Epidermal Growth Factor Receptor Transactivation Is Required for Mitogen-Activated Protein Kinase Activation by Muscarinic Acetylcholine Receptors in HaCaT Keratinocytes



Abstract
:1. Introduction
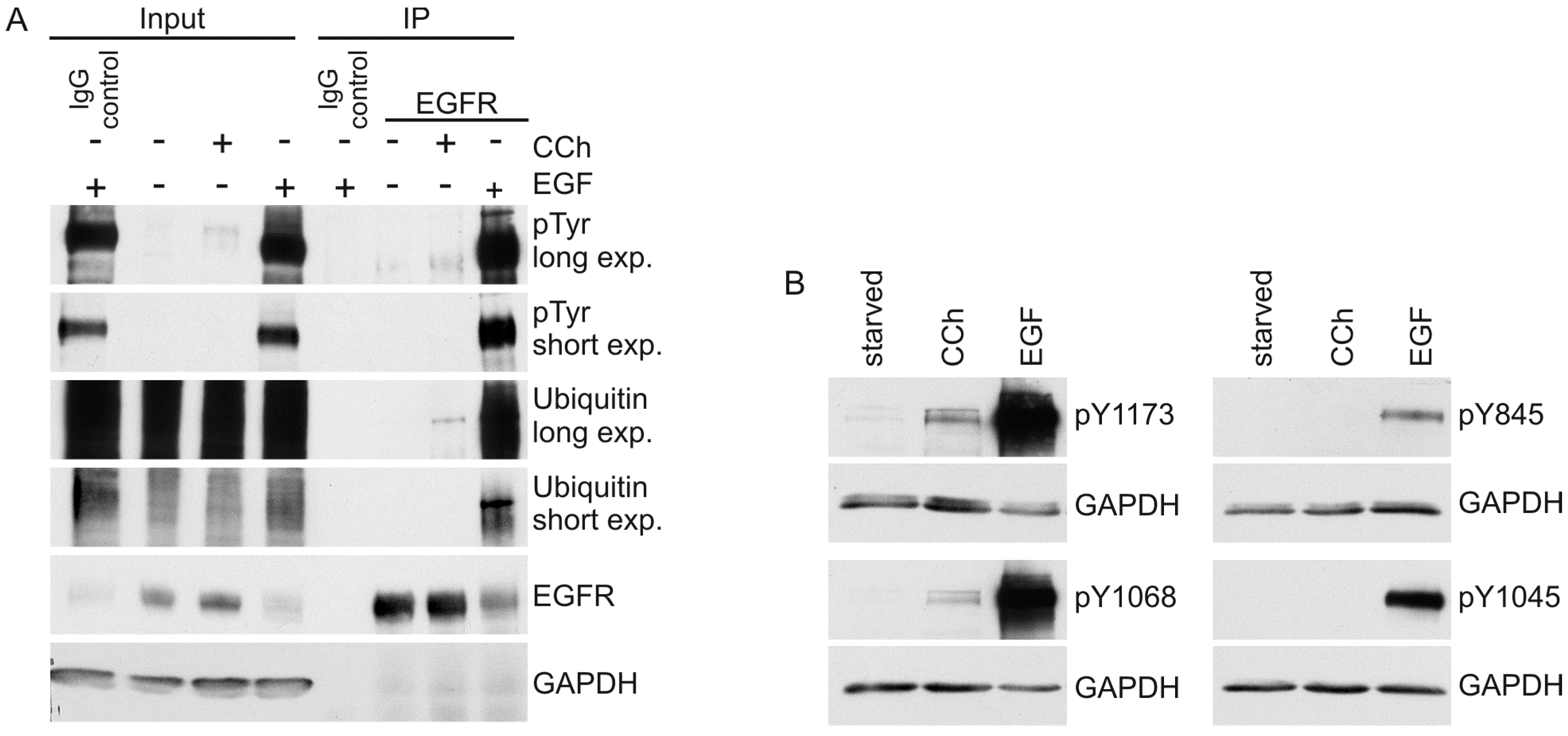
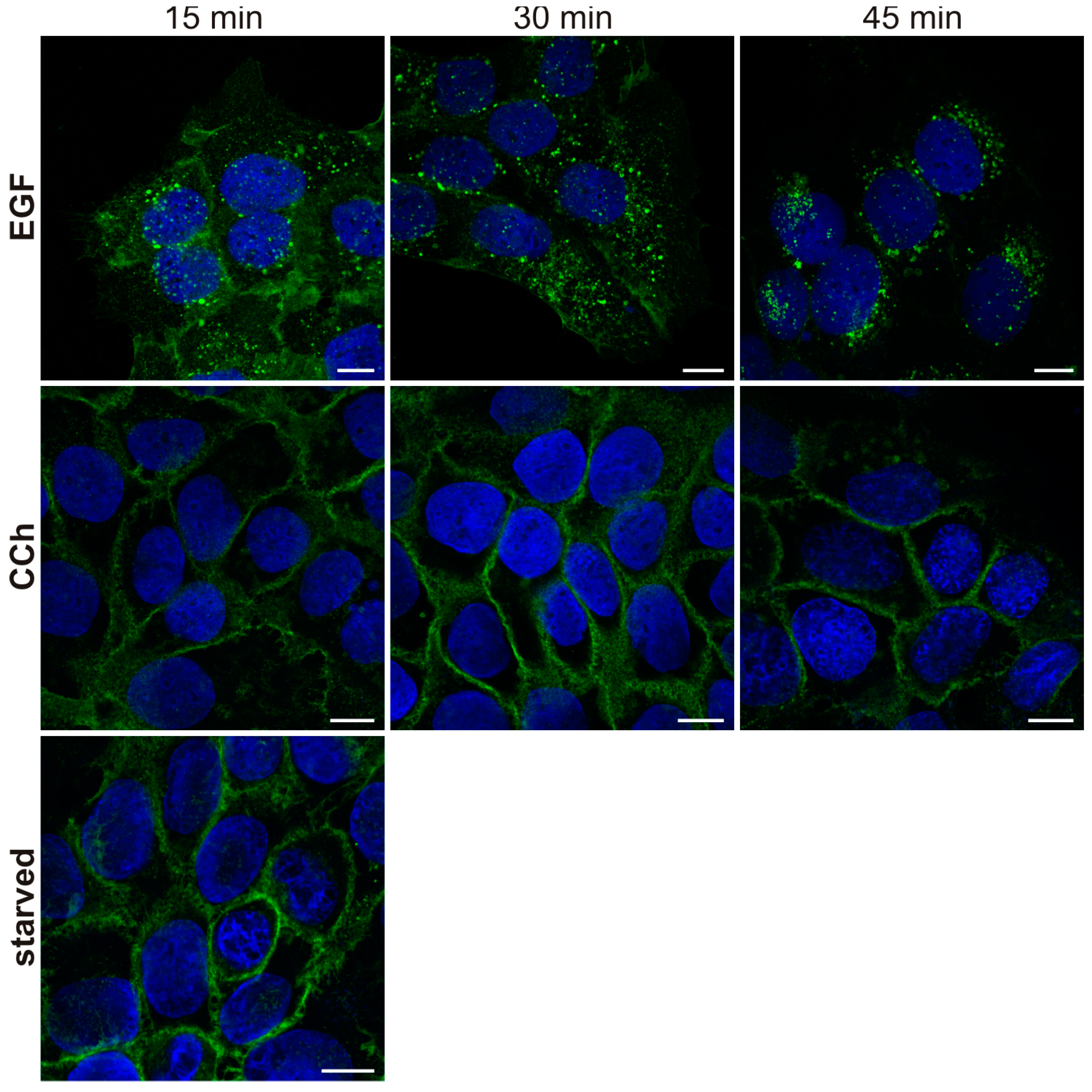
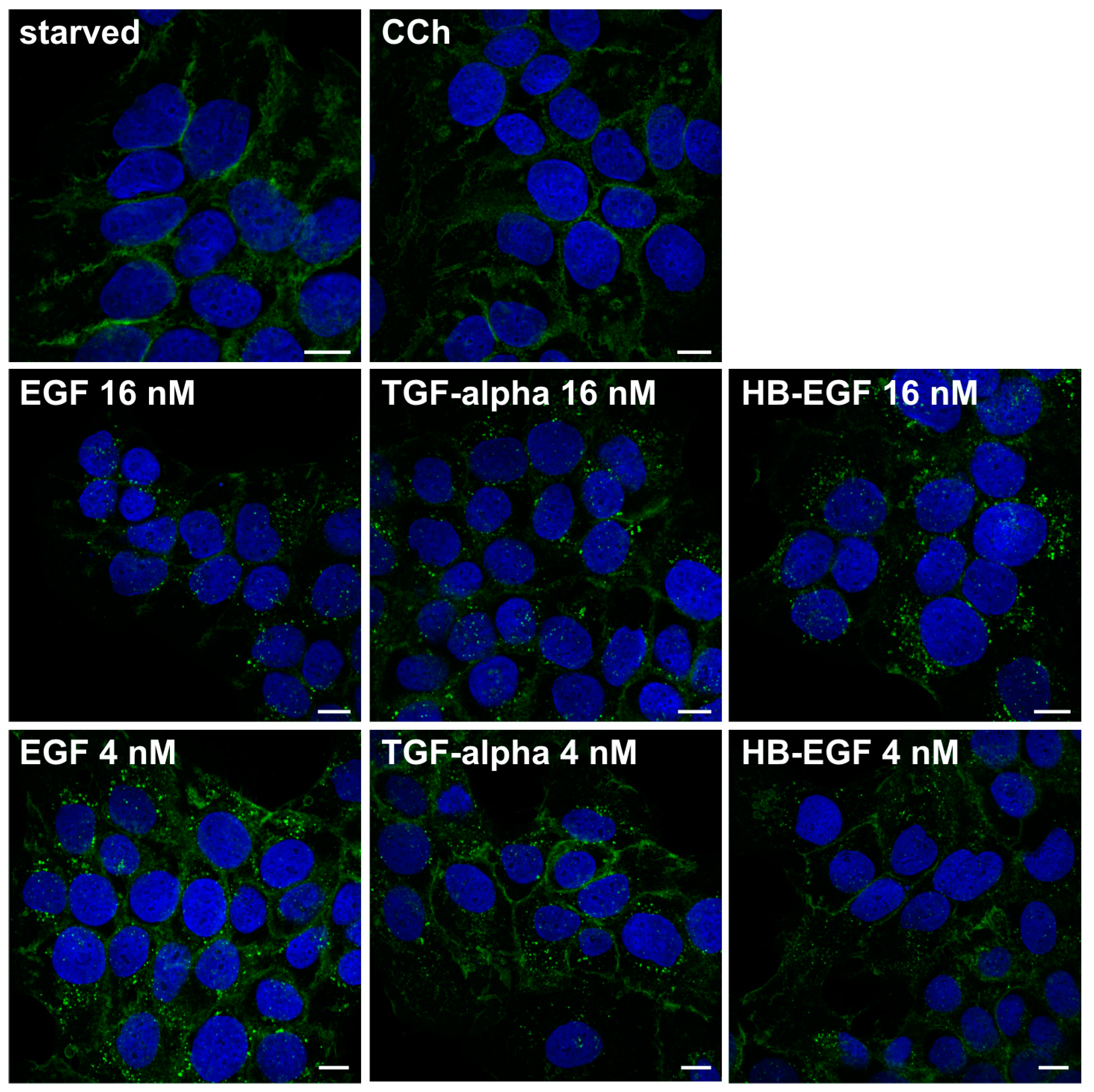
2. Results and Discussion
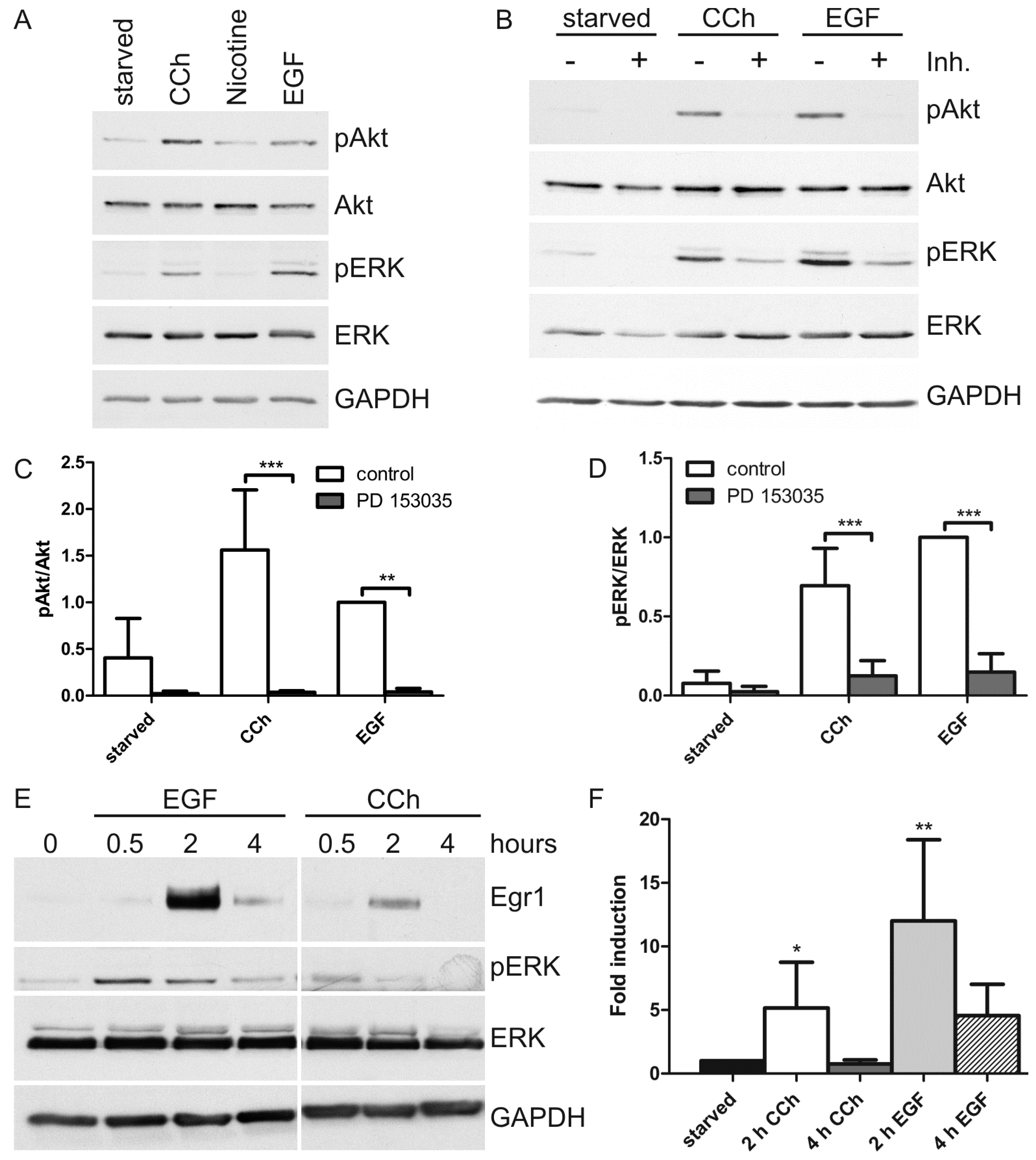
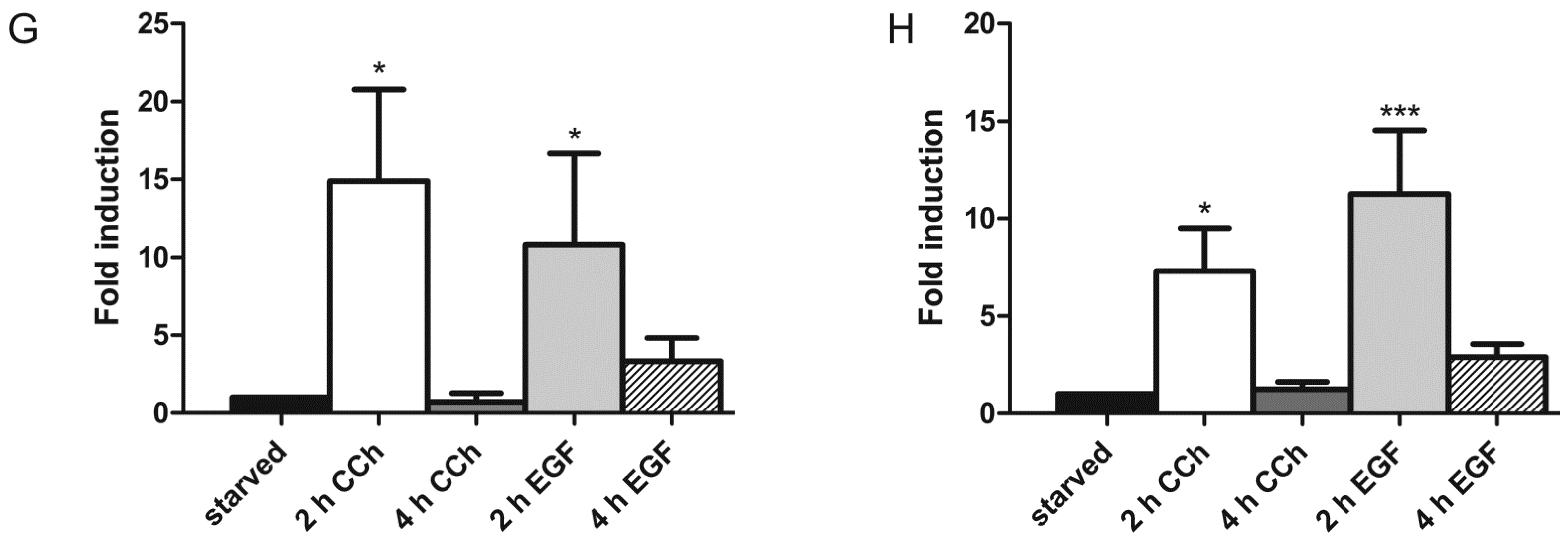
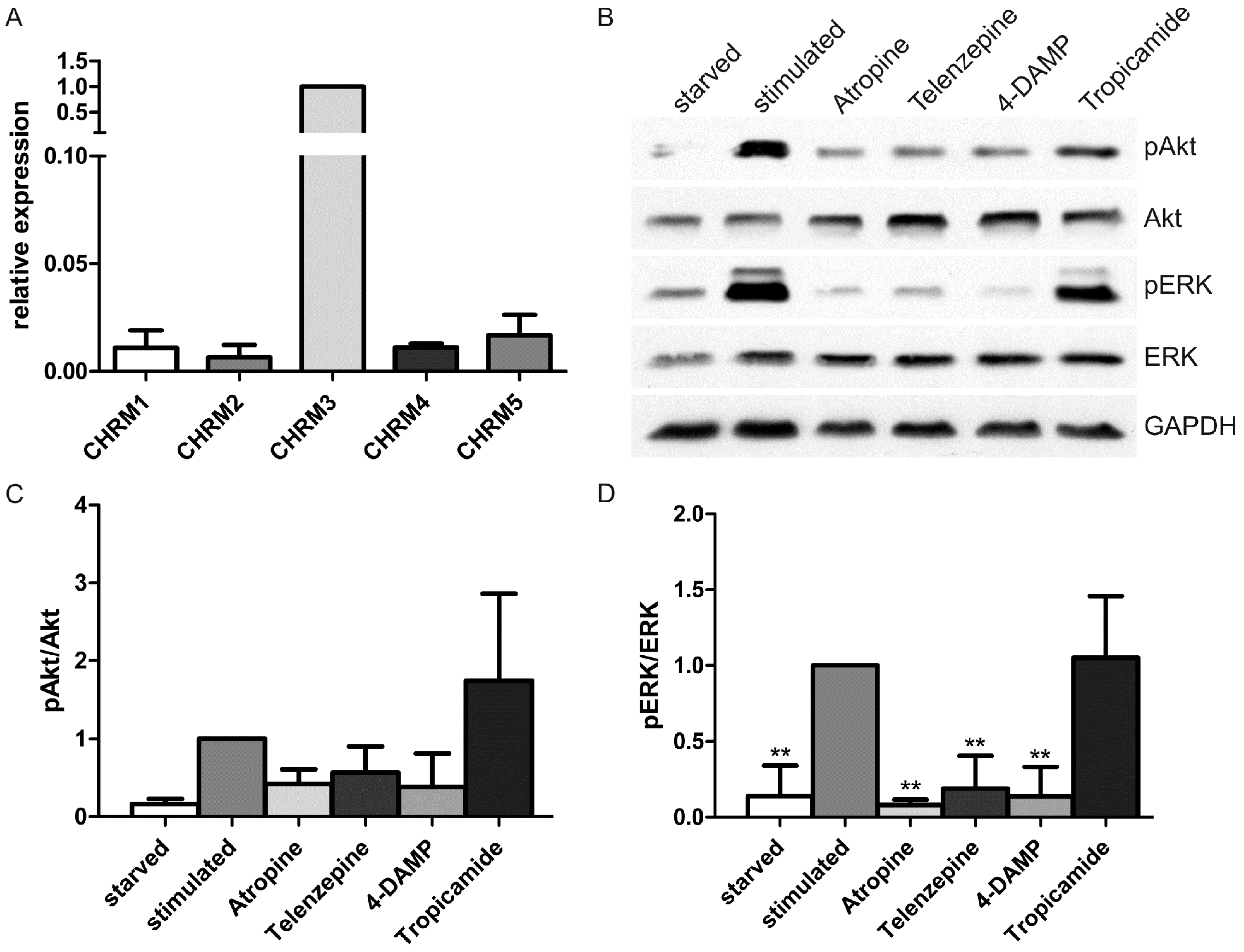
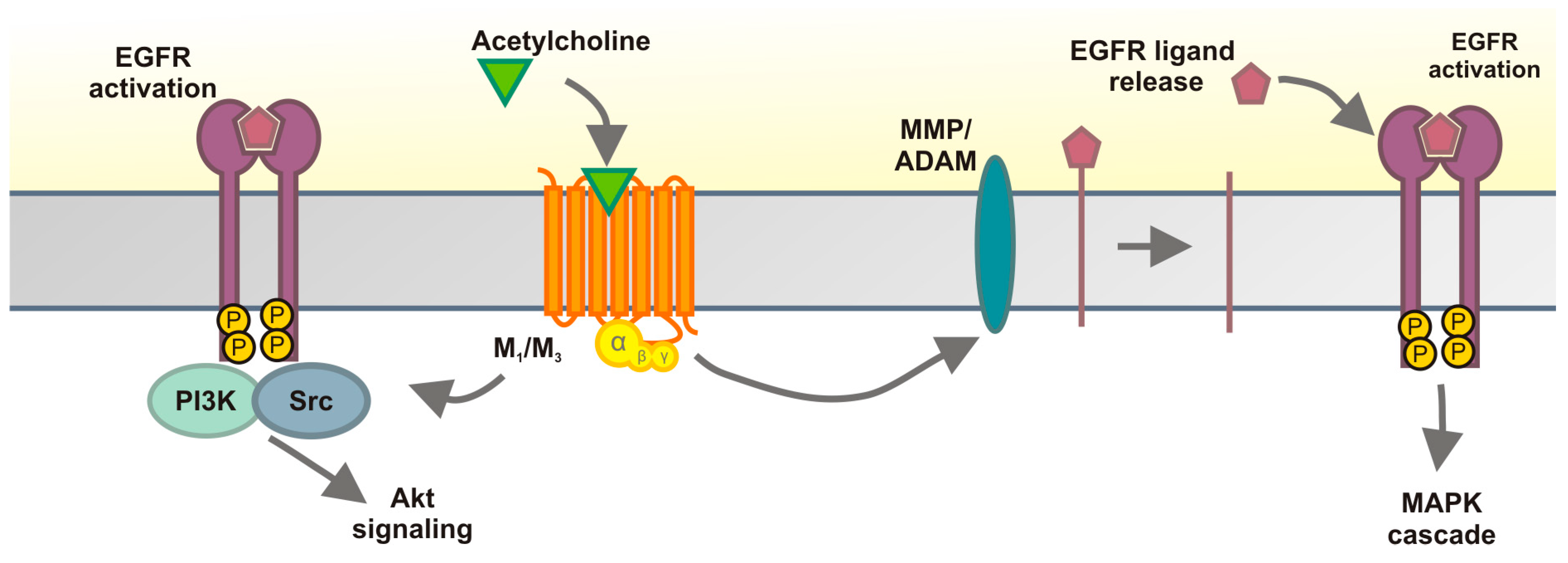
2.1. Results




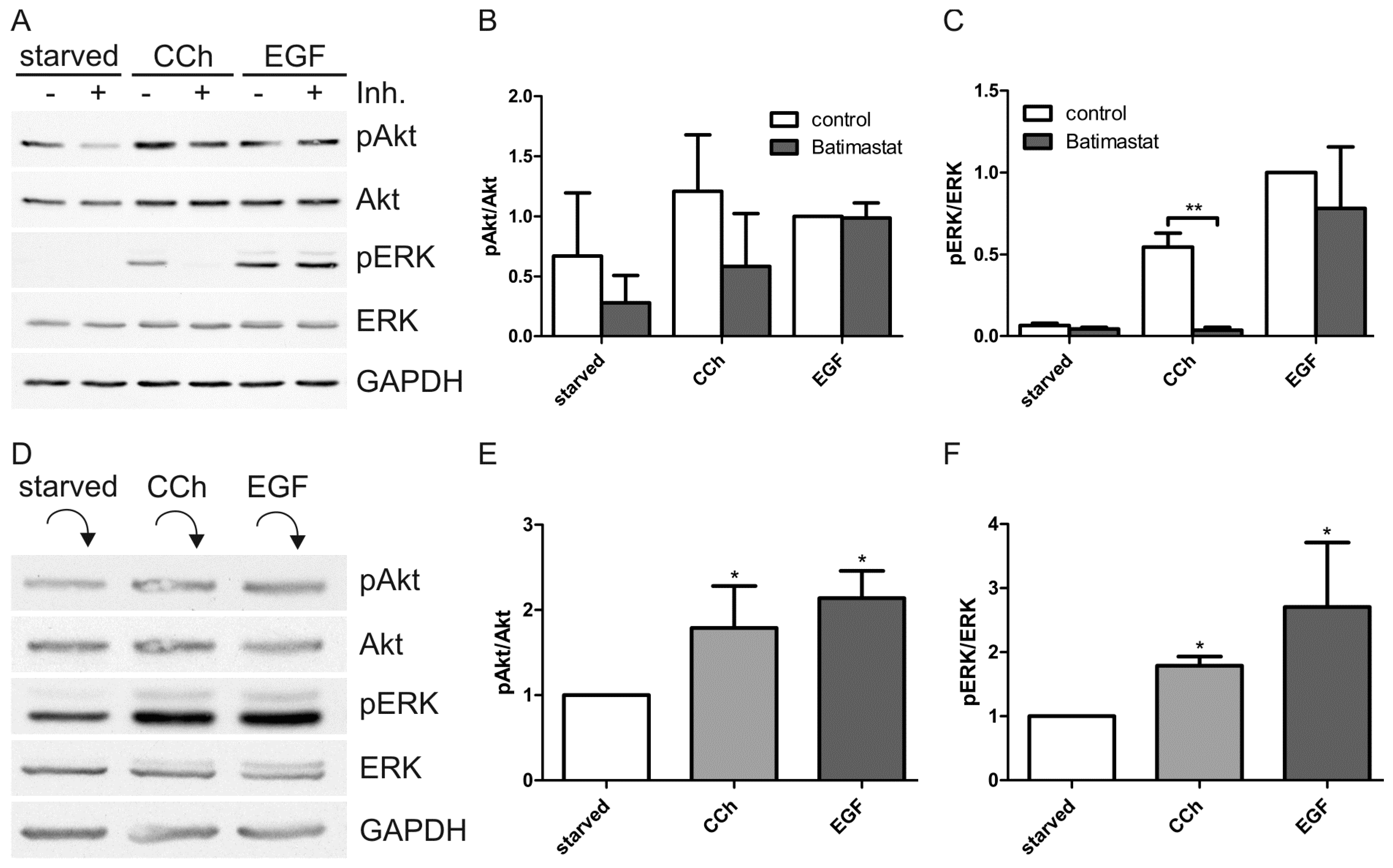



2.2. Discussion

3. Experimental Section
3.1. Reagents
3.2. Cell Culture
3.3. Cell Stimulation and Inhibitor Treatment
3.4. Antibodies
3.5. Cell Lysis, Gel Electrophoresis and Western Blot
3.6. Ligand Release Assay
3.7. Immunoprecipitation
3.8. RNA Isolation and RT-qPCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Forward | Primer Reverse |
|---|---|---|
| CHRM1 | CTGGTCAAGGAGAAGAAGGCGG | ACAGGGTCTCGGGAACACAGTC |
| CHRM2 | CAAGGGAGAAAGAGAACCGGCA | ACCTGTCGCTGGTTTCGCTC |
| CHRM3 | AGAAGAAAGCGGCCCAGACC | CACAGCCAGTAGCCCAGATTCC |
| CHRM4 | TCACCAAGCCTCTCACCTACCC | TCCGCTTACCCACCACAAACTG |
| CHRM5 | GAGAGGAAAGCAGCCCAGACAC | ACAACCAATAGCCCAAGTGCCA |
| cFos | TGGTGAAGACCGTGTCAGGAG | TGATCTGTCTCCGCTTGGAGTG |
| Dusp1 | GGAGGACAACCAGGCAGAC | AGGTAAGCAAGGCAGATGGTGG |
| Egr1 | TTCAACCCTCAGGCGGACAC | GTCTCCACCAGCACCTTCTCGT |
| GAPDH | CATCTTCCAGGAGCGAGATCCC | CCAGCCTTCTCCATGGTGGT |
| HPRT | GCAGTCCCAGGGTGCGTG | GGCCTCCCATCTCCTTCAT |
| Ywhaz | AGGTTGCCGCTGGTGATGAC | GGCCAGACCCAGTCTGATAGGA |
| Rpl13a | CCTGGAGGAGAAGAGGAAAGAGA | TTGAGGACCTCTGTGTATTTGTCAA |
3.9. Immunofluorescence
3.10. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kawashima, K.; Fujii, T. Basic and clinical aspects of non-neuronal acetylcholine: Overview of non-neuronal cholinergic systems and their biological significance. J. Pharmacol. Sci. 2008, 106, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Wessler, I.; Kirkpatrick, C.J. Acetylcholine beyond neurons: The non-neuronal cholinergic system in humans. Br. J. Pharmacol. 2008, 154, 1558–1571. [Google Scholar] [CrossRef] [PubMed]
