Multigenerational Study of Chemically Induced Cytotoxicity and Proliferation in Cultures of Human Proximal Tubular Cells

Abstract
:1. Introduction
2. Results
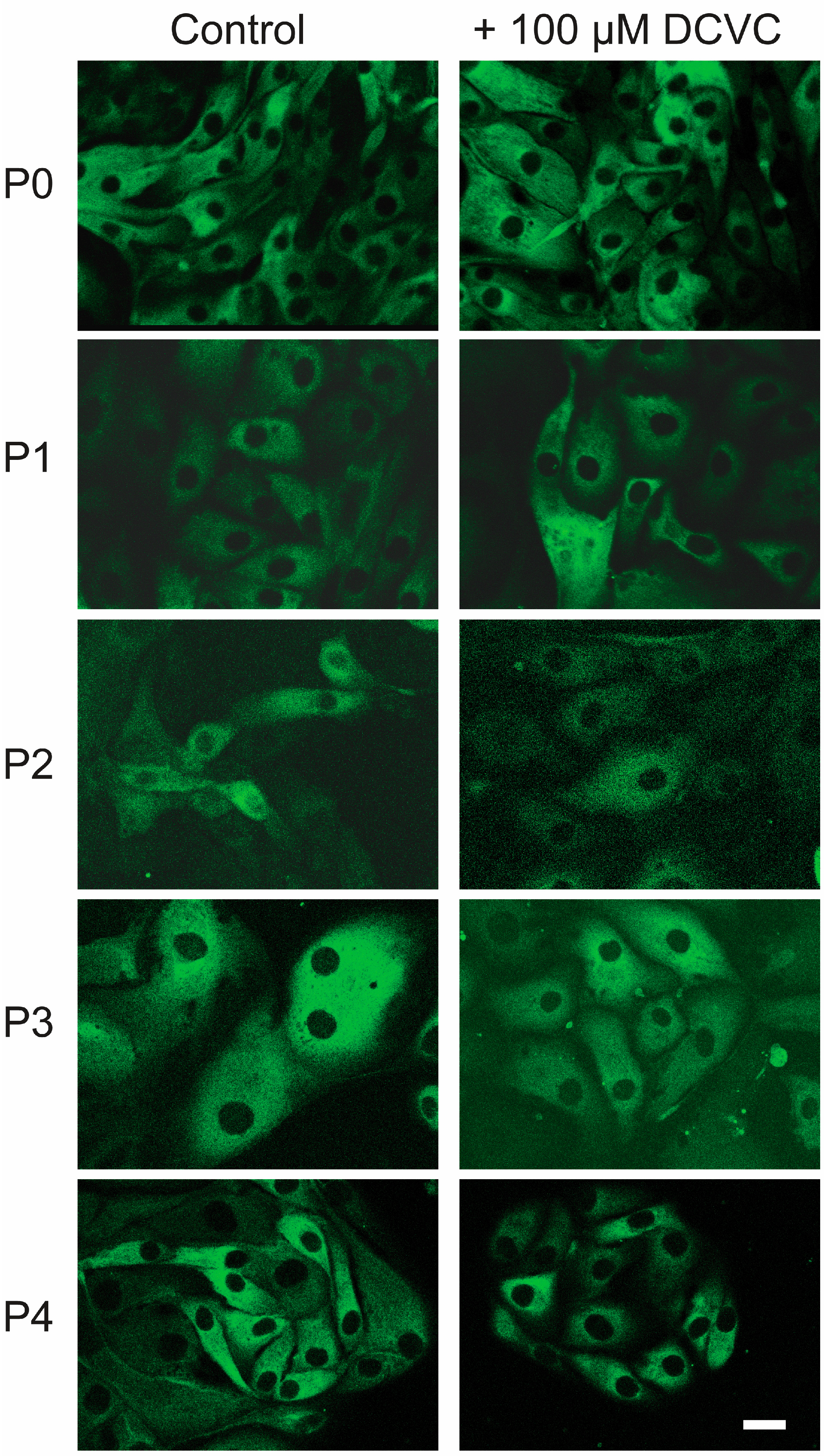
2.1. Cellular Growth and Morphology

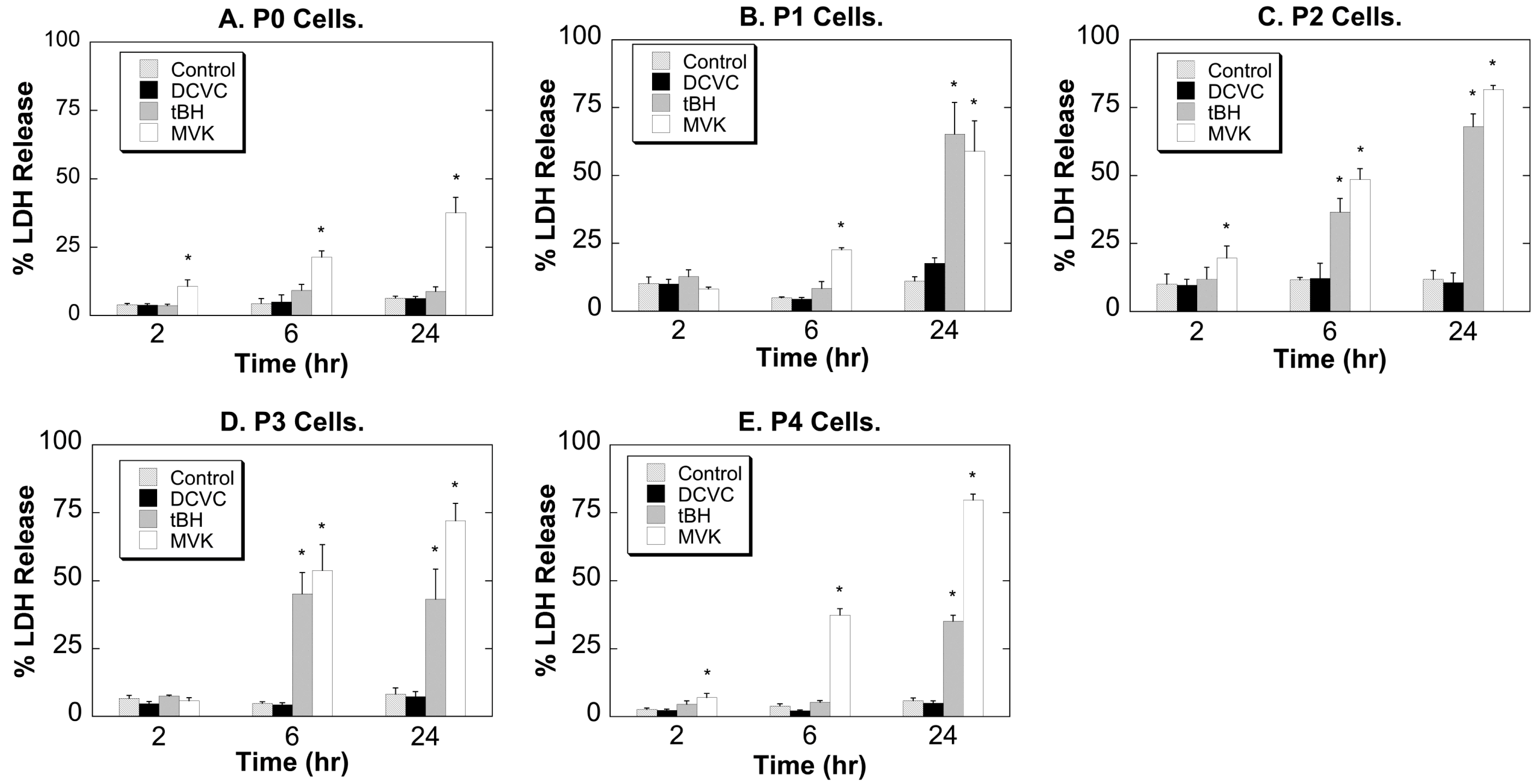
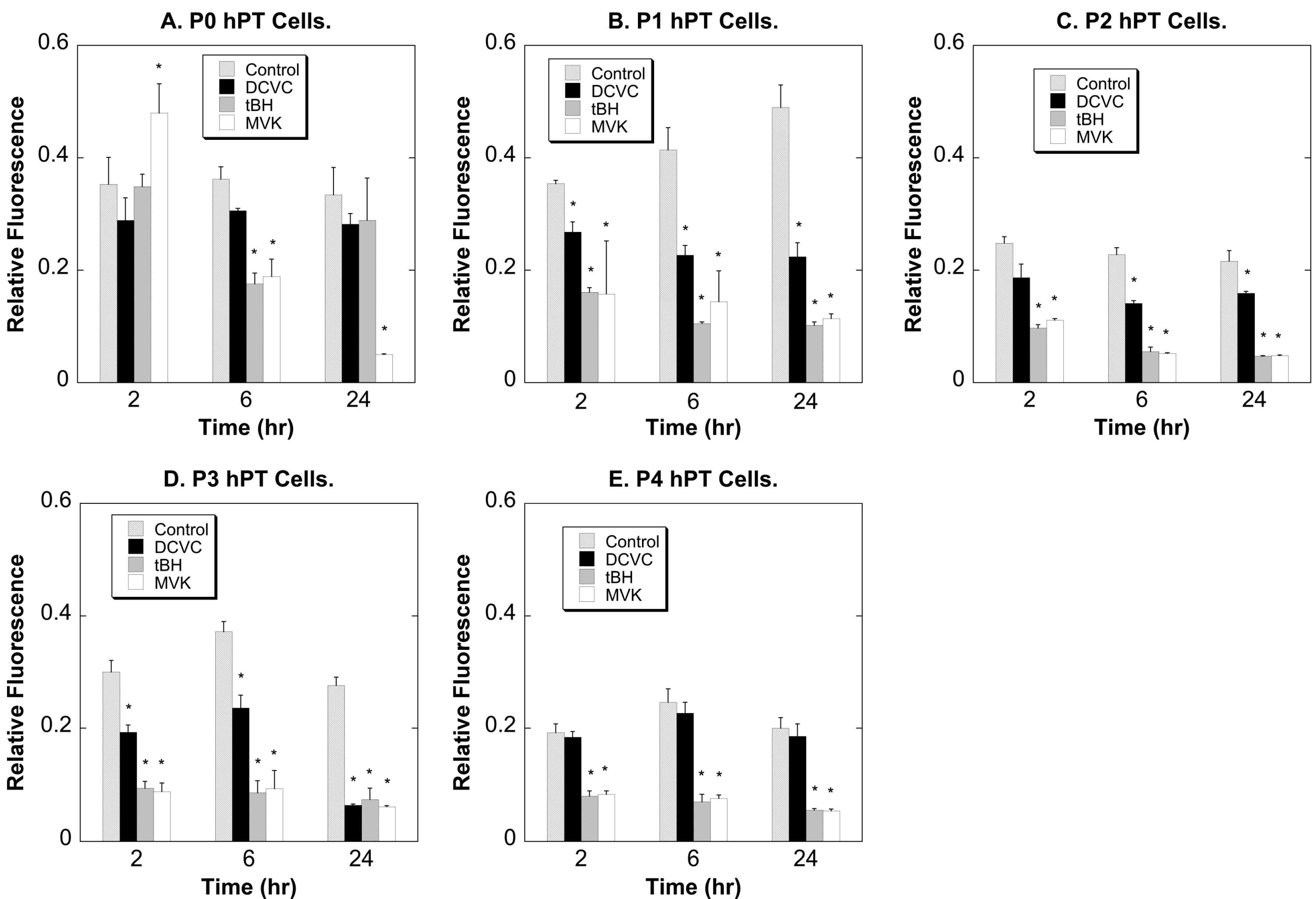
2.2. Comparative Cytotoxicity



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Passage Number | GPX Activity (mU/mg Protein) |
|---|---|
| P0 | 60.4 ± 11.0 |
| P1 | 37.9 ± 4.5 * |
| P2 | 50.0 ± 0.9 |
| P3 | 59.6 ± 7.9 |
| P4 | 46.3 ± 0.7 * |
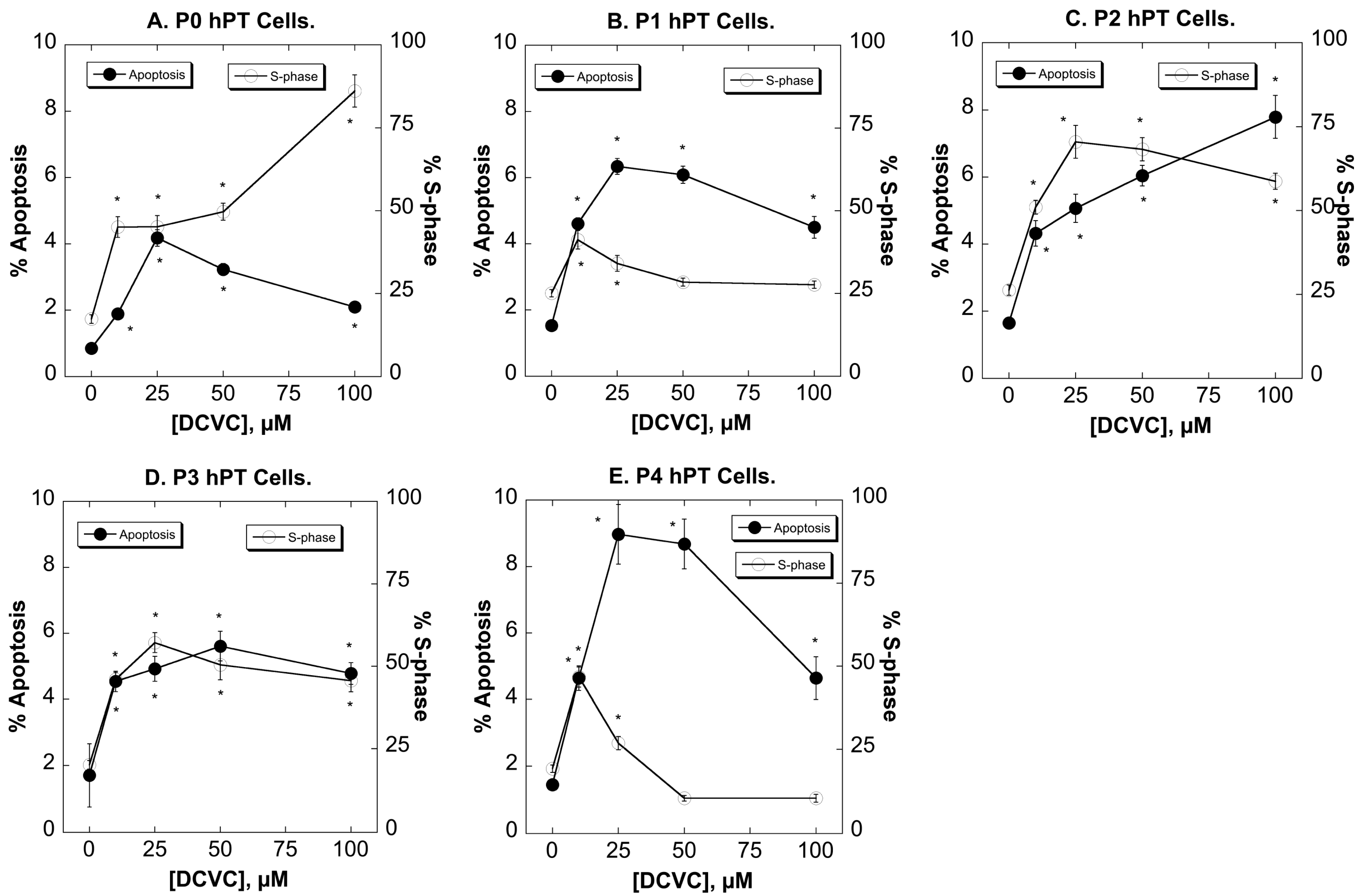
2.3. Effects of DCVC on Cell Cycle Distribution and Apoptosis

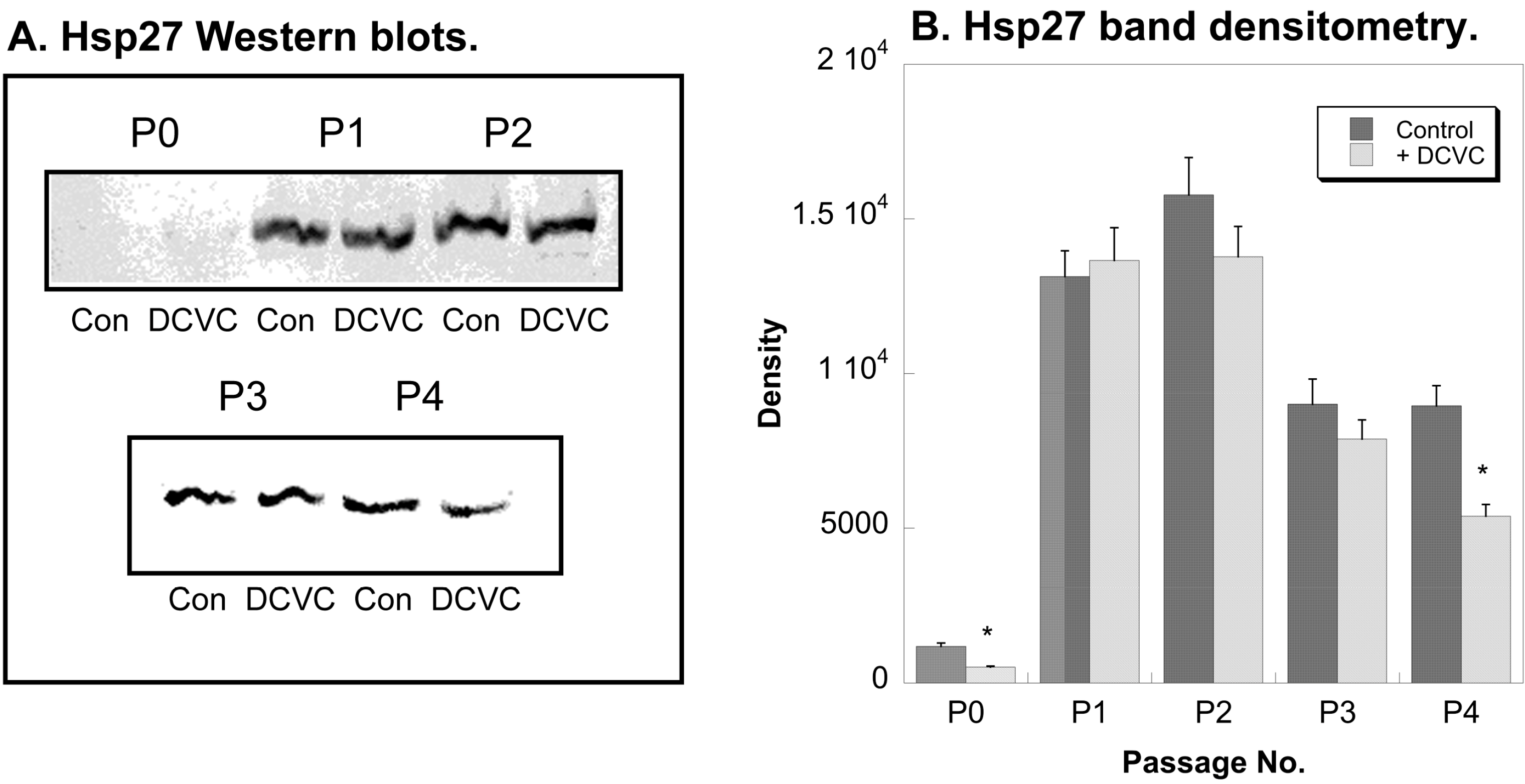
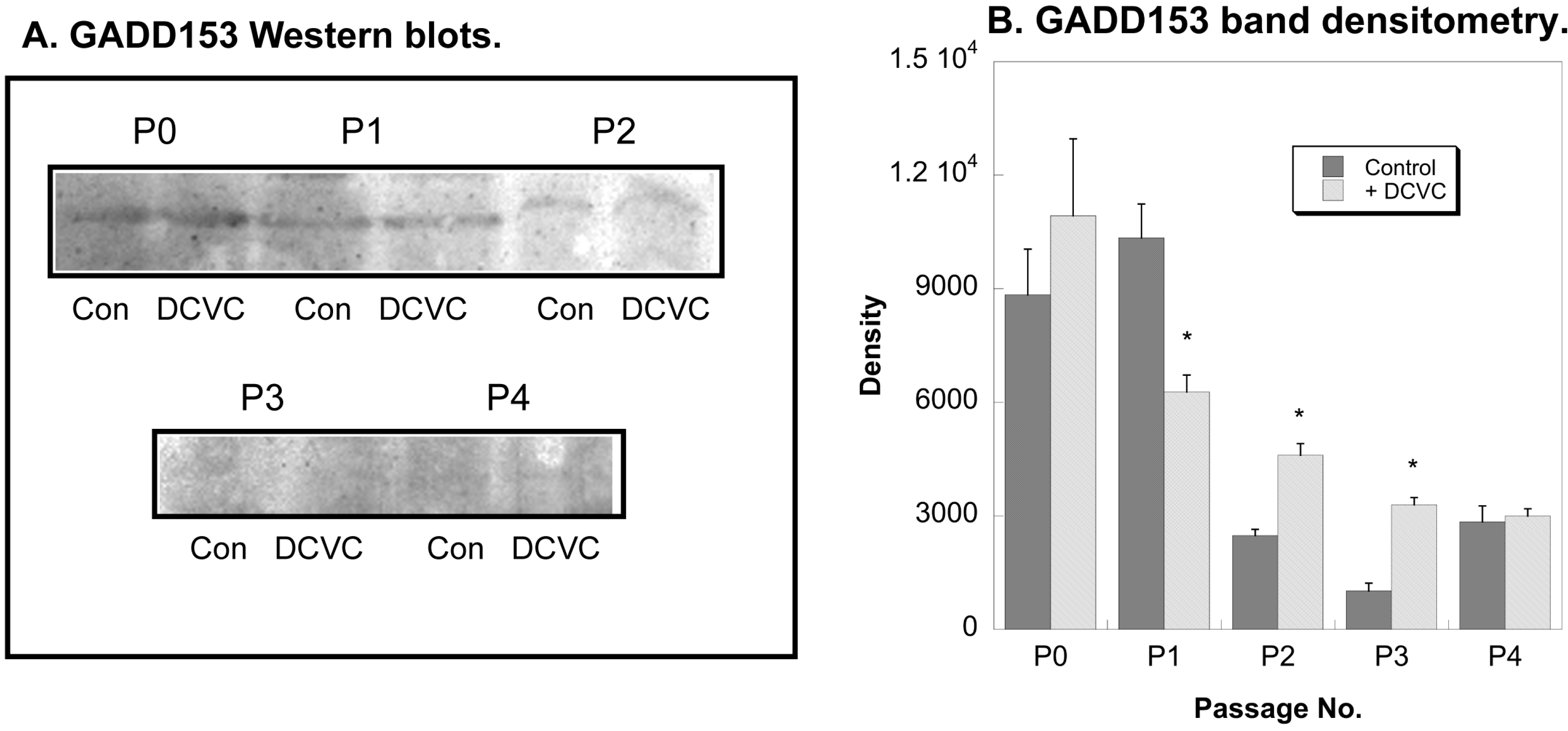
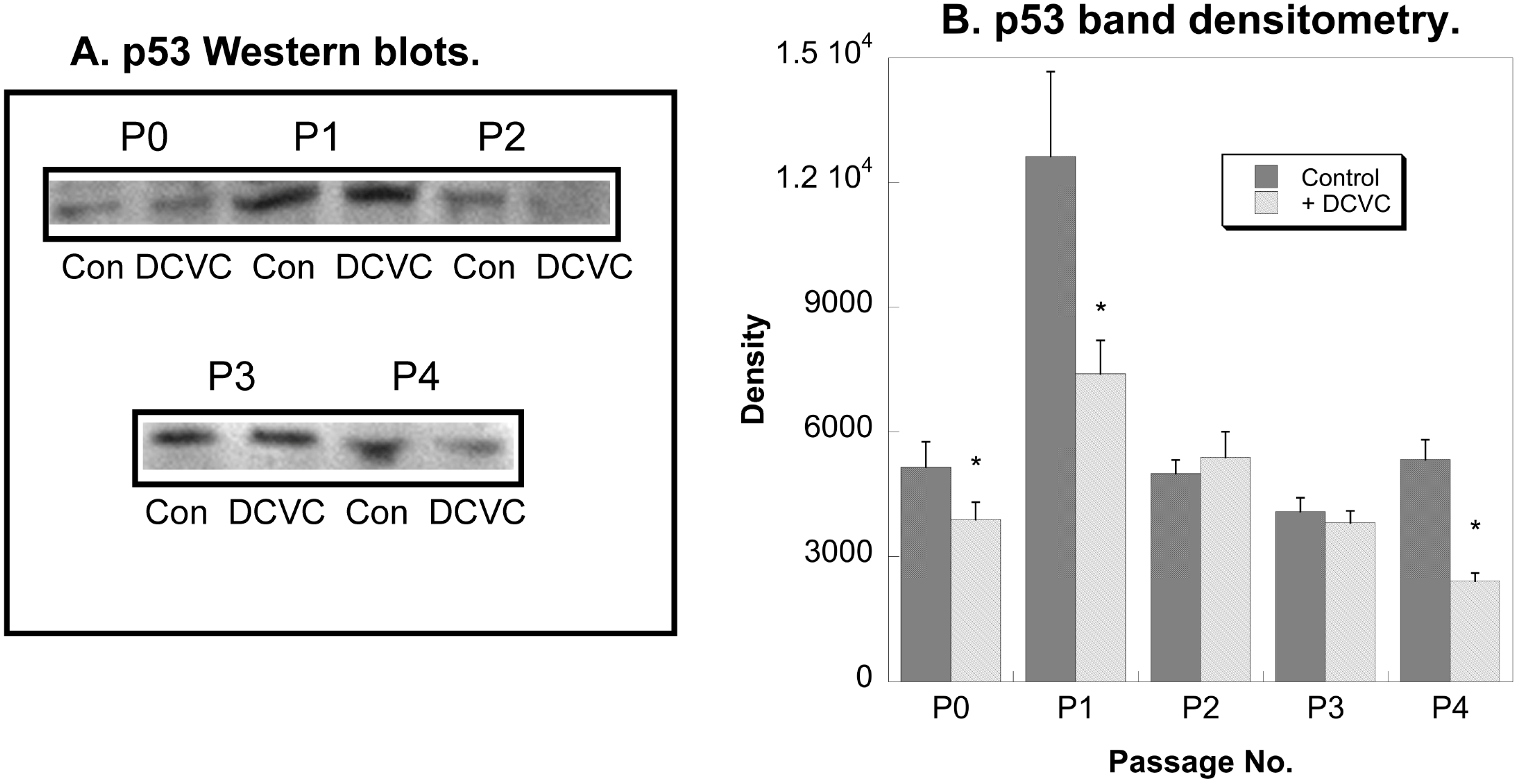
2.4. Effects of DCVC on Expression of Regulatory and Stress Response Proteins



3. Discussion
4. Materials and Methods
4.1. Experimental Design
4.2. Materials
4.3. Isolation and Primary Culture of hPT Cells
4.4. Passaging of hPT Cells
4.5. Immunocytochemical Staining for Cytokeratins
4.6. Measurement of Cell Death by Lactate Dehydrogenase (LDH) Release
4.7. Measurement of Cytotoxicity and Proliferation by MTT Assay
4.8. Flow Cytometry Assay of Cell Cycle Distribution and Apoptosis
4.9. Western Blot Analyses
4.10. Data Analysis
Acknowledgments
Author Contributions
Abbreviations
| DCVC | S-(1,2-dichlorovinyl)-l-cysteine |
| DMEM:F12 | Dulbecco’s Modified Eagle’s Medium:Ham’s F12 Medium |
| FITC | fluorescein isothiocyanate |
| GADD153 | growth and DNA damage protein153 |
| GPX | GSH peroxidase |
| GSH | glutathione |
| hPT | human proximal tubular |
| Hsp27 | heat shock protein 27 |
| LDH | lactate dehydrogenase |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| MVK | methyl vinyl ketone |
| P0 | primary cell cultures |
