anti-PrPc scFvs delayed the formation of proteinase K-resistant PrPsc and thus significantly reduced the PrPsc burden (Figure 3c,d). In summary, our work has demonstrated the potential for the use of the rAAV vector delivered anti-PrP scFvs in prion diseases.
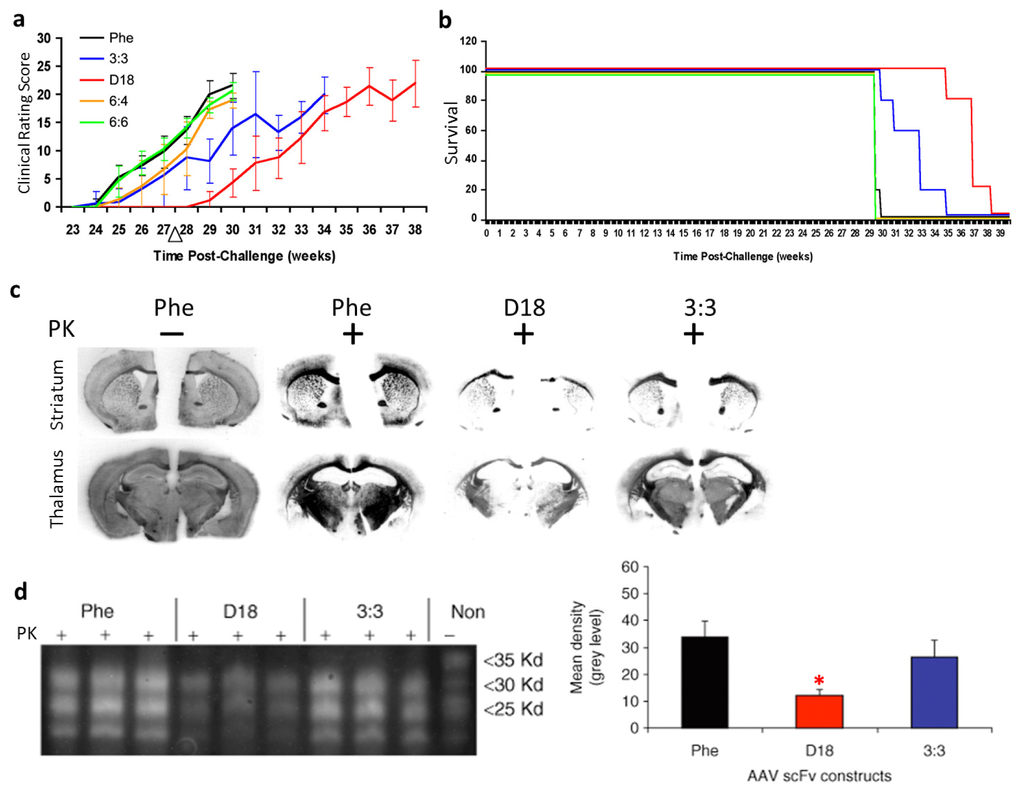
Figure 3.
Outcomes of recombinant adeno-associated virus (rAAV) delivered single-chain fragment variable (scFv) therapy for prion disease in transmissible spongiform encephalopathies (TSE) mouse model. (a) Clinical rating data show that the disease onset was delayed in mice administered rAAV2-scFvD18; (b) Gehan-Wilcoxon test data show that the incubation period of prion disease was delayed in mice treated with scFv3:3 and scFvD18; (c) Histoblot analyses of brain sections show decreased levels of proteinase K-resistant PrPsc in scFvD18 and scFv3:3 treated mice; (d) Immunoblots of brain homogenates (left panel) show a decreased level of proteinase K-resistant PrPsc in scFvD18 and scFv3:3 treated mice. Quantitative measurements (right panel) of the immunoblots indicate that the decrease in scFvD18 group is significant when compared with the Phe control (marked with “*”, p < 0.004, t-test). PK: proteinase K; “−”: no PK digestion; “+”: PK digested. Figures reproduced from Wuertzer et al., 2008 [39].
Another method of delivery for scFv is through scFv expressing cells. Donofrio et al. produced RD-4 rhabdomyosarcoma cells expressing anti-PrP 6H4 scFv [25]. Filesil et al. generated PC12 cells that express a secreted version of the 8H4 anti-prion scFv [26]. In both studies, the secreted anti-PrP scFv inhibited PrPsc formation. More recently, Fujita and colleagues established a Ra2 microglial cell line expressing anti-PrP 3S9scFv. Microglia are known to infiltrate the prion lesions. Delivery of the anti-PrP scFv by Ra2 microglial cells showed a small but significant increase in survival time when the microglial cells were injected into mice seven weeks after 22L scrapie prion infection [43].
A humanized scFv was developed based on the V5B2 anti-PrPsc IgG1 monoclonal antibody (mAb), which recognizes a synthetic peptide representing the C-terminus of the human PrP. The humanized V5B2 scFv was engineered with the aid of computer modeling. The resulting scFv had human amino acid residues and 13 mutations introduced at key positions compared with the original murine mAb, yet retained V5B2 mAb’s stability, specificity and affinity [42,45]. However, the effectiveness of V5B2 scFv has not been evaluated.
Most of the scFv antibodies established so far are either against PrPc or both PrPsc and PrPc, and are not conformation specific. The therapeutic potential of the mAbs or scFvs against different PrP conformations were first demonstrated by Petsch et al. [44]. In this study, a mAb specific to PrPsc (W261) and a conformation independent mAb against PrP (W226) were used to treat ScN2a cells. Only the conformation independent mAb showed a therapeutic benefit in the ScN2a model. This result agrees with the group’s earlier finding that the complementarity determining regions of W226, which specifically recognize PrPsc, did not have anti-prion activity [40]. However, mAb W226 or scFv W226 only showed marginal efficacy in vivo when they were used to treat scrapie-infected mice. This finding is inconsistent with an earlier study [51]. A recent study by Kubota et al. generated antibodies against the β-rich prion protein [47]. In this study, the conformation-defined recombinant PrP was generated and two human IgGs were established by screening a human scFv phage display library. These two antibodies recognize specifically the β-form but not the α-form of the recombinant PrP of human, bovine, sheep, and mouse. Using this antibody, the authors demonstrated that the β-form specific antibody could not inhibit the accumulation of PrPsc, and could only trigger weak apoptosis of prion-infected cells. On the other hand, an anti-PrP antibody recognizing all conformations could inhibit the accumulation of PrPsc and induce apoptotic cell death. In summary, these studies suggest that PrPc, instead of PrPsc, is the therapeutic target for prion diseases.
An alternative target for prion disease is the 37/67 kDa laminin receptor (LRP/LR), which is believed to act as a receptor for PrPc and PrPsc [52,53]. LRP/LR plays a vital role in prion infection [54,55]. It has been demonstrated to be responsible for bovine PrPsc internalization by human cells [56]. Expression of scFv against LRP/LR in scrappie-infected mice showed reduction of peripheral PrPsc levels but did not prolong survival [57,58].
3. scFv Therapy in Alzheimer’s Diseases
AD is the most common neurodegenerative disease affecting more than 36.5 million people worldwide [59,60]. The pathological features of AD include progressive neuronal loss, the accumulation of the Tau protein neurofibrillary tangles, and the formation of amyloid beta (Aβ) plaques. Passive immunotherapy has been demonstrated as the most promising therapeutics method to treat AD [21,61]. Currently efforts are primarily focused on the 4-kDa peptide Aβ, a major pathological peptide contributor to AD. Antibodies has been raised against the N-terminal, middle, and C-terminal of the monomeric Aβ as well as the soluble Aβ—containing oligomers, protofibrils and fibrils (reviewed in Robert et al., 2012 [21]). Like anti-prion antibodies, these antibodies are screened for their ability to prevent Aβ fibril formation or to disrupt existing fibrils. Many of those mAbs have entered clinical trials. Thus far, all clinical trials with these antibodies have failed to demonstrate efficacy.
As mentioned earlier, mAbs as a potential therapeutic reagent also suffer from disadvantages with regard to production, low tissue penetration, and potential serious adverse effects such as mass inflammatory reactions and cerebral amyloid angiopathy associated microhemorrhage [62,63]. These drawbacks may be improved through antibody engineering via the production of scFvs.