- Curtis, B.J.; Radek, K.A. Cholinergic regulation of keratinocyte innate immunity and permeability barrier integrity: New perspectives in epidermal immunity and disease. J. Investig. Dermatol. 2012, 132, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Kurzen, H.; Wessler, I.; Kirkpatrick, C.J.; Kawashima, K.; Grando, S.A. The non-neuronal cholinergic system of human skin. Horm. Metab. Res. 2007, 39, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Chernyavsky, A.I.; Arredondo, J.; Karlsson, E.; Wessler, I.; Grando, S.A. The Ras/Raf-1/MEK1/ERK signaling pathway coupled to integrin expression mediates cholinergic regulation of keratinocyte directional migration. J. Biol. Chem. 2005, 280, 39220–39228. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, S.; Maruo, M.; Yasuno, H.; Kimura, H.; Maeda, T. The regeneration of cholinesterase-positive structures in the process of burn wound healing in the skin of the guinea-pig. Burns Incl. Therm. Inj. 1982, 9, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, F.; Kurt, E.; Inan, U.U.; Emiroglu, L.; Ilker, S.S. The effects of acetylcholine and propolis extract on corneal epithelial wound healing in rats. Cornea 1999, 18, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Arredondo, J.; Nguyen, V.T.; Chernyavsky, A.I.; Bercovich, D.; Orr-Urtreger, A.; Kummer, W.; Lips, K.; Vetter, D.E.; Grando, S.A. Central role of α7 nicotinic receptor in differentiation of the stratified squamous epithelium. J. Cell. Biol. 2002, 159, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Kurzen, H.; Berger, H.; Jager, C.; Hartschuh, W.; Naher, H.; Gratchev, A.; Goerdt, S.; Deichmann, M. Phenotypical and molecular profiling of the extraneuronal cholinergic system of the skin. J. Investig. Dermatol. 2004, 123, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Ndoye, A.; Buchli, R.; Greenberg, B. Identification and mapping of keratinocyte muscarinic acetylcholine receptor subtypes in human epidermis. J. Investig. Dermatol. 1998, 111, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Klapproth, H.; Reinheimer, T.; Metzen, J.; Munch, M.; Bittinger, F.; Kirkpatrick, C.J.; Hohle, K.D.; Schemann, M.; Racke, K.; Wessler, I. Non-neuronal acetylcholine, a signalling molecule synthezised by surface cells of rat and man. Naunyn. Schmiedebergs Arch. Pharmacol. 1997, 355, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Ndoye, A.; Hall, L.L.; Zia, S.; Arredondo, J.; Chernyavsky, A.I.; Kist, D.A.; Zelickson, B.D.; Lawry, M.A.; Grando, S.A. Programmed cell death of keratinocytes culminates in apoptotic secretion of a humectant upon secretagogue action of acetylcholine. J. Cell. Sci. 2001, 114, 1189–1204. [Google Scholar] [PubMed]