| P1–P4 | cells passaged one to four generations |
| PBS | phosphate-buffered saline |
| rPT | rat proximal tubular |
| tBH | tert-butyl hydroperoxide |
| TCE | trichloroethylene |
| TTBS | tris-buffered saline containing Tween-20 |
Conflicts of Interest
References
- Lash, L.H. Human Proximal Tubular Cells as an in vitro Model for Drug Screening and Mechanistic Toxicology. Available online: http://alttox.org/human-proximal-tubular-cells-as-an-in-vitro-model-for-drug-screening-and-mechanistic-toxicology/ (accessed on 22 May 2012).
- Pelekis, M.; Krishnan, K. Assessing the relevance of rodent data on chemical interactions for health risk assessment purposes: A case study with dichloromethane-toluene mixture. Regul. Toxicol. Pharmacol. 1997, 25, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, A.D.; DeSesso, J.M. Have animal data been used inappropriately to estimate risks to humans from environmental trichloroethylene. Regul. Toxicol. Pharmacol. 1993, 18, 137–153. [Google Scholar] [CrossRef] [PubMed]
- National Research Council. Biologic Markers in Urinary Toxicology; National Academy Press: Washington, DC, USA, 1995. [Google Scholar]
- Cummings, B.S.; Lash, L.H. Metabolism and toxicity of trichloroethylene and S-(1,2-dichlorovinyl)-l-cysteine in freshly isolated human proximal tubular cells. Toxicol. Sci. 2000, 53, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Cummings, B.S.; Lasker, J.M.; Lash, L.H. Expression of glutathione-dependent enzymes and cytochrome P450s in freshly isolated and primary cultures of proximal tubular cells from human kidney. J. Pharmacol. Exp. Ther. 2000, 293, 677–685. [Google Scholar] [PubMed]