The first anti-Aβ scFv was produced by Frenkel et al. based on the variable regions of an anti-Aβ IgM 508 antibody [64]. This scFv, named 508F(Fv), could lead to the disaggregation of Aβ fibrils and prevent toxic effects in cultured PC-12 cells. By screening a naïve human scFv phage library with Aβ1–28 and Aβ1–40, Liu et al. selected two positive scFv clones, H1v2 and C1, which bound to the N-terminal or C-terminal of Aβ, respectively [65]. Liu et al. also showed that H1v2 but not C1 could inhibit Aβ aggregation in vitro [65]. Likewise, engineered scFv antibody based on mAb WO-2, which recognizes Aβ2–8 could inhibit Aβ fibril formation, disaggregate Aβ fibrils, and reduce Aβ oligomer toxicity [61].
Anti-Aβ scFvs can be easily delivered with AAV vectors. Tg2576 mouse AD model injected with rAAV2 expressing anti-Aβ scFv had detectable scFv in the hippocampal neurons after one year of injection. The rAAV2-CAscFv59 injected mice showed lower levels of amyloid deposits without obvious neurotoxicity [66]. Another study with AAV1 delivered anti-Aβ-16, Aβ40, and Aβ42 achieved widespread scFv expression in the mouse brain. The scFvs were cloned from established hybridomas mAb9, mAb40, and mAb42.2, respectively. Intracranial delivery of AAV1 expressing anti-Aβ scFvs did not show any adverse effect in CRND8 mice. About 20%–50% reduction in amyloid deposits were achieved with AAV1-scFv-Aβ in the CRND8 AD mouse model [67]. Wang et al. demonstrated that intramuscular delivery of rAAV2-scFv against Aβ was also safe and effectively reduced total Aβ levels in the brain [68].
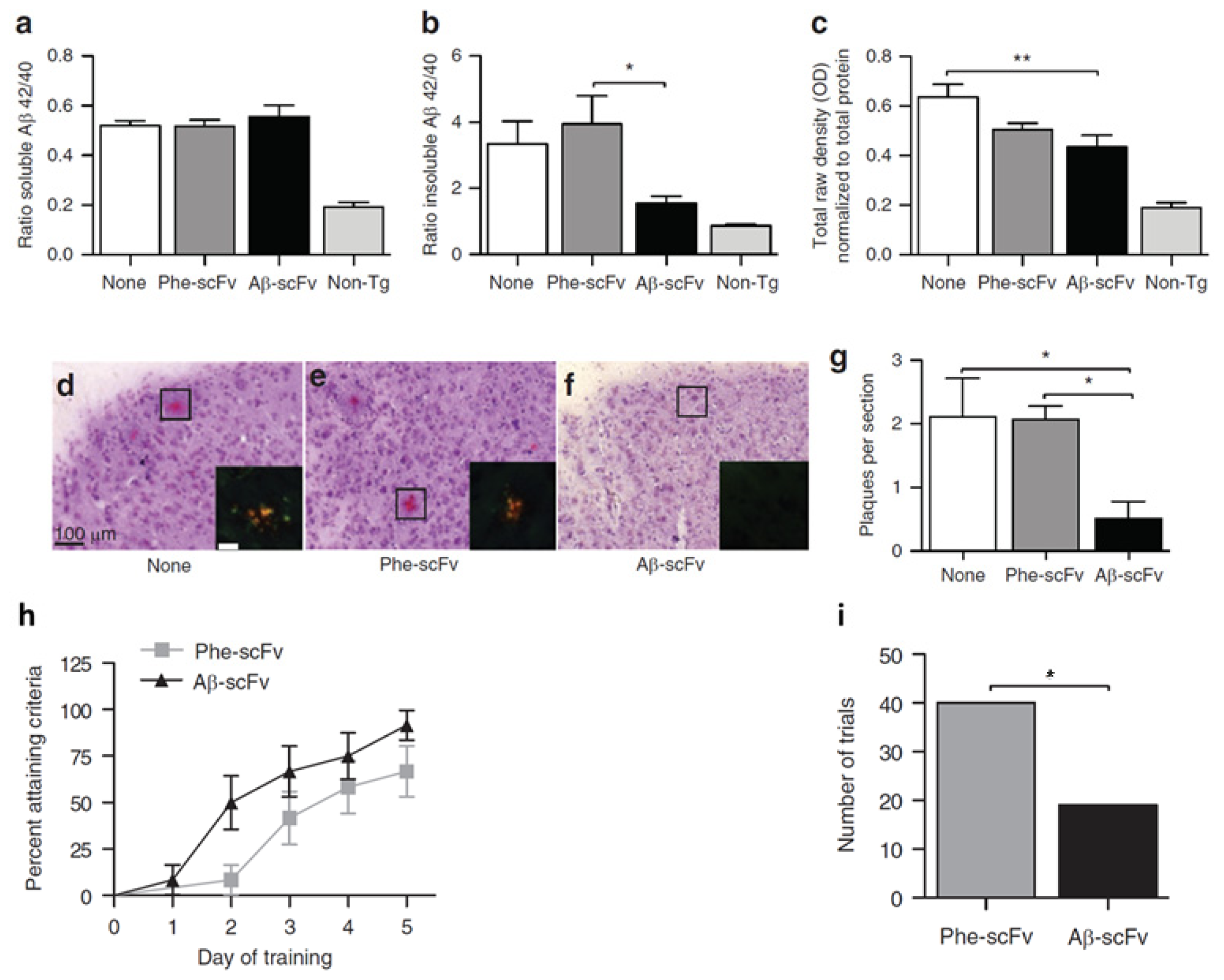
We previously showed that intrahippocampally delivered rAAV1-Aβ scFv was able to reduce Aβ and hyperphosphorylated tau levels and improve cognitive functions in a 3xTg-AD mouse model [69]. Compared to no injection control and Phe-scFv control, rAAV expressed anti-Aβ scFv significantly reduced insoluble Aβ and soluble oligomeric Aβ levels in mouse hippocampus, although the soluble Aβ levels remained unchanged (Figure 4). In addition, a > 75% decrease in Congo red-stained amyloid plaques was observed in Aβ-scFv treated 3xTg-AD mice when compared to the controls (Figure 4). Importantly, Aβ-scFv treatment improved spatial learning and memory abilities of the 3xTg-AD mice. In the Morris Water Maze test, 3xTg-AD mice with hippocampal injection of Aβ-scFv learned more quickly than the Phe-scFv controls (Figure 4). In this preclinical study, we validated rAAV-delivered anti-Aβ scFv as a potential therapeutic strategy to attenuate pathologic protein levels and ameliorate behavior defects.
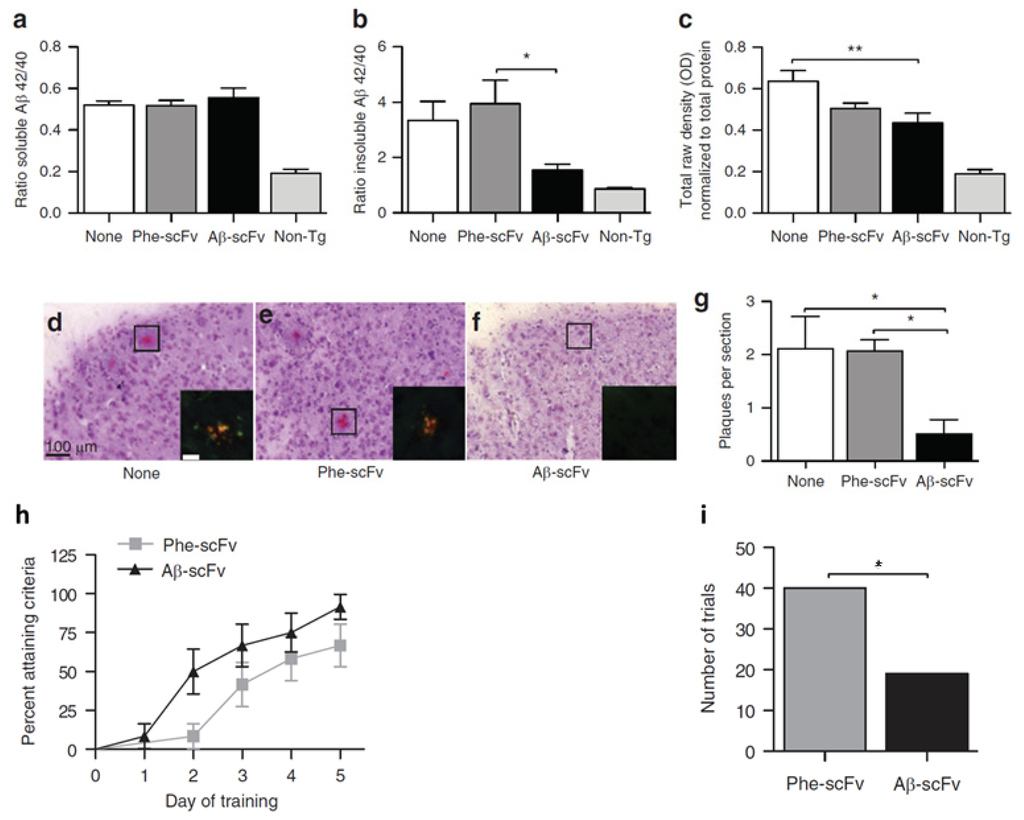
Figure 4.