- Arredondo, J.; Hall, L.L.; Ndoye, A.; Chernyavsky, A.I.; Jolkovsky, D.L.; Grando, S.A. Muscarinic acetylcholine receptors regulating cell cycle progression are expressed in human gingival keratinocytes. J. Periodontal Res. 2003, 38, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Grando, S.A.; Crosby, A.M.; Zelickson, B.D.; Dahl, M.V. Agarose gel keratinocyte outgrowth system as a model of skin re-epithelization: Requirement of endogenous acetylcholine for outgrowth initiation. J. Investig. Dermatol. 1993, 101, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Grando, S.A.; Horton, R.M.; Mauro, T.M.; Kist, D.A.; Lee, T.X.; Dahl, M.V. Activation of keratinocyte nicotinic cholinergic receptors stimulates calcium influx and enhances cell differentiation. J. Investig. Dermatol. 1996, 107, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Grando, S.A.; Zelickson, B.D.; Kist, D.A.; Weinshenker, D.; Bigliardi, P.L.; Wendelschafer-Crabb, G.; Kennedy, W.R.; Dahl, M.V. Keratinocyte muscarinic acetylcholine receptors: Immunolocalization and partial characterization. J. Investig. Dermatol. 1995, 104, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Sgard, F.; Charpantier, E.; Bertrand, S.; Walker, N.; Caput, D.; Graham, D.; Bertrand, D.; Besnard, F. A novel human nicotinic receptor subunit, α10, that confers functionality to the α9-subunit. Mol. Pharmacol. 2002, 61, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Ramachandran, J.; Capon, D.J. Acetylcholine analogue stimulates DNA synthesis in brain-derived cells via specific muscarinic receptor subtypes. Nature 1989, 340, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Belcheva, M.M.; Coscia, C.J. Diversity of g protein-coupled receptor signaling pathways to ERK/MAP kinase. Neurosignals 2002, 11, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Gutkind, J.S.; Novotny, E.A.; Brann, M.R.; Robbins, K.C. Muscarinic acetylcholine receptor subtypes as agonist-dependent oncogenes. Proc. Natl. Acad. Sci. USA 1991, 88, 4703–4707. [Google Scholar] [CrossRef] [PubMed]