- Lash, L.H.; Hueni, S.E.; Putt, D.A. Apoptosis, necrosis, and cell proliferation induced by S-(1,2-dichlorovinyl)-l-cysteine in primary cultures of human proximal tubular cells. Toxicol. Appl. Pharmacol. 2001, 177, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H.; Putt, D.A.; Hueni, S.E.; Krause, R.J.; Elfarra, A.A. Roles of necrosis, apoptosis, and mitochondrial dysfunction in S-(1,2-dichlorovinyl)-l-cysteine sulfoxide-induced cytotoxicity in primary cultures of human renal proximal tubular cells. J. Pharmacol. Exp. Ther. 2003, 305, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Krause, R.J.; Lash, L.H.; Elfarra, A.A. Human kidney flavin-containing monooxygenases and their potential roles in cysteine S-conjugate metabolism and nephrotoxicity. J. Pharmacol. Exp. Ther. 2003, 304, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H.; Putt, D.A.; Cai, H. Drug metabolism enzyme expression and activity in primary cultures of human proximal tubular cells. Toxicology 2008, 244, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H.; Putt, D.A.; Cai, H. Membrane transport function in primary cultures of human proximal tubular cells. Toxicology 2006, 228, 200–218. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H.; Putt, D.A.; Hueni, S.E.; Horwitz, B.P. Molecular markers of trichloroethylene-induced toxicity in human kidney cells. Toxicol. Appl. Pharmacol. 2005, 206, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H.; Putt, D.A.; Hueni, S.E.; Payton, S.G.; Zwickl, J. Interactive toxicity of inorganic mercury and trichloroethylene in rat and human proximal tubules: Effects on apoptosis, necrosis, and glutathione status. Toxicol. Appl. Pharmacol. 2007, 221, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Papanayotou, I.; Putt, D.A.; Wang, J.; Lash, L.H. Role of mitochondrial dysfunction in cellular responses to S-(1,2-dichlorovinyl)-l-cysteine in primary cultures of human proximal tubular cells. Biochem. Pharmacol. 2008, 76, 552–567. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Agrawal, A.K.; Putt, D.A.; Hashim, M.; Reddy, A.; Brodfuehrer, J.; Surendran, N.; Lash, L.H. Assessment of the renal toxicity of novel anti-inflammatory compounds using cynomolgus monkey and human kidney cells. Toxicology 2009, 258, 56–63. [Google Scholar] [CrossRef] [PubMed]
- McGoldrick, T.A.; Lock, E.A.; Rodilla, V.; Hawksworth, G.M. Renal cysteine conjugate C–S lyase mediated toxicity of halogenated alkenes in primary cultures of human and rat proximal tubular cells. Arch. Toxicol. 2003, 77, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Lock, E.A.; Barth, J.L.; Argraves, S.W.; Schnellmann, R.G. Changes in gene expression in human renal proximal tubule cells exposed to low concentrations of S-(1,2-dichlorovinyl)-l-cysteine, a metabolite of trichloroethylene. Toxicol. Appl. Pharmacol. 2006, 216, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.J.; Johnson, G.; Kirk, J.; Fuerstenberg, S.M.; Zager, R.A.; Torok-Storb, B. HK-2: An immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney Int. 1994, 45, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Shelden, E.A.; Borrelli, M.J.; Pollock, F.M.; Bonham, R. Heat shock protein 27 associated with basolateral cell boundaries in heat-shocked and ATP-depleted epithelial cells. J. Am. Soc. Nephrol. 2002, 13, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Van Why, S.K.; Mann, A.S.; Ardito, T.; Thulin, G.; Ferris, S.; Macleod, M.A.; Kashgarian, M.; Siegel, N.J. Hsp27 associated with actin and limits injury in energy-depleted renal epithelia. J. Am. Soc. Nephrol. 2003, 14, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Yu, K.; Holbrook, N.J.; Stevens, J.L. Activation of the growth arrest and DNA damage-inducible gene gadd 153 by nephrotoxic cysteine conjugates and dithiothreitol. J. Biol. Chem. 1992, 267, 8207–8212. [Google Scholar] [PubMed]
