[FeFe]-Hydrogenase Abundance and Diversity along a Vertical Redox Gradient in Great Salt Lake, USA

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Water Column Chemistry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Depth (m) | Latitude | Longitude | Salinity (ppt) | Dissolved | PAR (µmol·photons·m–2·s−1) | Temperature (°C) | pH |
|---|---|---|---|---|---|---|---|
| O2 (mg/L) | |||||||
| 0.0 | 41.1674600 | −112.6696117 | 149 | 5.46 | 2079 | 24.17 | 7.21 |
| 1.0 | 41.1674600 | −112.6696117 | 151 | 5.38 | 1219 | 24.16 | 7.46 |
| 4.0 | 41.1674717 | −112.6696117 | 143 | 5.13 | 175 | 23.77 | 7.83 |
| 6.0 | 41.1674717 | −112.6696417 | 151 | 2.32 | 75 | 23.37 | 8.07 |
| 6.5 | 41.1674833 | −112.6696200 | 203 | 1.68 | 44 | 20.20 b | 7.10 b |
| 8.0 | 41.1674817 | −112.6696083 | 247 | 0.98 | 0 | 16.59 | 5.95 |
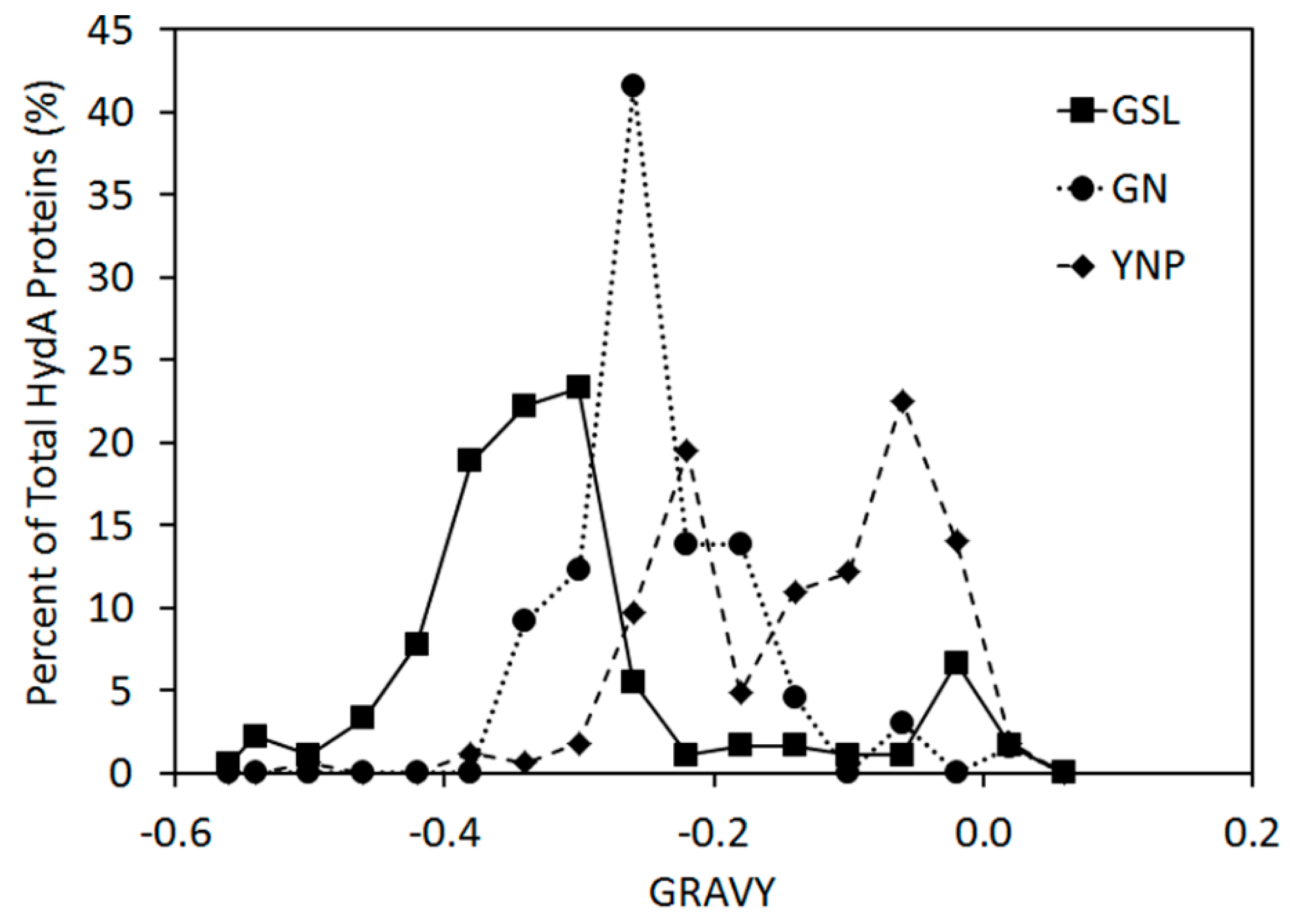
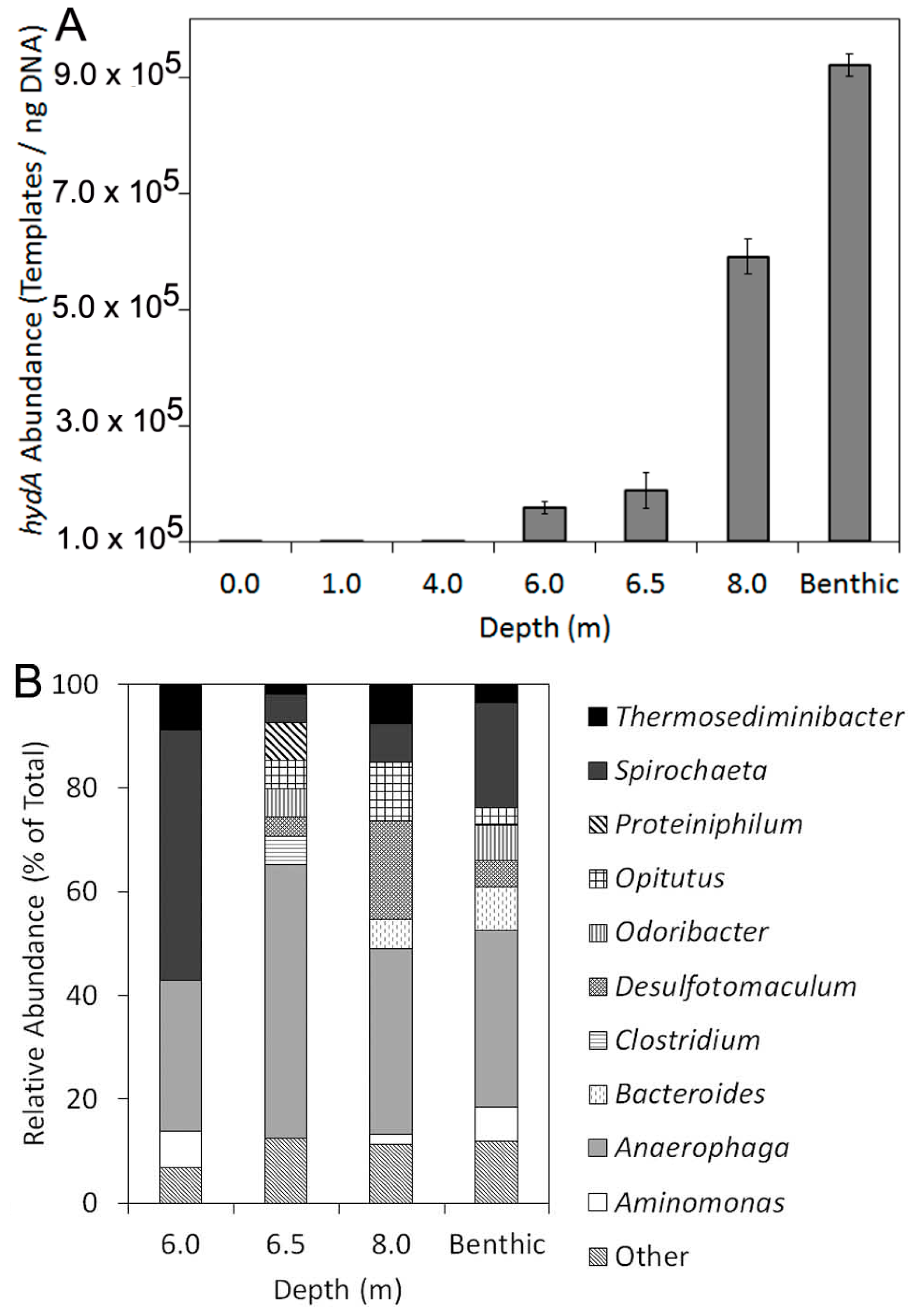
2.2. Abundance, Composition, and Diversity of hydA in GSL Water Column
| Depth (m) | Templates/ng DNA | SD | na | PD b |
|---|---|---|---|---|
| 0.0 | BD | − | − | − |
| 1.0 | BD | − | − | − |
| 4.0 | BD | − | − | − |
| 6.0 | 1.6 × 105 | 1.0 × 104 | 58 | 12.5 |