- Qian, N.X.; Russell, M.; Johnson, G.L. Acetylcholine muscarinic receptor regulation of the Ras/Raf/MAP kinase pathway. Life Sci. 1995, 56, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Daub, H.; Weiss, F.U.; Wallasch, C.; Ullrich, A. Role of transactivation of the egf receptor in signalling by G-protein-coupled receptors. Nature 1996, 379, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Daub, H.; Wallasch, C.; Lankenau, A.; Herrlich, A.; Ullrich, A. Signal characteristics of G protein-transactivated EGF receptor. EMBO J. 1997, 16, 7032–7044. [Google Scholar] [CrossRef] [PubMed]
- Prenzel, N.; Zwick, E.; Daub, H.; Leserer, M.; Abraham, R.; Wallasch, C.; Ullrich, A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 1999, 402, 884–888. [Google Scholar] [PubMed]
- Wallasch, C.; Crabtree, J.E.; Bevec, D.; Robinson, P.A.; Wagner, H.; Ullrich, A. Helicobacter pylori-stimulated EGF receptor transactivation requires metalloprotease cleavage of HB-EGF. Biochem. Biophys. Res. Commun. 2002, 295, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Amos, S.; Martin, P.M.; Polar, G.A.; Parsons, S.J.; Hussaini, I.M. Phorbol 12-myristate 13-acetate induces epidermal growth factor receptor transactivation via protein kinase Cδ/c-Src pathways in glioblastoma cells. J. Biol. Chem. 2005, 280, 7729–7738. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Xie, G.; Raufman, J.P. Matrix metalloproteinase-7-catalyzed release of HB-EGF mediates deoxycholyltaurine-induced proliferation of a human colon cancer cell line. Biochem. Pharmacol. 2007, 73, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Zimniak, P.; Raufman, J.P. Transactivation of the epidermal growth factor receptor mediates cholinergic agonist-induced proliferation of h508 human colon cancer cells. Cancer Res. 2003, 63, 6744–6750. [Google Scholar] [PubMed]
- Iwasaki, H.; Eguchi, S.; Marumo, F.; Hirata, Y. Endothelin-1 stimulates DNA synthesis of vascular smooth-muscle cells through transactivation of epidermal growth factor receptor. J. Cardiovasc. Pharmacol. 1998, 31 (Suppl. 1), S182–S184. [Google Scholar] [CrossRef]