Assessment of recombinant adeno-associated virus (rAAV) delivered single-chain fragment variable (scFv) therapy for Alzheimer’s disease (AD) in 3xTg AD mouse model. Hippocampal injections of rAAV1-Phe-scFv (Phe-scFv), rAAV-Aβ-scFv (Aβ-scFv), or no injection control (None) into three-month-old 3xTg-AD mice led to no change in hippocampal soluble Aβ. (a) Levels but significant decreases in insoluble Aβ; (b) Levels in the Aβ-scFv group; (c) The levels of soluble oligomeric Aβ in the Aβ-scFv treated mice was significantly decreased compared to mice with no injection; (d,e,f) Congo red staining showed reduction of amyloid plaques in Aβ-scFv injected mice; (g) Quantitative measurements of amyloid plagues. (*p < 0.05; **p < 0.01; One-way ANOVA with Bonferroni’s multiple comparison post-hoc test.); (h) Morris Water Maze test showed improved spatial learning in Aβ-scFv treated mice when compared to the Phe-scFv treated mice; (i) The total number of trials that Phe-scFv injected mice failed to meet the learning criteria was significantly higher than that of the Aβ-scFv injected mice (p = 0.0041, Fisher’s exact test). Figures reproduced from Ryan et al., 2010 [69].
All above studies demonstrated the safety and efficacy of AAV delivered long-term expression of anti-Aβ scFvs as a potentially viable AD treatment strategy.
Four conformation specific anti-Aβ scFvs have been identified from a naïve human scFv phage display library [70]. The four scFvs W8, W9, W20, and WC2 only bind to Aβ oligomers but not the other conformations. All four scFvs bind to the same region on the Aβ oligomer and can inhibit Aβ fibril formation and neurotoxicity [70]. Interestingly, scFvs W8 and W20 not only could recognize Aβ oligomers, but they could also recognize oligomers of other amyloid proteins such as SNCA and PrP. W8 and W20 could inhibit protein aggregation and reduce cytotoxicity of the amyloid protein oligomers [46].
4. scFv Therapy in Huntington’s Diseases
Huntington’s Disease (HD) is an autosomal dominant neurodegenerative disorder caused by the expansion of the polyglutamine (polyQ) tract in the Huntington (Htt) protein [71,72]. The expanded N-terminal of the Htt protein leads to pathological aggregation of the Htt protein in the patient brain and causes fatal motor disorder. Generally, patients carrying an htt gene with 35–39 polyQ repeats show late onset HD with incomplete penetrance. Patients carrying an htt gene with more than 40 polyQ repeats show early onset HD with 100% penetrance [20,73]. In the past decade, various intracellular anti-Htt immunotherapies have been developed as potential treatment options for HD [20], including anti-Htt scFvs.
By screening the human phage display library, a scFv (C4) recognizing the 17 N-terminal Htt was identified by Lecerf et al. [74]. Co-expression of this C4 scFv and the expanded repeat Htt-polyQ-GFP reduced the number of Htt aggregates in COS-7 cells [74]. In organotypic slice cultures, co-transfection of C4 scFv could reduce the malonate-induced cell death in mutant Htt-expressing cells [75]. Further study showed that C4 scFv specifically targets the soluble portion of the mutant Htt N-terminal fragment and therefore may shift the equilibrium of mutant monomeric vs. higher order Htt aggregates [76]. The C4 scFv had been tested in vivo with a Drosophila HD model that expresses Htt exon 1-Q93. Compared to only 30% of the transgenic flies surviving to adulthood, 100% of flies expressing elav-Gal4 driven UAS-C4 sFv expression reached adulthood. The C4 sFv expression also reduced neurodegeneration and prolonged the lifespan of the HD flies [77,78]. Striatum delivery of AAV2/1 expressing C4 scFv in B6.HDR6/1 mice reduced mHtt aggregates in neurons. Striatal expression of C4 scFv seemed to be well tolerated by mice and did not elicit adverse effects [79]. However, the effectiveness of the AAV delivered C4 scFv decreases over time and age of the animals. In an effort to improve the efficiency of C4 scFv, Butler and Messer generated a scFv fusion protein with a PEST domain. The C4 scFv-PEST fusion protein retained the anti-mutant-Htt function and has increased proteasomal degradation of the mutant Htt [80]. The above studies have established preclinical efficacy of C4 scFv for mutant Htt in vitro and in vivo.
Khoshnan and colleagues produced scFvs based on mAbs that recognize the polyQ (MW1, MW2) and the polyP (MW7) domains of the Htt exon 1. All three scFvs were found to bind to Htt in 293 cells expressing Htt exon 1 with 103 polyQ repeats. Interestingly, scFvs binding to the polyQ domain of Htt triggered pronounced cell death and Htt aggregation, whereas scFv binding to the polyP domain of Htt inhibited Htt aggregation and cytotoxicity [81]. These results suggest a similar scenario with regard to other amyloid proteins where the region of scFv binding can exhibit differential effects on protein aggregation and related toxicity.
5. scFv Therapy in Parkinson’s Diseases
Parkinson’s disease (PD) is a progressive neurodegenerative disordered caused by the loss of nigrostriatal dopaminergic neurons [82,83]. PD is a complex neuronal disease that involves both genetic and environmental factors [84,85]. Defects in many genes have been implemented in the pathogenesis of PD. Among these, SNCA is strongly associated with PD [86–89]. A hallmark of PD is the formation of aggregated SNCA-containing Lewy bodies in dopamine neurons located in the substantia nigra [88,89]. Therefore SNCA has become a major target for PD immunotherapy.
Our laboratory generated conformation-specific humanized scFvs against SNCA. These scFvs can be efficiently expressed in mammalian cells through transductions with HSV vector carrying the scFvs expression cassette [90]. Emadi et al. identified 10 anti-SNCA scFvs by screening naïve human phage display libraries. A strong scFv recognizing the C-terminal of SNCA could inhibit SNCA aggregation in vitro [91]. Co-expressing of anti-monomeric SNCA scFv and SNCA in mammalian cells prevented SNCA aggregation and rescued cellular toxicity caused by SNCA overexpression [92,93]. Similarly, scFv against oligomeric SNCA could also prevent SNCA aggregation and rescue SNCA-mediated cytotoxicity [94].
Although the use of scFv as a therapeutic intervention has been largely overlooked due to the complexity of human Parkinsonism and the uncertainty of disease pathogenesis, these studies provide an encouraging outlook that warrants further study of anti-SNCA scFv in PD models.
6. Improvements of scFv for Neuronal Disorder
Many efforts were directed towards improving the performance of the scFv in neurodegenerative diseases; these efforts include reduction of immunogenicity, enhanced scFv stability and solubility, improved avidity, and increased crossing of the blood-brain barrier (BBB). scFvs can be engineered into multimers such as diabodies or triabodies to increase antigen binding [22,95]. scFvs sequence optimization including the replacement of unpaired cysteine residue in the scFv fragment [64], coding region mutagenesis, computer-guided codon humanization [42,45], and addition of the PEST domain to the scFvs [80,96] has helped to increase scFv stability and decrease immune response against the scFvs. In certain cases, changes in scFvs may lead to reduced affinity against cognate antigens. Similarly, fusion protein strategies have been used to target scFvs to specific subcellular locations to enhance interdiction of pathogenic protein [29,96]. Many scFvs exhibit solubility issue when expressed in the cells. Linker length and amino acids composition is known to affect scFv solubility. This can be improved by the addition of charged residues to the linker sequence [97]. In addition, high-throughput selection strategy [98], peptide tag fusion strategy [99], and combination strategy with fusion tags and solubility-enhancing reagents have been developed to increase scFv solubility [100].
Although some scFv are believed to be able to cross the BBB, other scFvs failed in this regard. Two techniques have been developed to help scFv to cross the BBB. One takes advantage of the human insulin receptor (HIR). Boado and colleagues made a fusion protein of anti-Aβ scFv with a HIR mAb. The resulting fusion protein can be transported into the Rhesus monkey brain through binding to HIR [101]. Another approach uses the cell-penetrating peptide (CPP). In this case, the CPP replaced the linker peptide of scFv and the resulting scFv-CPP fusion protein was effective in crossing the BBB in mice [102].
