Structure–Biological Function Relationship Extended to Mitotic Arrest-Deficient 2-Like Protein Mad2 Native and Mutants-New Opportunity for Genetic Disorder Control




Abstract
:
1. Introduction
2. Results and Discussion
2.1. Results
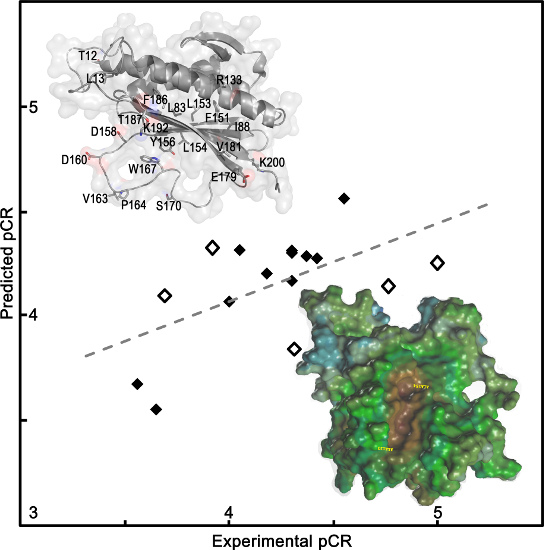
2.1.1. QSAR Models Predicted Mad2 Native and Mutants Binding to Cdc20

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| QSAR Model | q2 | r2 | RMSE | Cross-Validated RMSE | F test |
|---|---|---|---|---|---|
| QSAR model 1 | 0.53 | 0.82 | 0.15 | 0.27 | 13.22 |
| QSAR model 2 | 0.65 | 0.83 | 0.14 | 0.20 | 10.03 |
| QSAR model 3 | 0.60 | 0.90 | 0.10 | 0.25 | 10.23 |
| Mad2 Mutant | QSAR Model 1 | QSAR Model 2 | QSAR Model 3 | pCRexp | pK(dCdc20)exp | Kd (µM) |
|---|---|---|---|---|---|---|
| pK(dCdc20)pred | pK(dCdc20)pred | pCRpred | ||||
| R133A/L84A | 6.92 (−0.12) | 7.00 (−0.20) | 4.33 (−0.41) | 3.92 | 6.80 | 0.16 |
| R133A/I88A | 6.52 (0.18) | 6.58 (0.12) | 4.14 (0.62) | 4.76 | 6.70 | 0.20 |
| R133A/F151A | 6.52 (0.01) | 6.61 (−0.08) | 4.25 (0.75) | 5.00 | 6.53 | 0.29 |
| R133A/L154A | 5.93 (−0.02) | 5.97 (−0.06) | 4.29 (0.08) | 4.37 | 5.91 | 1.21 |
| R133A/D158A | 6.83 (0.05) | 6.90 (−0.02) | 4.17 (0.13) | 4.30 | 6.88 | 0.13 |
| R133A/V163A | 6.58 (0.05) | 6.63 (0.00) | 4.20 (−0.02) | 4.18 | 6.63 | 0.23 |
| R133A/S170A | 6.87 (0.22) | 6.95 (0.14) | 3.84 (0.47) | 4.31 | 7.09 | 0.081 |
| R133A/E179A | 6.86 (−0.07) | 6.88 (−0.09 ) | 4.32 (−0.02) | 4.30 | 6.79 | 0.16 |
| R133A/V181A | 6.19 (−0.19) | 6.27 (−0.27) | 4.27 (0.15) | 4.42 | 6.00 | 0.10 |
| R133A/K200A | 6.81 (−0.05) | 6.82 (−0.06) | 4.56 (−0.01) | 4.55 | 6.76 | 0.17 |
| R133A/L13A | 6.71 (0.21) | 6.79 (0.13) | Eq. not applied | NA | 6.92 | 0.12 |
| Native | 6.98 (0.02) | 7.05 (−0.05) | 5.76 | NA * | 7.00 | 0.1 |
| R133A | 7.66 (−0.81) | 7.76 (−0.91) | 4.30 (0.00) | 4.30 | 6.85 | 0.14 |
| R133A/L153A | 7.19 (−0.52) | 7.25 (−0.58) | Eq. not applied | NA | 6.67 | 0.21 |
| R133A/D160A | 7.23 (−0.65) | 7.32 (−0.74) | 4.07 (−0.07) | 4.00 | 6.58 | 0.26 |
| R133A/Y156A | 6.38 (−0.13) | 6.48 (−0.23) | Eq. not applied | NA | 6.25 | 0.56 |
| R133A/T12A | 5.94 (1.01) | 5.98 (0.97) | 3.55 (0.10) | 3.65 | 6.95 | 0.11 |
| L13A | 6.96 | 7.01 | Eq. not applied | NA * | ND | ND |
| R133A/P164A | 6.53 | 6.62 | 4.32 (−0.27) | 4.05 | ND | ND |
| R133A/T187A | 6.60 | 6.69 | 3.67 (−0.11) | 3.56 | ND | ND |
| R133A/K192A | 6.53 | 6.53 | 3.30 | ND | ND | ND |
| R133A/W167A | Eq. not applied | Eq. not applied | 4.09 (−0.40) | 3.96 | NBD | NBD |