- Kajiya, M.; Ichimonji, I.; Min, C.; Zhu, T.; Jin, J.O.; Yu, Q.; Almazrooa, S.A.; Cha, S.; Kawai, T. Muscarinic type 3 receptor induces cytoprotective signaling in salivary gland cells through epidermal growth factor receptor transactivation. Mol. Pharmacol. 2012, 82, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Keely, S.J.; Uribe, J.M.; Barrett, K.E. Carbachol stimulates transactivation of epidermal growth factor receptor and mitogen-activated protein kinase in T 84 cells. J. Biol. Chem. 1998, 273, 27111–27117. [Google Scholar] [CrossRef] [PubMed]
- Maretzky, T.; Evers, A.; Zhou, W.; Swendeman, S.L.; Rafii, S.; Reiss, K.; Blobel, C.P. Migration of FGF7-stimulated epithelial cells and VEGF-A-stimulated HUVECs depends on EGFR transactivation by ADAM17. Nat. Commun. 2011, 2, 1–25. [Google Scholar] [CrossRef]
- McCole, D.F.; Keely, S.J.; Coffey, R.J.; Barrett, K.E. Transactivation of the epidermal growth factor receptor in colonic epithelial cells by carbachol requires extracellular release of transforming growth factor-α. J. Biol. Chem. 2002, 277, 42603–42612. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.; Morielli, A.D.; Peralta, E.G. The M1 muscarinic acetylcholine receptor transactivates the EGF receptor to modulate ion channel activity. EMBO J. 1997, 16, 4597–4605. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell. Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Maas-Szabowski, N.; Starker, A.; Fusenig, N.E. Epidermal tissue regeneration and stromal interaction in hacat cells is initiated by TGF-α. J. Cell. Sci. 2003, 116, 2937–2948. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.; Just, L.; Boss, A.; Drews, U. Identification and functional characterization of the muscarinic receptor M3 in the human keratinocyte cell line hacat. Cells Tissues Organs 2005, 180, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Goebeler, M.; Posern, G.; Feller, S.M.; Seitz, C.S.; Brocker, E.B.; Rapp, U.R.; Ludwig, S. Ras-independent activation of the Raf/MEK/ERK pathway upon calcium-induced differentiation of keratinocytes. J. Biol. Chem. 2000, 275, 41011–41017. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.M.; Martinez, N.; Rodriguez-Garcia, I.; Alonso, A. EGFR family-mediated signal transduction in the human keratinocyte cell line hacat. Exp. Cell. Res. 1999, 252, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Ilasaca, M.; Crespo, P.; Pellici, P.G.; Gutkind, J.S.; Wetzker, R. Linkage of G protein-coupled receptors to the MAPK signaling pathway through PI 3-kinase γ. Science 1997, 275, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Slack, B.E. The