Moreover, more appropriate disease targeting scFvs can be generated based on available mAb panels or natural occurring autoimmune antibodies that bind to aggregated proteins. A comprehensive collection of anti-PrPc mAb has been developed against different epitopes located across the entire mouse PrPc protein [38]. Similar comprehensive mAb panels can be developed as diagnostics and candidate therapeutics. It is also possible to engineer scFvs based on natural occurring antibodies directed against amyloid proteins. Human autoantibodies have been shown to inhibit PrP aggregation and relieve neurotoxicity [22,103].
7. Right Target at the Right Place
Amyloid proteins exist in different conformations. In most cases, monomer, oligomer, protofibril, and fibril forms of the amyloid proteins coexist. In recent years, more evidence has shown that neurotoxicity is caused primarily by the monomeric and/or oligomeric forms of the disease-associated amyloid proteins; the formation of the aggregates is likely to be a protective process [2,104–106]. For example, SNCA fibrils are mostly present in Lewy bodies and are considered as structurally stable with low toxicity [89]. In contrast, oligomer forms of SNCA have been detected in diseased brain and believed to change neuronal membrane permeability [107,108], damage mitochondria [109], disrupt microtubules [110] (reviewed by Lashuel et al. and Brown [106,111]). The monomeric or oligomeric forms of PrP have been implicated in neurotoxicity in vitro and in vivo [112–115]. In addition, much evidence supports that Aβ oligomers are neurotoxic (reviewed by Gilbert et al. [2]).
Although the debate over whether the oligomer or the fibril aggregate is not yet settled, we believe scFvs targeting the monomeric and oligomeric forms represent the most important therapeutic opportunities for AD, PD, HD, and prion diseases. Removal of soluble amyloid proteins eliminates the substrates for toxic oligomer/protofibril formation. This is clearly demonstrated in Prnp0/0 mice, which are resistant to prion infections [116]. scFvs can also change the conversion dynamics between different forms of these proteins. By removing the monomeric and oligomeric forms of the amyloid proteins, scFvs change the equilibrium, disaggregate proteins and potentially reduce neurotoxicity. Indeed, as we have summarized in the previous sections, scFvs against PrP, Aβ, SNCA monomers or oligomers have demonstrated neuroprotection [40,43,46,47,61,70,91,93,94,117]. Conformation specific scFvs against the fibril forms of the proteins are not effective in reducing protein aggregates and neurotoxicity [33,42,44,47,81].
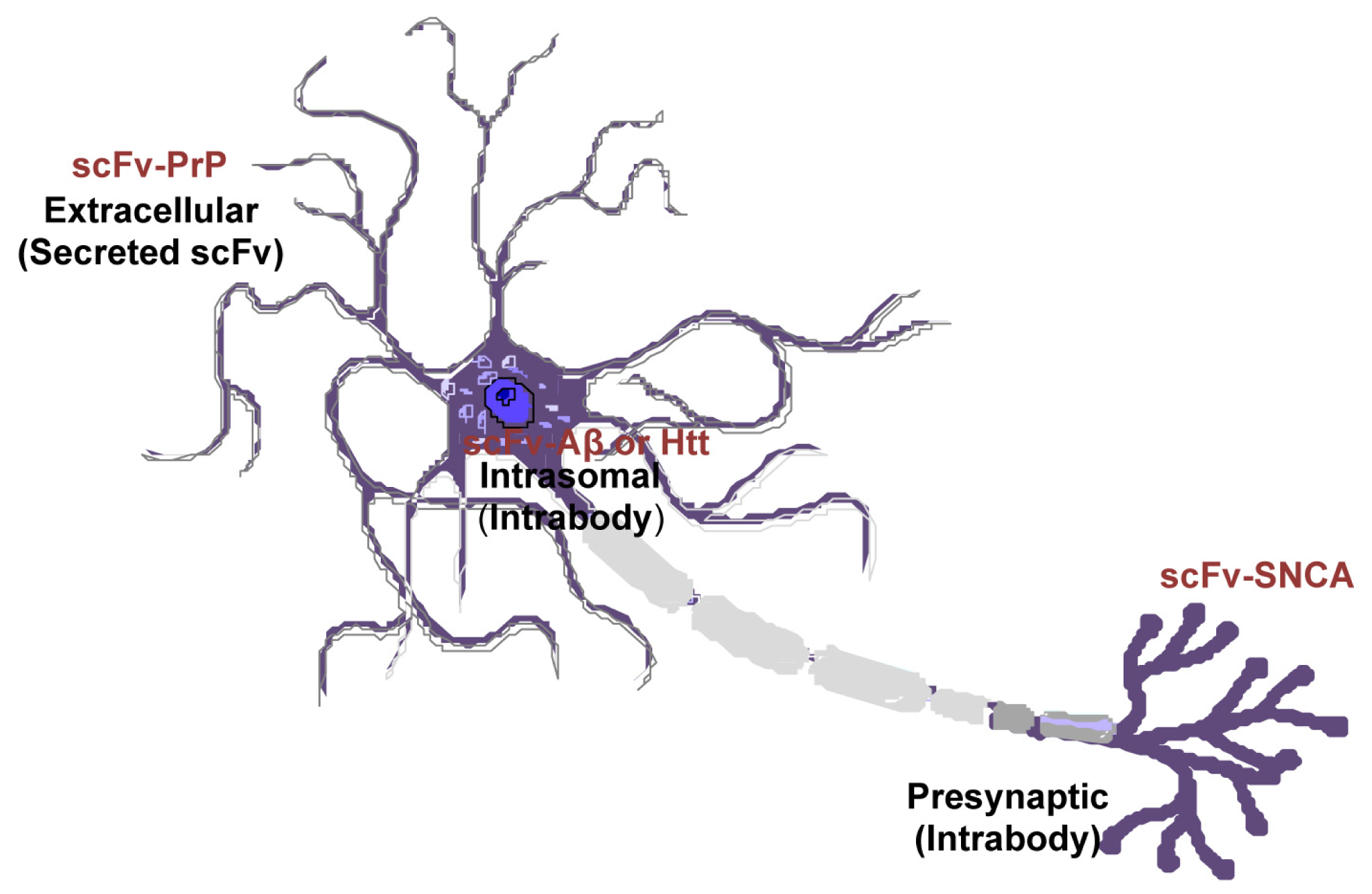
Amyloid proteins are located at a variety of locations: PrP has an extracelluar domain; Aβ is both intracellular and extracellular; SNCA is present on the membrane and within cells; Htt is mostly cytoplasmic. However, the subcellular location of native proteins and their pathological forms can be different. SNCA fibrils are mostly resided in the neuronal cell body whereas the oligomeric forms are predominantly located at the axon and synapses [118–121]. Mutant Htt in diseased neurons changes its localization from cytoplasm to the nucleus [122]. The scFvs can be engineered to localize to desired subcellular regions (intrabodies), or configured as a secreted molecule to engage membrane-bound or extracellular targeted proteins (Figure 5). Previous studies have found endoplasmic reticulum-targeted [29], secreted [25,26,43], or intracellular expressed [81] scFvs are effective in disaggregate amyloids depending on the types of the proteins or the pathological state. Future work is needed to elucidate the subcellular locations where amyloid proteins produce greatest toxicity. With this information, more targeted and effective therapeutics are envisaged.
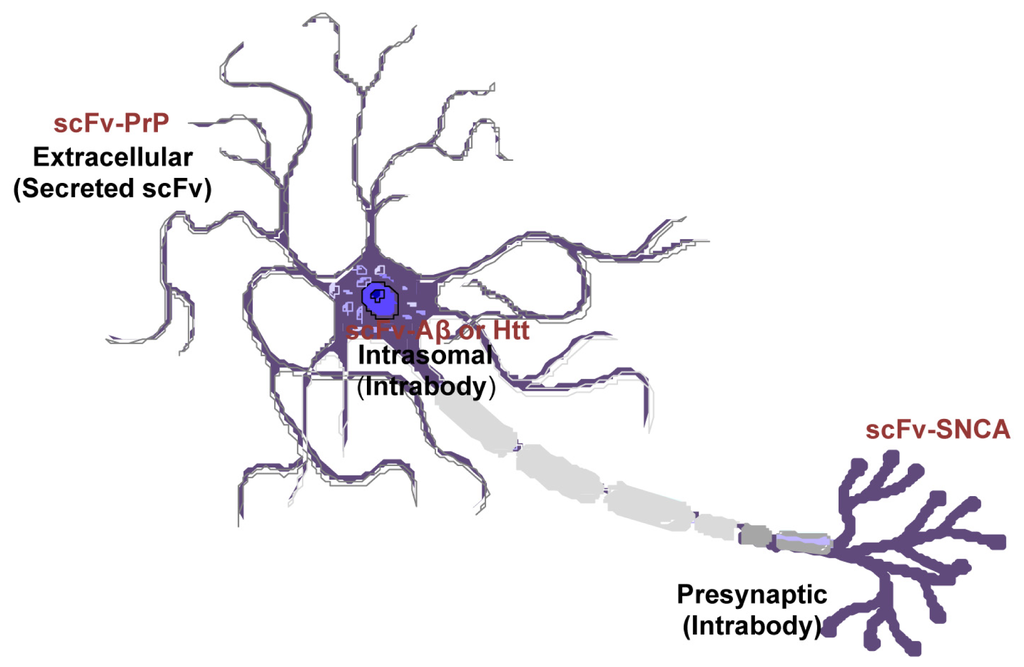
Figure 5.