| Mad2 Mutants | QSAR Model 1 | QSAR Model 2 | pK(dCdc20)exp |
|---|---|---|---|
| pK(dCdc20)pred | pK(dCdc20)pred | ||
| Templates | |||
| F186A | 5.77 | 5.96 | NBD |
| R133A/F186A | 5.34 | 5.44 | NBD |
| de novo Mad2 Mutants | |||
| F186M | 7.37 | 7.44 | ND * |
| F186S | 6.22 | 6.30 | ND * |
| F186T | 6.67 | 6.71 | ND * |
| F186W | 7.40 | 7.46 | ND * |
| F186N | 6.40 | 6.51 | ND * |
| R133A/F186M | 7.11 | 7.24 | ND * |
| R133A/F186S | 5.92 | 6.01 | ND * |
| R133A/F186T | 6.30 | 6.40 | ND * |
| R133A/F186N | 5.64 | 5.73 | ND * |
2.1.2. QSAR Model Predicted Mad2 Native and Mutants Function Expressed as O-Mad2–C-Mad2 Interconversion Rate
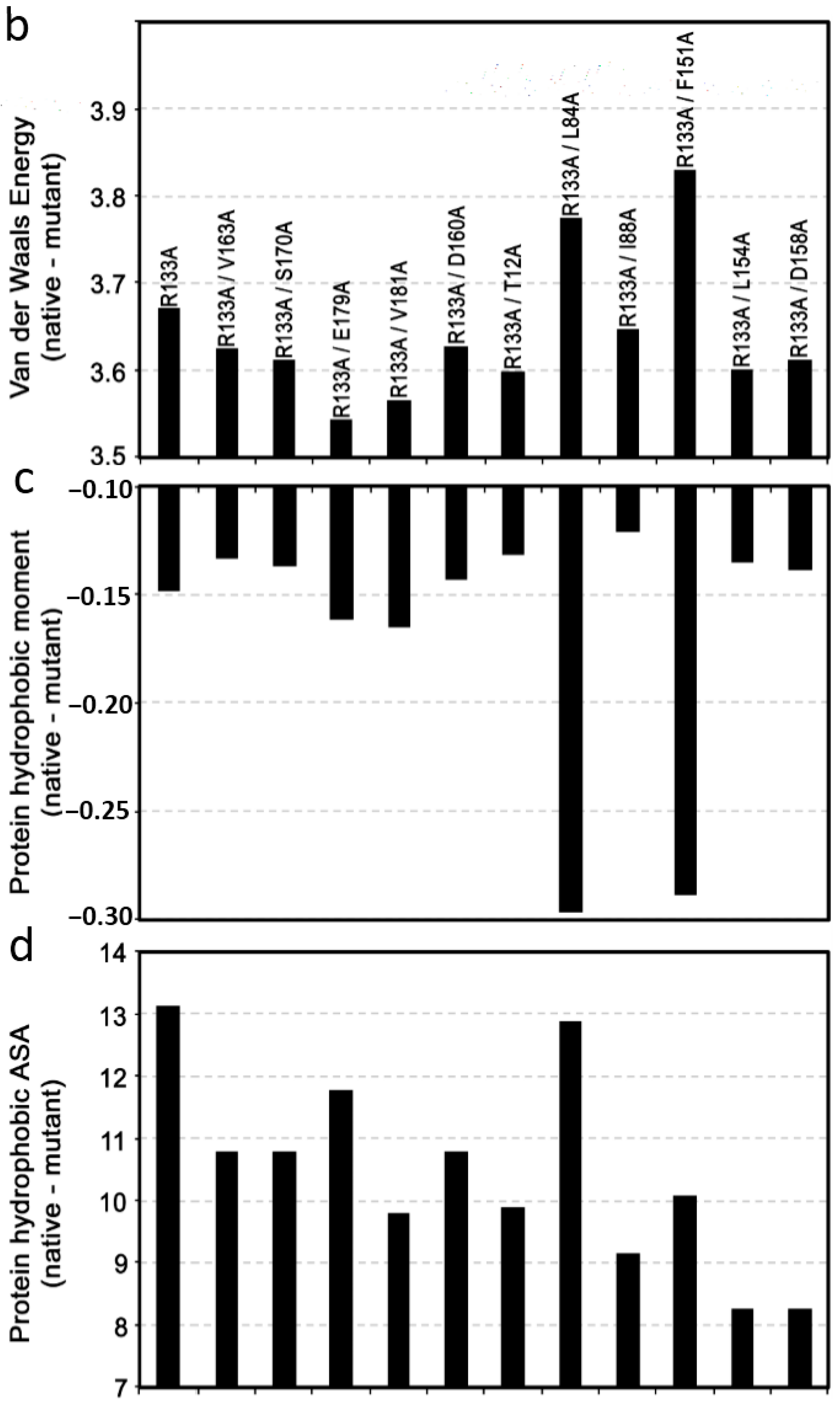
2.1.3. Structure–Function Relationship Model of Mad2 Native and Mutants at C-Terminal Domain Residues


2.2. Discussion
2.2.1. Power of QSAR Model to Predict of Mad2 Native and Its Mutants Binding against Cdc20

2.2.2. Power of the QSAR Model to Predict Mad2 Native and Its Mutants in O-Mad2–C-Mad2 Interconversion Rate
2.2.3. Power of QSAR Model to Predict Mad2 de Novo Mutants Binding to Cdc20
2.2.4. SAR Analysis of Mad2 Native and Mutants at C-Terminal Domain Residues
3. Experimental Section
3.1. Dataset for Analysis
3.2. Rational Design of de Novo Mad2 F186 and Mad2 R133/F186 Mutants with Possible Non-CIN Functions
3.3. Modeling of Native and Mutant Mad2 Proteins and Their Minimum Energy Calculation Strategy
3.4. QSAR Methodology
3.4.1. Descriptors Calculations
3.4.2. Chemometric Analyses
3.4.3. Training and Testing Sets
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yu, H. Structural activation of Mad2 in the mitotic spindle checkpoint: The two-state Mad2 model versus the Mad2 template model. J. Cell Biol. 2006, 173, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Musacchio, A. Spindle assembly checkpoint: The third decade. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3595–3604. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Kim, C.; Mao, Y. New insights into the mechanism for chromosome alignment in metaphase. Int. Rev. Cell. Mol. Biol. 2013, 303, 237–262. [Google Scholar] [PubMed]
- Kim, S.; Sun, H.; Tomchick, D.R.; Yu, H.; Luo, X. Structure of human Mad1 C-terminal domain reveals its involvement in kinetochore targeting. Proc. Natl. Acad. Sci. USA 2012, 109, 6549–6554. [Google Scholar] [CrossRef] [PubMed]