M3 muscarinic acetylcholine receptor is coupled to mitogen-activated protein kinase via protein kinase C and epidermal growth factor receptor kinase. Biochem. J. 2000, 348, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Stirnweiss, J.; Valkova, C.; Ziesche, E.; Drube, S.; Liebmann, C. Muscarinic M2 receptors mediate transactivation of EGF receptor through Fyn kinase and without matrix metalloproteases. Cell Signal. 2006, 18, 1338–1349. [Google Scholar] [CrossRef] [PubMed]
- Haglund, K.; Dikic, I. The role of ubiquitylation in receptor endocytosis and endosomal sorting. J. Cell. Sci. 2012, 125, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Grovdal, L.M.; Stang, E.; Sorkin, A.; Madshus, I.H. Direct interaction of Cbl with pTyr 1045 of the EGF receptor (EGFR) is required to sort the EGFR to lysosomes for degradation. Exp. Cell. Res. 2004, 300, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Sato, A.; Aoto, M.; Fukami, Y. c-Src phosphorylates epidermal growth factor receptor on tyrosine 845. Biochem. Biophys. Res. Commun. 1995, 215, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Roepstorff, K.; Grandal, M.V.; Henriksen, L.; Knudsen, S.L.; Lerdrup, M.; Grovdal, L.; Willumsen, B.M.; van Deurs, B. Differential effects of egfr ligands on endocytic sorting of the receptor. Traffic 2009, 10, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Gschwind, A.; Hart, S.; Fischer, O.M.; Ullrich, A. Tace cleavage of proamphiregulin regulates gpcr-induced proliferation and motility of cancer cells. EMBO J. 2003, 22, 2411–2421. [Google Scholar] [CrossRef] [PubMed]
- Wetzker, R.; Bohmer, F.D. Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat. Rev. Mol. Cell. Biol. 2003, 4, 651–657. [Google Scholar] [CrossRef] [PubMed]
- McCole, D.F.; Truong, A.; Bunz, M.; Barrett, K.E. Consequences of direct versus indirect activation of epidermal growth factor receptor in intestinal epithelial cells are dictated by protein-tyrosine phosphatase 1B. J. Biol. Chem. 2007, 282, 13303–13315. [Google Scholar] [CrossRef] [PubMed]