Differential cellular location of single-chain fragment variable (scFv) that targets specific pathogenic antigen in different neurodegenerative diseases. Recombinant adeno-associated virus (rAAV)-scFv can be engineered to localized to desired subcellular regions (intrabodies), or as a secreted peptide to target membrane-bound or extracellular proteins.
8. Conclusions
As reviewed above, the scFvs have been developed as a passive immunotherapy and evaluated in models of neurodegenerative disorders caused by pathogenic misfolded proteins. This potential therapeutic has unique advantages although additional work on scFv-based therapy is required to improve solubility, tissue retention, rapid turnover, lower avidity, scFv-antigen complex clearance from brain, and potential adverse effects due to the targeting of normal functions of self-antigens. Careful toxicity study is required for any scFv prior to its clinical use. Accordingly, it is prudent to refine scFvs therapeutic action in animal models and judiciously extend a candidate in clinical trials in a neurodegenerative disease where unmet medical need is great.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar]
- Gilbert, B.J. The role of amyloid beta in the pathogenesis of Alzheimer’s disease. J. Clin. Pathol 2013, 66, 362–366. [Google Scholar]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. (Berl) 2003, 81, 678–699. [Google Scholar]
- Polymenidou, M.; Cleveland, D.W. Prion-like spread of protein aggregates in neurodegeneration. J. Exp. Med 2012, 209, 889–893. [Google Scholar]
- El-Agnaf, O.M.; Irvine, G.B. Review: Formation and properties of amyloid-like fibrils derived from alpha-synuclein and related proteins. J. Struct. Biol 2000, 130, 300–309. [Google Scholar]
- Arrasate, M.; Finkbeiner, S. Protein aggregates in Huntington’s disease. Exp. Neurol 2012, 238, 1–11. [Google Scholar]
- Gadad, B.S.; Britton, G.B.; Rao, K.S. Targeting oligomers in neurodegenerative disorders: Lessons from alpha-synuclein, tau, and amyloid-beta peptide. J. Alzheimers Dis 2011, 24, 223–232. [Google Scholar]
- Ghadge, G.D.; Pavlovic, J.D.; Koduvayur, S.P.; Kay, B.K.; Roos, R.P. Single chain variable fragment antibodies block aggregation and toxicity induced by familial ALS-linked mutant forms of SOD1. Neurobiol. disease 2013, 56, 74–78. [Google Scholar]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar]
- McKinley, M.P.; Bolton, D.C.; Prusiner, S.B. A protease-resistant protein is a structural component of the scrapie prion. Cell 1983, 35, 57–62. [Google Scholar]
- Fernandez-Busquets, X.; de Groot, N.S.; Fernandez, D.; Ventura, S. Recent structural and computational insights into conformational diseases. Curr. Med. Chem 2008, 15, 1336–1349. [Google Scholar]
- Invernizzi, G.; Papaleo, E.; Sabate, R.; Ventura, S. Protein aggregation: Mechanisms and functional consequences. Int. J. Biochem. Cell Biol 2012, 44, 1541–1554. [Google Scholar]
- Peretz, D.; Williamson, R.A.; Kaneko, K.; Vergara, J.; Leclerc, E.; Schmitt-Ulms, G.; Mehlhorn, I.R.; Legname, G.; Wormald, M.R.; Rudd, P.M.; et al. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 2001, 412, 739–743. [Google Scholar]
- Zhu, M.; Li, J.; Fink, A.L. The association of alpha-synuclein with membranes affects bilayer structure, stability, and fibril formation. J. Biol. Chem 2003, 278, 40186–40197. [Google Scholar]
- Walsh, D.M.; Selkoe, D.J. Oligomers on the brain: The emerging role of soluble protein aggregates in neurodegeneration. Protein Pept. Lett 2004, 11, 213–228. [Google Scholar]
- Miller, T.W.; Messer, A. Intrabody applications in neurological disorders: Progress and future prospects. Mol. Ther 2005, 12, 394–401. [Google Scholar]
- Federoff, H.J. Development of vaccination approaches for the treatment of neurological diseases. J. Comp. Neurol 2009, 515, 4–14. [Google Scholar]
- Wang, Y.J.; Zhou, H.D.; Zhou, X.F. Modified immunotherapies against Alzheimer’s disease: Toward safer and effective amyloid clearance. J. Alzheimers Dis 2010, 21, 1065–1075. [Google Scholar]
- Ahmad, Z.A.; Yeap, S.K.; Ali, A.M.; Ho, W.Y.; Alitheen, N.B.; Hamid, M. scFv antibody: Principles and clinical application. Clin. Dev. Immunol 2012, 2012, 980250. [Google Scholar]
- Butler, D.C.; McLear, J.A.; Messer, A. Engineered antibody therapies to counteract mutant huntingtin and related toxic intracellular proteins. Prog. Neurobiol 2012, 97, 190–204. [Google Scholar]
- Robert, R.; Wark, K.L. Engineered antibody approaches for Alzheimer’s disease immunotherapy. Arch. Biochem. Biophys 2012, 526, 132–138. [Google Scholar]
- Roettger, Y.; Du, Y.; Bacher, M.; Zerr, I.; Dodel, R.; Bach, J.P. Immunotherapy in prion disease. Nat. Rev. Neurol 2013, 9, 98–105. [Google Scholar]
- Malone, J.; Sullivan, M.A. Analysis of antibody selection by phage display utilizing anti-phenobarbital antibodies. J. Mol. Recognit 1996, 9, 738–745. [Google Scholar]
- Campana, V.; Zentilin, L.; Mirabile, I.; Kranjc, A.; Casanova, P.; Giacca, M.; Prusiner, S.B.; Legname, G.; Zurzolo, C. Development of antibody fragments for immunotherapy of prion diseases. Biochem. J 2009, 418, 507–515. [Google Scholar]
- Donofrio, G.; Heppner, F.L.; Polymenidou, M.; Musahl, C.; Aguzzi, A. Paracrine inhibition of prion propagation by anti-PrP single-chain Fv miniantibodies. J. Virol 2005, 79, 8330–8338. [Google Scholar]
- Filesi, I.; Cardinale, A.; Mattei, S.; Biocca, S. Selective re-routing of prion protein to proteasomes and alteration of its vesicular secretion prevent PrP(Sc) formation. J. Neurochem 2007, 101, 1516–1526. [Google Scholar]
- Verma, R.; Boleti, E.; George, A.J. Antibody engineering: Comparison of bacterial, yeast, insect and mammalian expression systems. J. Immunol. Methods 1998, 216, 165–181. [Google Scholar]
- Prusiner, S.B. Shattuck lecture-neurodegenerative diseases and prions. N. Engl. J. Med 2001, 344, 1516–1526. [Google Scholar]
- Cardinale, A.; Filesi, I.; Vetrugno, V.; Pocchiari, M.; Sy, M.S.; Biocca, S. Trapping prion protein in the endoplasmic reticulum impairs PrPC maturation and prevents PrPSc accumulation. J. Biol. Chem 2005, 280, 685–694. [Google Scholar]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E.; et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar]
- Turk, E.; Teplow, D.B.; Hood, L.E.; Prusiner, S.B. Purification and properties of the cellular and scrapie hamster prion proteins. Eur. J. Biochem 1988, 176, 21–30. [Google Scholar]
- Leclerc, E.; Liemann, S.; Wildegger, G.; Vetter, S.W.; Nilsson, F. Selection and characterization of single chain Fv fragments against murine recombinant prion protein from a synthetic human antibody phage display library. Hum. Antibodies 2000, 9, 207–214. [Google Scholar]
- Heppner, F.L.; Musahl, C.; Arrighi, I.; Klein, M.A.; Rulicke, T.; Oesch, B.; Zinkernagel, R.M.; Kalinke, U.; Aguzzi, A. Prevention of scrapie pathogenesis by transgenic expression of anti-prion protein antibodies. Science 2001, 294, 178–182. [Google Scholar]