- Dmitrieva, N.; Michea, L.; Burg, M. p53 protects renal inner medullary cells from hypertonic stress by restricting DNA replication. Am. J. Physiol. 2001, 281, F522–F530. [Google Scholar]
- Healy, E.; Dempsey, M.; Lally, C.; Ryan, M.P. Apoptosis and necrosis: Mechanisms of cell death induced by cyclosporin A in a renal proximal tubular cell line. Kidney Int. 1998, 54, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Megyesi, J.; Andrade, L.; Vieira, J.M., Jr.; Safirstein, R.L.; Price, P.M. Coordination of the cell cycle is an important determinant of the syndrome of acute renal failure. Am. J. Physiol. 2002, 283, F810–F816. [Google Scholar]
- Hoey, J.G.; Garrett, S.H.; Sens, M.A.; Todd, J.H.; Sens, D.A. Expression of MT-3 mRNA in human kidney, proximal tubule cell cultures, and renal cell carcinoma. Toxicol. Lett. 1997, 92, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Garrett, S.H.; Somji, S.; Todd, J.H.; Sens, D.A. Exposure of human proximal tubule cells to Cd2+, Zn2+, and Cu2+ induces metallothionein protein accumulation but not metallothionein isoform 2 mRNA. Environ. Health Perspect. 1998, 106, 587–595. [Google Scholar] [PubMed]
- Garrett, S.H.; Somji, S.; Todd, J.H.; Sens, M.A.; Sens, D.A. Differential expression of human metallothionein isoform I mRNA in human proximal tubule cells exposed to metals. Environ. Health Perspect. 1998, 106, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Somji, S.; Todd, J.H.; Sens, M.A.; Garrett, S.H.; Sens, D.A. Expression of the constitutive and inducible forms of heat shock protein 70 in human proximal tubule cells exposed to heat, sodium arsenite, and CdCl2. Environ. Health Perspect. 1999, 107, 887–893. [Google Scholar] [PubMed]