- Ross, K.E.; Arighi, C.N.; Ren, J.; Natale, D.A.; Huang, H.; Wu, C.H. Use of the protein ontology for multi-faceted analysis of biological processes: A case study of the spindle checkpoint. Front. Genet. 2013, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Kim, S.; Yu, H. Tracking spindle checkpoint signals from kinetochores to APC/C. Trends Biochem. Sci. 2013, 38, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Li, B.; Warrington, R.; Tomchick, D.R.; Yu, H.; Luo, X. Structural analysis of human Cdc20 supports multisite degron recognition by APC/C. Proc. Natl. Acad. Sci. USA 2012, 109, 18419–18424. [Google Scholar] [CrossRef] [PubMed]
- Funabiki, H.; Wynne, D.J. Making an effective switch at the kinetochore by phosphorylation and dephosphorylation. Chromosoma 2013, 122, 135–158. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Yu, H. Protein metamorphosis: The two-state behavior of Mad2. Structure 2008, 16, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, B.; Liu, C.J.; Tomchick, D.R.; Machius, M.; Rizo, J.; Yu, H.; Luo, X. Insights into mad2 regulation in the spindle checkpoint revealed by the crystal structure of the symmetric mad2 dimer. PLoS Biol. 2008, 6, e50. [Google Scholar] [CrossRef] [PubMed]
- Han, J.S.; Holland, A.J.; Fachinetti, D.; Kulukian, A.; Cetin, B.; Cleveland, D.W. Catalytic assembly of the mitotic checkpoint inhibitor BubR1-Cdc20 by a Mad2-induced functional switch in Cdc20. Mol. Cell 2013, 51, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Yu, H. Mutual regulation between the spindle checkpoint and APC/C. Semin. Cell Dev. Biol. 2011, 22, 551–558. [Google Scholar]
- Magrane, M.; Consortium, U. UniProt Knowledgebase: A hub of integrated protein data. Database (Oxford) 2011, 2011, bar009. [Google Scholar] [CrossRef]
- Kim, S.; Sun, H.; Ball, H.L.; Wassmann, K.; Luo, X.; Yu, H. Phosphorylation of the spindle checkpoint protein Mad2 regulates its conformational transition. Proc. Natl. Acad. Sci. USA 2010, 107, 19772–19777. [Google Scholar] [CrossRef] [PubMed]
- Sironi, L.; Mapelli, M.; Knapp, S.; de Antoni, A.; Jeang, K.T.; Musacchio, A. Crystal structure of the tetrameric Mad1–Mad2 core complex: implications of a “safety belt” binding mechanism for the spindle checkpoint. EMBO J. 2002, 21, 2496–2506. [Google Scholar] [CrossRef] [PubMed]
- Mapelli, M.; Massimiliano, L.; Santaguida, S.; Musacchio, A. The Mad2 conformational dimer: Structure and implications for the spindle assembly checkpoint. Cell 2007, 131, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Tang, Z.; Xia, G.; Wassmann, K.; Matsumoto, T.; Rizo, J.; Yu, H. The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat. Struct. Mol. Biol. 2004, 11, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Kulukian, A.; Han, J.S.; Cleveland, D.W. Unattached kinetochores catalyze production of an anaphase inhibitor that requires a Mad2 template to prime Cdc20 for BubR1 binding. Dev. Cell 2009, 16, 105–117. [Google Scholar] [CrossRef]