- Luginbuhl, B.; Kanyo, Z.; Jones, R.M.; Fletterick, R.J.; Prusiner, S.B.; Cohen, F.E.; Williamson, R.A.; Burton, D.R.; Pluckthun, A. Directed evolution of an anti-prion protein scFv fragment to an affinity of 1 pM and its structural interpretation. J. Mol. Biol 2006, 363, 75–97. [Google Scholar]
- Flego, M.; Ascione, A.; Zamboni, S.; Dupuis, M.L.; Imperiale, V.; Cianfriglia, M. Generation of human scFvs antibodies recognizing a prion protein epitope expressed on the surface of human lymphoblastoid cells. BMC Biotechnol 2007, 7, 38. [Google Scholar]
- Miyamoto, K.; Shimamoto, T.; Aosasa, M.; Kimura, S.; Nakamura, N.; Okubo, Y.; Yokoyama, T.; Horiuchi, H.; Furusawa, S.; Matsuda, H. Development of recombinant chicken IgY from single chain fragment of variable region for diagnosis of BSE. Biologicals 2007, 35, 31–34. [Google Scholar]
- Padiolleau-Lefevre, S.; Alexandrenne, C.; Dkhissi, F.; Clement, G.; Essono, S.; Blache, C.; Couraud, J.Y.; Wijkhuisen, A.; Boquet, D. Expression and detection strategies for an scFv fragment retaining the same high affinity than Fab and whole antibody: Implications for therapeutic use in prion diseases. Mol. Immunol 2007, 44, 1888–1896. [Google Scholar]
- Polymenidou, M.; Moos, R.; Scott, M.; Sigurdson, C.; Shi, Y.Z.; Yajima, B.; Hafner-Bratkovic, I.; Jerala, R.; Hornemann, S.; Wuthrich, K.; et al. The POM monoclonals: A comprehensive set of antibodies to non-overlapping prion protein epitopes. PLoS One 2008, 3, e3872. [Google Scholar]
- Wuertzer, C.A.; Sullivan, M.A.; Qiu, X.; Federoff, H.J. CNS delivery of vectored prion-specific single-chain antibodies delays disease onset. Mol. Ther 2008, 16, 481–486. [Google Scholar]
- Muller-Schiffmann, A.; Petsch, B.; Leliveld, S.R.; Muyrers, J.; Salwierz, A.; Mangels, C.; Schwarzinger, S.; Riesner, D.; Stitz, L.; Korth, C. Complementarity determining regions of an anti-prion protein scFv fragment orchestrate conformation specificity and antiprion activity. Mol. Immunol 2009, 46, 532–540. [Google Scholar]
- Shimizu, Y.; Kaku-Ushiki, Y.; Iwamaru, Y.; Muramoto, T.; Kitamoto, T.; Yokoyama, T.; Mohri, S.; Tagawa, Y. A novel anti-prion protein monoclonal antibody and its single-chain fragment variable derivative with ability to inhibit abnormal prion protein accumulation in cultured cells. Microbiol. Immunol 2010, 54, 112–121. [Google Scholar]
- Skrlj, N.; Serbec, V.C.; Dolinar, M. Single-chain Fv antibody fragments retain binding properties of the monoclonal antibody raised against peptide P1 of the human prion protein. Appl. Biochem. Biotechnol 2010, 160, 1808–1821. [Google Scholar]
- Fujita, K.; Yamaguchi, Y.; Mori, T.; Muramatsu, N.; Miyamoto, T.; Yano, M.; Miyata, H.; Ootsuyama, A.; Sawada, M.; Matsuda, H.; et al. Effects of a brain-engraftable microglial cell line expressing anti-prion scFv antibodies on survival times of mice infected with scrapie prions. Cell Mol. Neurobiol 2011, 31, 999–1008. [Google Scholar]
- Petsch, B.; Muller-Schiffmann, A.; Lehle, A.; Zirdum, E.; Prikulis, I.; Kuhn, F.; Raeber, A.J.; Ironside, J.W.; Korth, C.; Stitz, L. Biological effects and use of PrPSc- and PrP-specific antibodies generated by immunization with purified full-length native mouse prions. J. Virol 2011, 85, 4538–4546. [Google Scholar]
- Skrlj, N.; Vranac, T.; Popovic, M.; Curin Serbec, V.; Dolinar, M. Specific binding of the pathogenic prion isoform: Development and characterization of a humanized single-chain variable antibody fragment. PLoS One 2011, 6, e15783. [Google Scholar]
- Zhang, X.; Sun, X.X.; Xue, D.; Liu, D.G.; Hu, X.Y.; Zhao, M.; Yang, S.G.; Yang, Y.; Xia, Y.J.; Wang, Y.; Liu, R.T. Conformation-dependent scFv antibodies specifically recognize the oligomers assembled from various amyloids and show colocalization of amyloid fibrils with oligomers in patients with amyloidoses. Biochim. Biophys. Acta 2011, 1814, 1703–1712. [Google Scholar]
- Kubota, T.; Hamazoe, Y.; Hashiguchi, S.; Ishibashi, D.; Akasaka, K.; Nishida, N.; Katamine, S.; Sakaguchi, S.; Kuroki, R.; Nakashima, T.; et al. Direct evidence of generation and accumulation of beta-sheet-rich prion protein in scrapie-infected neuroblastoma cells with human IgG1 antibody specific for beta-form prion protein. J. Biol. Chem 2012, 287, 14023–14039. [Google Scholar]
- Moda, F.; Vimercati, C.; Campagnani, I.; Ruggerone, M.; Giaccone, G.; Morbin, M.; Zentilin, L.; Giacca, M.; Zucca, I.; Legname, G.; Tagliavini, F. Brain delivery of AAV9 expressing an anti-PrP monovalent antibody delays prion disease in mice. Prion 2012, 6, 383–390. [Google Scholar]
- Kalinke, U.; Krebber, A.; Krebber, C.; Bucher, E.; Pluckthun, A.; Zinkernagel, R.M.; Hengartner, H. Monovalent single-chain Fv fragments and bivalent miniantibodies bound to vesicular stomatitis virus protect against lethal infection. Eur. J. Immunol 1996, 26, 2801–2806. [Google Scholar]
- Krebber, A.; Bornhauser, S.; Burmester, J.; Honegger, A.; Willuda, J.; Bosshard, H.R.; Pluckthun, A. Reliable cloning of functional antibody variable domains from hybridomas and spleen cell repertoires employing a reengineered phage display system. J. Immunol. Methods 1997, 201, 35–55. [Google Scholar]
- White, A.R.; Enever, P.; Tayebi, M.; Mushens, R.; Linehan, J.; Brandner, S.; Anstee, D.; Collinge, J.; Hawke, S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 2003, 422, 80–83. [Google Scholar]
- Gauczynski, S.; Hundt, C.; Leucht, C.; Weiss, S. Interaction of prion proteins with cell surface receptors, molecular chaperones, and other molecules. Advan. Prot. Chem 2001, 57, 229–272. [Google Scholar]
- Gauczynski, S.; Nikles, D.; El-Gogo, S.; Papy-Garcia, D.; Rey, C.; Alban, S.; Barritault, D.; Lasmezas, C.I.; Weiss, S. The 37-kDa/67-kDa laminin receptor acts as a receptor for infectious prions and is inhibited by polysulfated glycanes. J. Infect. Dis 2006, 194, 702–709. [Google Scholar]
- Ludewigs, H.; Zuber, C.; Vana, K.; Nikles, D.; Zerr, I.; Weiss, S. Therapeutic approaches for prion disorders. Expert Rev. Anti. Infect. Ther 2007, 5, 613–630. [Google Scholar]
- Vana, K.; Weiss, S. A trans-dominant negative 37kDa/67kDa laminin receptor mutant impairs PrP(Sc) propagation in scrapie-infected neuronal cells. J. Mol. Biol. 2006, 358, 57–66. [Google Scholar]
- Morel, E.; Andrieu, T.; Casagrande, F.; Gauczynski, S.; Weiss, S.; Grassi, J.; Rousset, M.; Dormont, D.; Chambaz, J. Bovine prion is endocytosed by human enterocytes via the 37 kDa/67 kDa laminin receptor. Amer. J. Pathol 2005, 167, 1033–1042. [Google Scholar]
- Zuber, C.; Knackmuss, S.; Rey, C.; Reusch, U.; Rottgen, P.; Frohlich, T.; Arnold, G.J.; Pace, C.; Mitteregger, G.; Kretzschmar, H.A.; et al. Single chain Fv antibodies directed against the 37 kDa/67 kDa laminin receptor as therapeutic tools in prion diseases. Mol. Immunol 2008, 45, 144–151. [Google Scholar]