- Keely, S.J.; Calandrella, S.O.; Barrett, K.E. Carbachol-stimulated transactivation of epidermal growth factor receptor and mitogen-activated protein kinase in T(84) cells is mediated by intracellular Ca2+, PYK-2, and p60(src). J. Biol. Chem. 2000, 275, 12619–12625. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Sinnett-Smith, J.; Rozengurt, E. Carbachol induces p70s6k1 activation through an ERK-dependent but Akt-independent pathway in human colonic epithelial cells. Biochem. Biophys. Res. Commun. 2009, 387, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Batzer, A.G.; Rotin, D.; Urena, J.M.; Skolnik, E.Y.; Schlessinger, J. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol. Cell. Biol. 1994, 14, 5192–5201. [Google Scholar] [PubMed]
- Margolis, B.L.; Lax, I.; Kris, R.; Dombalagian, M.; Honegger, A.M.; Howk, R.; Givol, D.; Ullrich, A.; Schlessinger, J. All autophosphorylation sites of epidermal growth factor (EGF) receptor and HER2/neu are located in their carboxyl-terminal tails. Identification of a novel site in EGF receptor. J. Biol. Chem. 1989, 264, 10667–10671. [Google Scholar] [PubMed]
- Levkowitz, G.; Waterman, H.; Ettenberg, S.A.; Katz, M.; Tsygankov, A.Y.; Alroy, I.; Lavi, S.; Iwai, K.; Reiss, Y.; Ciechanover, A.; et al. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol. Cell. 1999, 4, 1029–1040. [Google Scholar] [CrossRef] [PubMed]
- Levkowitz, G.; Waterman, H.; Zamir, E.; Kam, Z.; Oved, S.; Langdon, W.Y.; Beguinot, L.; Geiger, B.; Yarden, Y. c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 1998, 12, 3663–3674. [Google Scholar] [CrossRef] [PubMed]
- Kasina, S.; Scherle, P.A.; Hall, C.L.; Macoska, J.A. Adam-mediated amphiregulin shedding and EGFR transactivation. Cell. Prolif. 2009, 42, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Coffey, R.J., Jr.; Derynck, R.; Wilcox, J.N.; Bringman, T.S.; Goustin, A.S.; Moses, H.L.; Pittelkow, M.R. Production and auto-induction of transforming growth factor-α in human keratinocytes. Nature 1987, 328, 817–820. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Higashiyama, S.; Asada, H.; Hashimura, E.; Kobayashi, T.; Sudo, K.; Nakagawa, T.; Damm, D.; Yoshikawa, K.; Taniguchi, N. Heparin-binding epidermal growth factor-like growth factor is an autocrine growth factor for human keratinocytes. J. Biol. Chem. 1994, 269, 20060–20066. [Google Scholar] [PubMed]
- Shirakata, Y.; Komurasaki, T.; Toyoda, H.; Hanakawa, Y.; Yamasaki, K.; Tokumaru, S.; Sayama, K.; Hashimoto, K. Epiregulin, a novel member of the epidermal growth factor family, is an autocrine growth factor in normal human keratinocytes. J. Biol. Chem. 2000, 275, 5748–5753. [Google Scholar] [CrossRef] [PubMed]