- Yu, L.; Liu, S.; Guo, W.; Zhang, B.; Liang, Y.; Feng, Q. Upregulation of Mad2 facilitates in vivo and in vitro osteosarcoma progression. Oncol. Rep. 2012, 28, 2170–2176. [Google Scholar] [PubMed]
- Schvartzman, J.M.; Duijf, P.H.; Sotillo, R.; Coker, C.; Benezra, R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell 2011, 19, 701–714. [Google Scholar] [CrossRef]
- Yu, L.; Guo, W.; Zhao, S.; Tang, J.; Liu, J. Knockdown of Mad2 induces osteosarcoma cell apoptosis-involved Rad21 cleavage. J. Orthop. Sci. 2011, 16, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Schuyler, S.C.; Wu, Y.F.; Kuan, V.J. The Mad1–Mad2 balancing act—a damaged spindle checkpoint in chromosome instability and cancer. J. Cell Sci. 2012, 125, 4197–4206. [Google Scholar] [CrossRef]
- Morishita, M.; Sumi, T.; Nakano, Y.; Teramae, M.; Fukuda, T.; Nobeyama, H.; Yoshida, H.; Matsumoto, Y.; Yasui, T.; Ishiko, O. Expression of mitotic-arrest deficiency 2 predicts the efficacy of neoadjuvant chemotherapy for locally advanced uterine cervical cancer. Exp. Ther. Med. 2012, 3, 341–346. [Google Scholar] [PubMed]
- Calborean, O.; Mernea, M.; Avram, S.; Mihailescu, D.F. Pharmacological descriptors related to the binding of Gp120 to CD4 corresponding to 60 representative HIV-1 strains. J. Enzyme Inhib. Med. Chem. 2013, 28, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Avram, S.; Buiu, C.; Borcan, F.; Milac, A.L. More effective antimicrobial mastoparan derivatives, generated by 3D-QSAR-Almond and computational mutagenesis. Mol. Biosyst. 2012, 8, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Avram, S.; Duda-Seiman, D.; Borcan, F.; Radu, B.; Duda-Seiman, C.; Mihailescu, D. Evaluation of antimicrobial activity of new mastoparan derivatives using QSAR and computational mutagenesis. Int. J. Pept. Res. Ther. 2011, 17, 7–17. [Google Scholar] [CrossRef]
- Avram, S.; Mihailescu, D.; Borcan, F.; Milac, A.-L. Prediction of improved antimicrobial mastoparan derivatives by 3D-QSAR-CoMSIA/CoMFA and computational mutagenesis. Monatsh. Chem. 2012, 143, 535–543. [Google Scholar] [CrossRef]
- SYBYL Molecular Modeling Suite, version 7.0; Tripos Inc.: St. Louis, MO, USA, 2004.