- Zuber, C.; Mitteregger, G.; Schuhmann, N.; Rey, C.; Knackmuss, S.; Rupprecht, W.; Reusch, U.; Pace, C.; Little, M.; Kretzschmar, H.A.; et al. Delivery of single-chain antibodies (scFvs) directed against the 37/67 kDa laminin receptor into mice via recombinant adeno-associated viral vectors for prion disease gene therapy. J. Gen. Virol 2008, 89, 2055–2061. [Google Scholar]
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement 2007, 3, 186–191. [Google Scholar]
- Sosa-Ortiz, A.L.; Acosta-Castillo, I.; Prince, M.J. Epidemiology of dementias and Alzheimer’s disease. Arch. Med. Res 2012, 43, 600–608. [Google Scholar]
- Robert, R.; Dolezal, O.; Waddington, L.; Hattarki, M.K.; Cappai, R.; Masters, C.L.; Hudson, P.J.; Wark, K.L. Engineered antibody intervention strategies for Alzheimer’s disease and related dementias by targeting amyloid and toxic oligomers. Protein Eng. Des. Sel 2009, 22, 199–208. [Google Scholar]
- Pfeifer, M.; Boncristiano, S.; Bondolfi, L.; Stalder, A.; Deller, T.; Staufenbiel, M.; Mathews, P.M.; Jucker, M. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science 2002, 298, 1379. [Google Scholar]
- Racke, M.M.; Boone, L.I.; Hepburn, D.L.; Parsadainian, M.; Bryan, M.T.; Ness, D.K.; Piroozi, K.S.; Jordan, W.H.; Brown, D.D.; Hoffman, W.P.; et al. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J. Neurosci 2005, 25, 629–636. [Google Scholar]
- Frenkel, D.; Katz, O.; Solomon, B. Immunization against Alzheimer’s beta -amyloid plaques via EFRH phage administration. Proc. Natl. Acad. Sci. USA 2000, 97, 11455–11459. [Google Scholar]
- Liu, R.; Yuan, B.; Emadi, S.; Zameer, A.; Schulz, P.; McAllister, C.; Lyubchenko, Y.; Goud, G.; Sierks, M.R. Single chain variable fragments against beta-amyloid (Abeta) can inhibit Abeta aggregation and prevent abeta-induced neurotoxicity. Biochemistry 2004, 43, 6959–6967. [Google Scholar]
- Fukuchi, K.; Tahara, K.; Kim, H.D.; Maxwell, J.A.; Lewis, T.L.; Accavitti-Loper, M.A.; Kim, H.; Ponnazhagan, S.; Lalonde, R. Anti-Abeta single-chain antibody delivery via adeno-associated virus for treatment of Alzheimer’s disease. Neurobiol. Dis 2006, 23, 502–511. [Google Scholar]
- Levites, Y.; Jansen, K.; Smithson, L.A.; Dakin, R.; Holloway, V.M.; Das, P.; Golde, T.E. Intracranial adeno-associated virus-mediated delivery of anti-pan amyloid beta, amyloid beta40, and amyloid beta42 single-chain variable fragments attenuates plaque pathology in amyloid precursor protein mice. J. Neurosci 2006, 26, 11923–11928. [Google Scholar]
- Wang, Y.J.; Pollard, A.; Zhong, J.H.; Dong, X.Y.; Wu, X.B.; Zhou, H.D.; Zhou, X.F. Intramuscular delivery of a single chain antibody gene reduces brain Abeta burden in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2009, 30, 364–376. [Google Scholar]
- Ryan, D.A.; Mastrangelo, M.A.; Narrow, W.C.; Sullivan, M.A.; Federoff, H.J.; Bowers, W.J. Abeta-directed single-chain antibody delivery via a serotype-1 AAV vector improves learning behavior and pathology in Alzheimer’s disease mice. Mol. Ther 2010, 18, 1471–1481. [Google Scholar]
- Wang, X.P.; Zhang, J.H.; Wang, Y.J.; Feng, Y.; Zhang, X.; Sun, X.X.; Li, J.L.; Du, X.T.; Lambert, M.P.; Yang, S.G.; Zhao, M.; Klein, W.L.; Liu, R.T. Conformation-dependent single-chain variable fragment antibodies specifically recognize beta-amyloid oligomers. FEBS Lett 2009, 583, 579–584. [Google Scholar]
- Finkbeiner, S. Huntington’s Disease. Cold Spring Harb. Perspect. Biol 2011, 3, a007476. [Google Scholar]
- La Spada, A.R.; Taylor, J.P. Repeat expansion disease: Progress and puzzles in disease pathogenesis. Nat. Rev. Genet 2010, 11, 247–258. [Google Scholar]
- Wexler, N.S.; Lorimer, J.; Porter, J.; Gomez, F.; Moskowitz, C.; Shackell, E.; Marder, K.; Penchaszadeh, G.; Roberts, S.A.; Gayan, J.; et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc. Natl. Acad. Sci. USA 2004, 101, 3498–3503. [Google Scholar]
- Lecerf, J.M.; Shirley, T.L.; Zhu, Q.; Kazantsev, A.; Amersdorfer, P.; Housman, D.E.; Messer, A.; Huston, J.S. Human single-chain Fv intrabodies counteract in situ huntingtin aggregation in cellular models of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 4764–4769. [Google Scholar]
- Murphy, R.C.; Messer, A. A single-chain Fv intrabody provides functional protection against the effects of mutant protein in an organotypic slice culture model of Huntington’s disease. Mol. Brain Res 2004, 121, 141–145. [Google Scholar]
- Miller, T.W.; Zhou, C.; Gines, S.; MacDonald, M.E.; Mazarakis, N.D.; Bates, G.P.; Huston, J.S.; Messer, A. A human single-chain Fv intrabody preferentially targets amino-terminal Huntingtin’s fragments in striatal models of Huntington’s disease. Neurobiol. Dis 2005, 19, 47–56. [Google Scholar]
- Bortvedt, S.F.; McLear, J.A.; Messer, A.; Ahern-Rindell, A.J.; Wolfgang, W.J. Cystamine and intrabody co-treatment confers additional benefits in a fly model of Huntington’s disease. Neurobiol. Dis 2010, 40, 130–134. [Google Scholar]
- Wolfgang, W.J.; Miller, T.W.; Webster, J.M.; Huston, J.S.; Thompson, L.M.; Marsh, J.L.; Messer, A. Suppression of Huntington’s disease pathology in Drosophila by human single-chain Fv antibodies. Proc. Natl. Acad. Sci. USA 2005, 102, 11563–11568. [Google Scholar]
- Snyder-Keller, A.; McLear, J.A.; Hathorn, T.; Messer, A. Early or late-stage anti-N-terminal Huntingtin intrabody gene therapy reduces pathological features in B6.HDR6/1 mice. J. Neuropathol. Exp. Neurol 2010, 69, 1078–1085. [Google Scholar]
- Butler, D.C.; Messer, A. Bifunctional anti-huntingtin proteasome-directed intrabodies mediate efficient degradation of mutant huntingtin exon 1 protein fragments. PLoS One 2011, 6, e29199. [Google Scholar]
- Khoshnan, A.; Ko, J.; Patterson, P.H. Effects of intracellular expression of anti-huntingtin antibodies of various specificities on mutant huntingtin aggregation and toxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 1002–1007. [Google Scholar]
- Hornykiewicz, O. Biochemical aspects of Parkinson’s disease. Neurology 1998, 51, S2–S9. [Google Scholar]
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol 1996, 55, 259–272. [Google Scholar]
- Vance, J.M.; Ali, S.; Bradley, W.G.; Singer, C.; Di Monte, D.A. Gene-environment interactions in Parkinson’s disease and other forms of parkinsonism. Neurotoxicology 2010, 31, 598–602. [Google Scholar]
- Gao, H.M.; Hong, J.S. Gene-environment interactions: Key to unraveling the mystery of Parkinson’s disease. Prog. Neurobiol 2011, 94, 1–19. [Google Scholar]
- Dickson, D.W. Parkinson’s disease and parkinsonism: Neuropathology. Cold Spring Harb. Perspect. Med 2012, 2, a009258. [Google Scholar]
- Kalia, L.V.; Kalia, S.K.; McLean, P.J.; Lozano, A.M.; Lang, A.E. alpha-Synuclein oligomers and clinical implications for Parkinson disease. Ann. Neurol 2013, 73, 155–169. [Google Scholar]
- Mezey, E.; Dehejia, A.M.; Harta, G.; Tresser, N.; Suchy, S.F.; Nussbaum, R.L.; Brownstein, M.J.; Polymeropoulos, M.H. Alpha synuclein is present in Lewy bodies in sporadic Parkinson’s disease. Mol. Psychiatry 1998, 3, 493–499. [Google Scholar]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar]