- Somji, S.; Sens, D.A.; Garrett, S.H.; Sens, M.A.; Todd, J.H. Heat shock protein 27 expression in human proximal tubule cells exposed to lethal and sublethal concentrations of CdCI2. Environ. Health Perspect. 1999, 107, 545–552. [Google Scholar] [PubMed]
- Garrett, S.H.; Phillips, V.; Somji, S.; Sens, M.A.; Dutta, R.; Park, S.; Kim, D.; Sens, D.A. Transient induction of metallothionein isoform 3 (MT-3), c-fos, c-jun and c-myc in human proximal tubule cells exposed to cadmium. Toxicol. Lett. 2002, 126, 89–101. [Google Scholar] [CrossRef]
- Kim, D.; Garrett, S.H.; Sens, M.A.; Somji, S.; Sens, D.A. Metallothionein isoform 3 and proximal tubule vectorial active transport. Kidney Int. 2002, 61, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Somji, S.; Garrett, S.H.; Sens, M.A.; Gurel, V.; Sens, D.A. Expression of metallothionein isoform 3 (MT-3) determines the choice between apoptotic or necrotic cell death in Cd2+-exposed human proximal tubule cells. Toxicol. Sci. 2002, 80, 358–366. [Google Scholar] [CrossRef]
- Bathula, C.S.; Garrett, S.H.; Zhou, X.D.; Sens, M.A.; Sens, D.A.; Somji, S. Cadmium, vectorial active transport, and MT-3–dependent regulation of cadherin expression in human proximal tubular cells. Toxicol. Sci. 2008, 102, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.; Schumacher, K.M.; Tasnim, F.; Kandasamy, K.; Schumacher, A.; Ni, M.; Gao, S.; Zink, D.; Ying, J.Y. Human embryonic stem cells differentiate into functional renal proximal tubular-like cells. Kidney Int. 2013, 83, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Nivet, E.; Sancho-Martinez, I.; Gallegos, T.; Suzuki, K.; Okamura, D.; Wu, M.-Z.; Dubova, I.; Esteban, C.R.; Montserrat, N.; et al. Directed differentiation of human pluripotent cells to ureteric bud kidney progenitor-like cells. Nat. Cell Biol. 2013, 15, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Han, Y.