- Cook, P.W.; Mattox, P.A.; Keeble, W.W.; Pittelkow, M.R.; Plowman, G.D.; Shoyab, M.; Adelman, J.P.; Shipley, G.D. A heparin sulfate-regulated human keratinocyte autocrine factor is similar or identical to amphiregulin. Mol. Cell. Biol. 1991, 11, 2547–2557. [Google Scholar] [PubMed]
- Pastore, S.; Mascia, F.; Mariani, V.; Girolomoni, G. The epidermal growth factor receptor system in skin repair and inflammation. J. Investig. Dermatol. 2008, 128, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Chernyavsky, A.I.; Arredondo, J.; Wess, J.; Karlsson, E.; Grando, S.A. Novel signaling pathways mediating reciprocal control of keratinocyte migration and wound epithelialization through M3 and M4 muscarinic receptors. J. Cell. Biol. 2004, 166, 261–272. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ockenga, W.; Kühne, S.; Bocksberger, S.; Banning, A.; Tikkanen, R. Epidermal Growth Factor Receptor Transactivation Is Required for Mitogen-Activated Protein Kinase Activation by Muscarinic Acetylcholine Receptors in HaCaT Keratinocytes. Int. J. Mol. Sci. 2014, 15, 21433-21454. https://doi.org/10.3390/ijms151121433
Ockenga W, Kühne S, Bocksberger S, Banning A, Tikkanen R. Epidermal Growth Factor Receptor Transactivation Is Required for Mitogen-Activated Protein Kinase Activation by Muscarinic Acetylcholine Receptors in HaCaT Keratinocytes. International Journal of Molecular Sciences. 2014; 15(11):21433-21454. https://doi.org/10.3390/ijms151121433
Chicago/Turabian StyleOckenga, Wymke, Sina Kühne, Simone Bocksberger, Antje Banning, and Ritva Tikkanen. 2014. "Epidermal Growth Factor Receptor Transactivation Is Required for Mitogen-Activated Protein Kinase Activation by Muscarinic Acetylcholine Receptors in HaCaT Keratinocytes" International Journal of Molecular Sciences 15, no. 11: 21433-21454. https://doi.org/10.3390/ijms151121433
APA StyleOckenga, W., Kühne, S., Bocksberger, S., Banning, A., & Tikkanen, R. (2014). Epidermal Growth Factor Receptor Transactivation Is Required for Mitogen-Activated Protein Kinase Activation by Muscarinic Acetylcholine Receptors in HaCaT Keratinocytes. International Journal of Molecular Sciences, 15(11), 21433-21454. https://doi.org/10.3390/ijms151121433