- Gellert, A.; Salanki, K.; Naray-Szabo, G.; Balazs, E. Homology modelling and protein structure based functional analysis of five cucumovirus coat proteins. J. Mol. Graph. Model. 2006, 24, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Henriques, J.; Costa, P.J.; Calhorda, M.J.; Machuqueiro, M. Charge parametrization of the DvH-c3 heme group: Validation using constant-(pH, E) molecular dynamics simulations. J. Phys. Chem. B 2013, 117, 70–82. [Google Scholar] [CrossRef] [PubMed]
- The Molecular Operating Environment (MOE), version 2012.10; Chemical Computing Group Inc.: Montreal, QC, Canada, 2012.
- Oprea, T.I.; Waller, C.L. Theoretical and practical aspects of three-dimensional quantitative structure-activity relationships. In Review in Computational. Chemistry; Lipkowitz, K.B., Boyd, D.B., Eds.; Wiley-VCH: New York, NY, USA, 2007; Volume 11, pp. 127–182. [Google Scholar]
- Ghafourian, T.; Amin, Z. QSAR models for the prediction of plasma protein binding. Bioimpacts 2013, 3, 21–27. [Google Scholar] [PubMed]
- Tsai, C.F.; Lee, K.J. A comparative study of the second-order hydrophobic moments for globular proteins: The consensus scale of hydrophobicity and the CHARMM partial atomic charges. Int. J. Mol. Sci. 2011, 12, 8449–8465. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Sumi, T.; Morishita, M.; Fukuda, T.; Nobeyama, H.; Yoshida, H.; Matsumoto, Y.; Yasui, T.; Ishiko, O. Mitotic arrest deficiency 2 induces carcinogenesis in mucinous ovarian tumors. Oncol. Lett. 2012, 3, 281–286. [Google Scholar] [PubMed]
- Lee, Y.; Jana, S.; Acharya, G.; Lee, C.H. Computational analysis and predictive modeling of polymorph descriptors. Chem. Cent. J. 2013, 7, 23. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avram, S.; Milac, A.; Mernea, M.; Mihailescu, D.; Putz, M.V.; Buiu, C. Structure–Biological Function Relationship Extended to Mitotic Arrest-Deficient 2-Like Protein Mad2 Native and Mutants-New Opportunity for Genetic Disorder Control. Int. J. Mol. Sci. 2014, 15, 21381-21400. https://doi.org/10.3390/ijms151121381
Avram S, Milac A, Mernea M, Mihailescu D, Putz MV, Buiu C. Structure–Biological Function Relationship Extended to Mitotic Arrest-Deficient 2-Like Protein Mad2 Native and Mutants-New Opportunity for Genetic Disorder Control. International Journal of Molecular Sciences. 2014; 15(11):21381-21400. https://doi.org/10.3390/ijms151121381
Chicago/Turabian StyleAvram, Speranta, Adina Milac, Maria Mernea, Dan Mihailescu, Mihai V. Putz, and Catalin Buiu. 2014. "Structure–Biological Function Relationship Extended to Mitotic Arrest-Deficient 2-Like Protein Mad2 Native and Mutants-New Opportunity for Genetic Disorder Control" International Journal of Molecular Sciences 15, no. 11: 21381-21400. https://doi.org/10.3390/ijms151121381
APA StyleAvram, S., Milac, A., Mernea, M., Mihailescu, D., Putz, M. V., & Buiu, C. (2014). Structure–Biological Function Relationship Extended to Mitotic Arrest-Deficient 2-Like Protein Mad2 Native and Mutants-New Opportunity for Genetic Disorder Control. International Journal of Molecular Sciences, 15(11), 21381-21400. https://doi.org/10.3390/ijms151121381