-M. Differentiation of human pluripotent stem cells into nephron progenitor cells in a serum and feeder free system. PLoS One 2014, 9, e94888. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.Q.; Freedman, B.S.; Morizane, R.; Lerou, P.H.; Valerius, M.T.; Bonventre, J.V. Rapid and efficient differentiation of human pluripotent stem cells into intermediate mesoderm that forms tubules expressing kidney proximal tubular markers. J. Am. Soc. Nephrol. 2014, 25, 1211–1225. [Google Scholar] [CrossRef] [PubMed]
- Takasato, M.; Er, P.X.; Becroft, M.; Vanslambrouck, J.M.; Stanley, E.G.; Elefanty, A.G.; Little, M.H. Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nat. Cell Biol. 2014, 16, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Angelotti, M.L.; Ronconi, E.; Ballerini, L.; Peired, A.; Mazzinghi, B.; Sagrinati, C.; Parente, E.; Gacci, M.; Carini, M.; Rotondi, M.; et al. Characterization of renal progenitors committed toward tubular lineage and their regenerative potential in renal tubular injury. Stem Cells 2012, 30, 1714–1725. [Google Scholar] [CrossRef] [PubMed]
- Bi, B.; Schmitt, R.; Israilova, M.; Nishio, H.; Cantley, L.G. Stromal cells protect against acute tubular injury via an endocrine effect. J. Am. Soc. Nephrol. 2007, 18, 2486–2496. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.-Y.; Chien, Y.; Chiou, G.-Y.; Lin, C.-H.; Chiou, C.-H.; Tarng, D.-C. Induced pluripotent stem cells without c-Myc attenuate acute kidney injury via downregulating the signaling of oxidative stress and inflammation in ischemia-reperfusion rats. Cell Transplant. 2012, 21, 2569–2585. [Google Scholar] [CrossRef] [PubMed]
- Tiong, H.Y.; Huang, P.; Xiong, P.; Xiong, S.; Li, Y.; Vathsala, A.; Zink, D. Drug-induced nephrotoxicity: Clinical impact and preclinical in vitro models. Mol. Pharm. 2014, 11, 1933–1948. [Google Scholar] [CrossRef] [PubMed]