- Maguire-Zeiss, K.A.; Yehling, E.; Giuliano, R.; Sullivan, M.; J.F.H. HSV Amplicon Expression of Single Chain Antibodies Directed Against α-Synuclein Conformers. Mol. Ther 2004, 9, S86. [Google Scholar]
- Emadi, S.; Liu, R.; Yuan, B.; Schulz, P.; McAllister, C.; Lyubchenko, Y.; Messer, A.; Sierks, M.R. Inhibiting aggregation of alpha-synuclein with human single chain antibody fragments. Biochemistry 2004, 43, 2871–2878. [Google Scholar]
- Lynch, S.M.; Zhou, C.; Messer, A. An scFv intrabody against the nonamyloid component of alpha-synuclein reduces intracellular aggregation and toxicity. J. Mol. Biol 2008, 377, 136–147. [Google Scholar]
- Zhou, C.; Emadi, S.; Sierks, M.R.; Messer, A. A human single-chain Fv intrabody blocks aberrant cellular effects of overexpressed alpha-synuclein. Mol. Ther 2004, 10, 1023–1031. [Google Scholar]
- Emadi, S.; Barkhordarian, H.; Wang, M.S.; Schulz, P.; Sierks, M.R. Isolation of a human single chain antibody fragment against oligomeric alpha-synuclein that inhibits aggregation and prevents alpha-synuclein-induced toxicity. J. Mol. Biol 2007, 368, 1132–1144. [Google Scholar]
- Hudson, P.J.; Kortt, A.A. High avidity scFv multimers; diabodies and triabodies. J. Immunol. Methods 1999, 231, 177–189. [Google Scholar]
- Joshi, S.N.; Butler, D.C.; Messer, A. Fusion to a highly charged proteasomal retargeting sequence increases soluble cytoplasmic expression and efficacy of diverse anti-synuclein intrabodies. mAbs 2012, 4, 686–693. [Google Scholar]
- Whitlow, M.; Bell, B.A.; Feng, S.L.; Filpula, D.; Hardman, K.D.; Hubert, S.L.; Rollence, M.L.; Wood, J.F.; Schott, M.E.; Milenic, D.E.; et al. An improved linker for single-chain Fv with reduced aggregation and enhanced proteolytic stability. Protein Eng 1993, 6, 989–995. [Google Scholar]
- Fisher, A.C.; DeLisa, M.P. Efficient isolation of soluble intracellular single-chain antibodies using the twin-arginine translocation machinery. J. Mol. Biol 2009, 385, 299–311. [Google Scholar]
- Chames, P.; Fieschi, J.; Baty, D. Production of a soluble and active MBP-scFv fusion: Favorable effect of the leaky tolR strain. FEBS Lett 1997, 405, 224–228. [Google Scholar]
- Sun, W.; Xie, J.; Lin, H.; Mi, S.; Li, Z.; Hua, F.; Hu, Z. A combined strategy improves the solubility of aggregation-prone single-chain variable fragment antibodies. Protein Expr. Purif 2012, 83, 21–29. [Google Scholar]
- Boado, R.J.; Lu, J.Z.; Hui, E.K.; Pardridge, W.M. IgG-single chain Fv fusion protein therapeutic for Alzheimer’s disease: Expression in CHO cells and pharmacokinetics and brain delivery in the rhesus monkey. Biotechnol. Bioeng 2010, 105, 627–635. [Google Scholar]
- Skrlj, N.; Drevensek, G.; Hudoklin, S.; Romih, R.; Curin Serbec, V.; Dolinar, M. Recombinant single-chain antibody with the Trojan peptide penetratin positioned in the linker region enables cargo transfer across the blood-brain barrier. Appl. Biochem. Biotechnol 2013, 169, 159–169. [Google Scholar]
- Wei, X.; Roettger, Y.; Tan, B.; He, Y.; Dodel, R.; Hampel, H.; Wei, G.; Haney, J.; Gu, H.; Johnstone, B.H.; et al. Human anti-prion antibodies block prion peptide fibril formation and neurotoxicity. J. Biol. Chem 2012, 287, 12858–12866. [Google Scholar]
- Hoffner, G.; Soues, S.; Djian, P. Aggregation of expanded huntingtin in the brains of patients with Huntington disease. Prion 2007, 1, 26–31. [Google Scholar]
- Huang, P.; Lian, F.; Wen, Y.; Guo, C.; Lin, D. Prion protein oligomer and its neurotoxicity. Acta Biochim. Biophys. Sin. (Shanghai) 2013, 45, 442–451. [Google Scholar]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of alpha-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci 2013, 14, 38–48. [Google Scholar]
- Danzer, K.M.; Haasen, D.; Karow, A.R.; Moussaud, S.; Habeck, M.; Giese, A.; Kretzschmar, H.; Hengerer, B.; Kostka, M. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J. Neurosci 2007, 27, 9220–9232. [Google Scholar]
- Volles, M.J.; Lee, S.J.; Rochet, J.C.; Shtilerman, M.D.; Ding, T.T.; Kessler, J.C.; Lansbury, P.T., Jr. Vesicle permeabilization by protofibrillar alpha-synuclein: Implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry 2001, 40, 7812–7819. [Google Scholar]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Amer. J. Pathol 2000, 157, 401–410. [Google Scholar]
- Alim, M.A.; Ma, Q.L.; Takeda, K.; Aizawa, T.; Matsubara, M.; Nakamura, M.; Asada, A.; Saito, T.; Kaji, H.; Yoshii, M.; et al. Demonstration of a role for alpha-synuclein as a functional microtubule-associated protein. J. Alzheimer’s Dis 2004, 6, 435–442. [Google Scholar]
- Brown, D.R. Oligomeric alpha-synuclein and its role in neuronal death. IUBMB Life 2010, 62, 334–339. [Google Scholar]
- Kudo, W.; Lee, H.P.; Zou, W.Q.; Wang, X.; Perry, G.; Zhu, X.; Smith, M.A.; Petersen, R.B.; Lee, H.G. Cellular prion protein is essential for oligomeric amyloid-beta-induced neuronal cell death. Hum. Mol. Genet 2012, 21, 1138–1144. [Google Scholar]
- Kudo, W.; Petersen, R.B.; Lee, H.G. Cellular prion protein and Alzheimer disease: Link to oligomeric amyloid-beta and neuronal cell death. Prion 2013, 7, 114–116. [Google Scholar]
- Silveira, J.R.; Raymond, G.J.; Hughson, A.G.; Race, R.E.; Sim, V.L.; Hayes, S.F.; Caughey, B. The most infectious prion protein particles. Nature 2005, 437, 257–261. [Google Scholar]
- Simoneau, S.; Rezaei, H.; Sales, N.; Kaiser-Schulz, G.; Lefebvre-Roque, M.; Vidal, C.; Fournier, J.G.; Comte, J.; Wopfner, F.; Grosclaude, J.; et al. In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog 2007, 3, e125. [Google Scholar]
- Bueler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar]
- Nannenga, B.L.; Zameer, A.; Sierks, M.R. Anti-oligomeric single chain variable domain antibody differentially affects huntingtin and alpha-synuclein aggregates. FEBS Lett 2008, 582, 517–522. [Google Scholar]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010, 65, 66–79. [Google Scholar]
- Lundblad, M.; Decressac, M.; Mattsson, B.; Bjorklund, A. Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc. Nat. Acad. Sci. USA 2012, 109, 3213–3219. [Google Scholar]
- Bellucci, A.; Zaltieri, M.; Navarria, L.; Grigoletto, J.; Missale, C.; Spano, P. From alpha-synuclein to synaptic dysfunctions: New insights into the pathophysiology of Parkinson’s disease. Brain Res 2012, 1476, 183–202. [Google Scholar]
- Bellucci, A.; Navarria, L.; Zaltieri, M.; Missale, C.; Spano, P. alpha-Synuclein synaptic pathology and its implications in the development of novel therapeutic approaches to cure Parkinson’s disease. Brain Res 2012, 1432, 95–113. [Google Scholar]
- Atwal, R.S.; Desmond, C.R.; Caron, N.; Maiuri, T.; Xia, J.; Sipione, S.; Truant, R. Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat. Chem. Biol 2011, 7, 453–460. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).