- Elfarra, A.A.; Jakobson, I.; Anders, M.W. Mechanism of S-(1,2-dichlorovinyl)gluta-thione-induced nephrotoxicity. Biochem. Pharmacol. 1986, 35, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.H.; McMartin, K.E.; Sens, D.A. Enzymatic isolation and serum-free culture of human renal cells. In Methods in Molecular Medicine; Jones, G.E., Ed.; Human Cell Culture Protocols, Humana Press, Inc.: Totowa, NJ, USA, 1996. [Google Scholar]
- Lash, L.H.; Tokarz, J.J.; Pegouske, D.M. Susceptibility of primary cultures of proximal tubular and distal tubular cells from rat kidney to chemically induced toxicity. Toxicology 1995, 103, 85–103. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lash, L.H.; Putt, D.A.; Benipal, B. Multigenerational Study of Chemically Induced Cytotoxicity and Proliferation in Cultures of Human Proximal Tubular Cells. Int. J. Mol. Sci. 2014, 15, 21348-21365. https://doi.org/10.3390/ijms151121348
Lash LH, Putt DA, Benipal B. Multigenerational Study of Chemically Induced Cytotoxicity and Proliferation in Cultures of Human Proximal Tubular Cells. International Journal of Molecular Sciences. 2014; 15(11):21348-21365. https://doi.org/10.3390/ijms151121348
Chicago/Turabian StyleLash, Lawrence H., David A. Putt, and Bavneet Benipal. 2014. "Multigenerational Study of Chemically Induced Cytotoxicity and Proliferation in Cultures of Human Proximal Tubular Cells" International Journal of Molecular Sciences 15, no. 11: 21348-21365. https://doi.org/10.3390/ijms151121348
APA StyleLash, L. H., Putt, D. A., & Benipal, B. (2014). Multigenerational Study of Chemically Induced Cytotoxicity and Proliferation in Cultures of Human Proximal Tubular Cells. International Journal of Molecular Sciences, 15(11), 21348-21365. https://doi.org/10.3390/ijms151121348