| 6.5 | 1.9 × 105 | 3.0 × 104 | 55 | 12.5 |
| 8.0 | 5.9 × 105 | 3.1 × 104 | 53 | 13.9 |
| Benthic c | 9.2 × 105 | 1.9 × 104 | 59 | 16.0 |

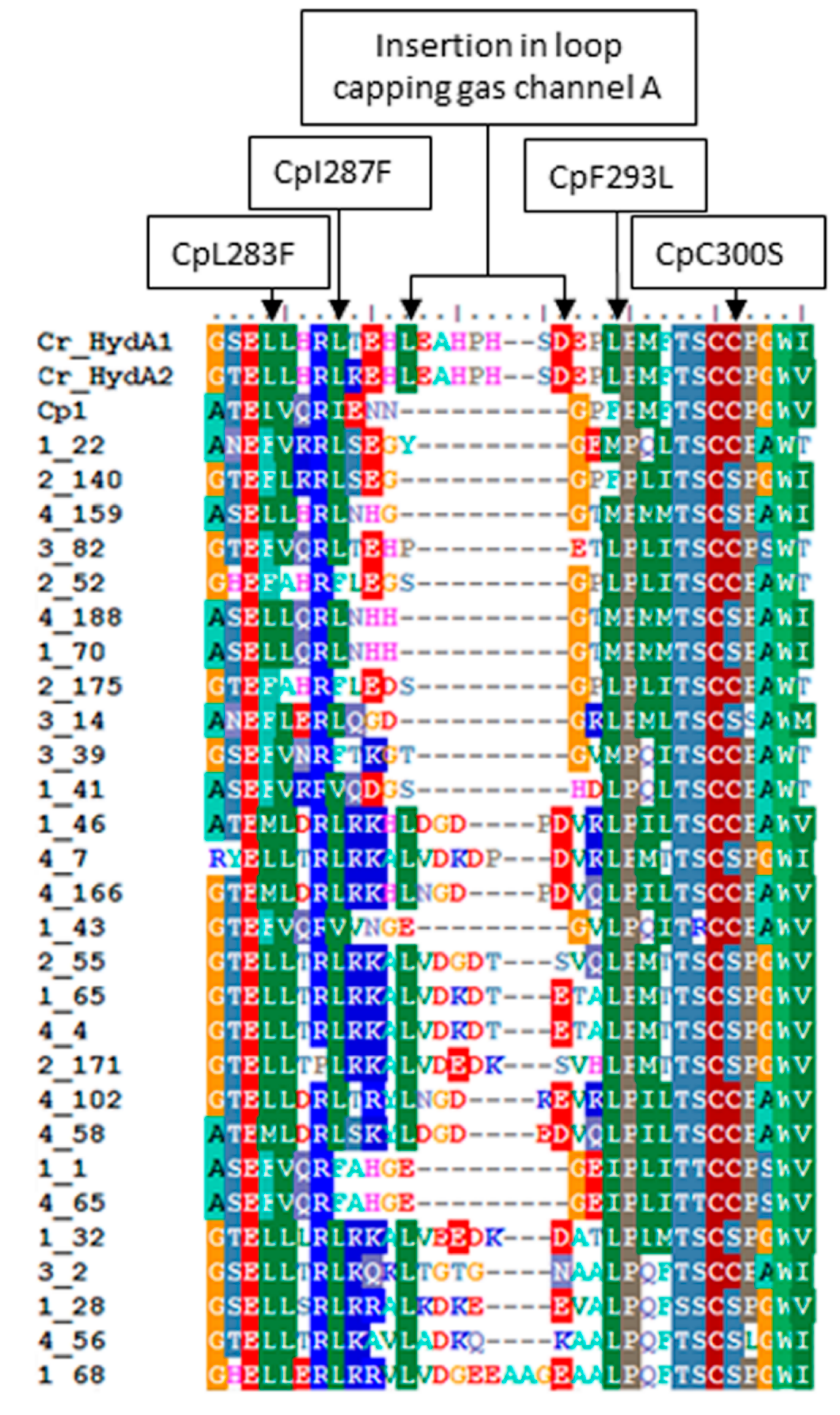
2.3. Variation in H-Cluster Binding Motifs and Putative Gas Channels

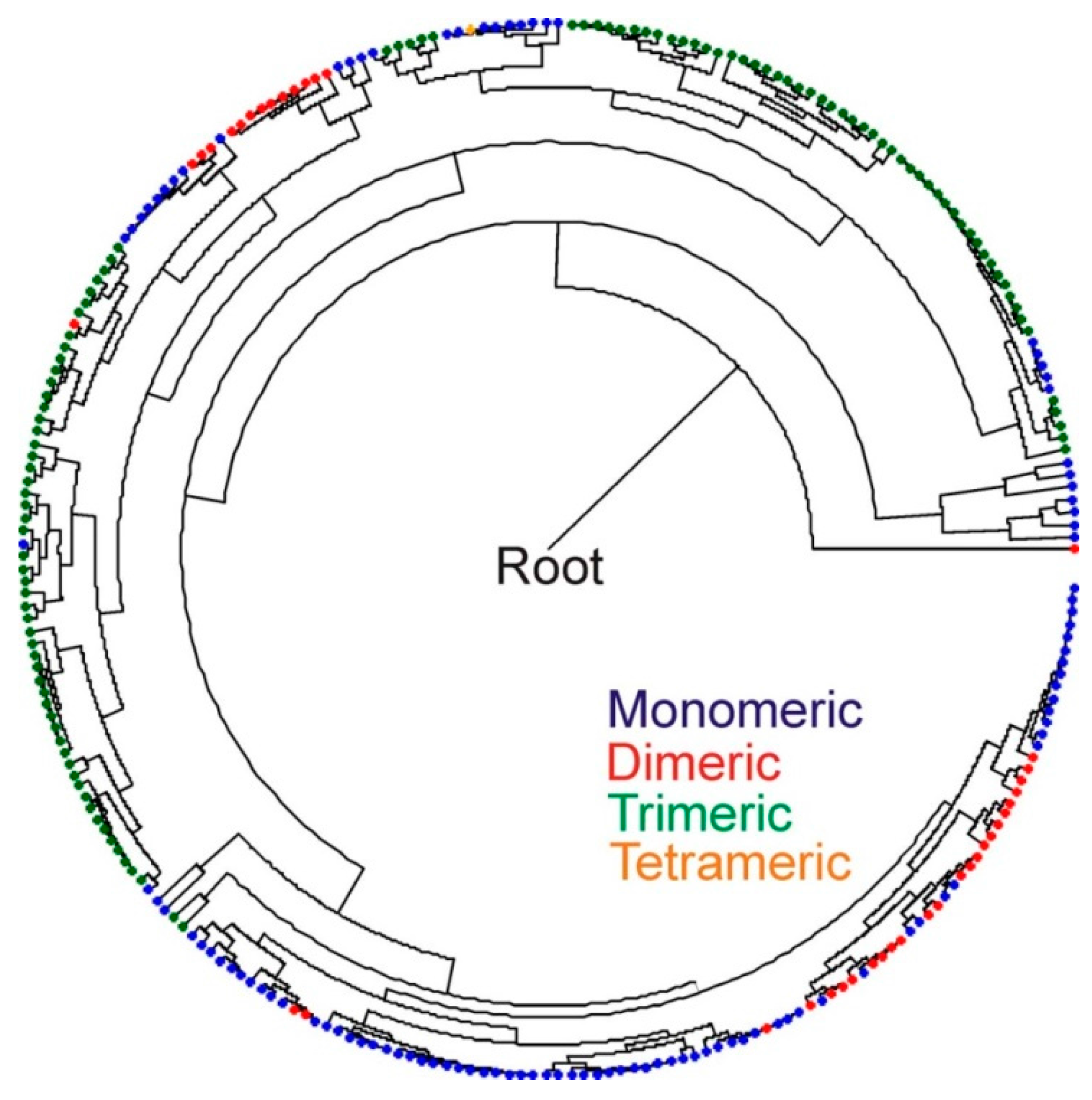
2.4. Inferred Structural Variation of GSL HydA


3. Discussion

4. Experimental Section
4.1. Site Description and Sample Collection
4.2. Physical and Chemical Analysis
4.3. DNA Extraction, Amplification, Cloning, and Sequencing of hydA
4.4. Primary Sequence Analysis
4.5. Prediction of Environmental HydA Accessary Cluster Composition
4.6. hydA Quantitative PCR (qPCR)
4.7. HydA Phylogenetic Diversity
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McInerney, M.J.; Sieber, J.R.; Gunsalus, R.P. Syntrophy in anaerobic global carbon cycles. Curr. Opin. Biotechnol. 2009, 20, 623–632. [Google Scholar]
- Schink, B. Energetics of syntrophic cooperation in methanogenic degradation. Microbiol. Mol. Biol. Rev. 1997, 61, 262–280. [Google Scholar]
- Meyer, J. [FeFe] hydrogenases and their evolution: A genomic perspective. Cell Mol. Life Sci. 2007, 64, 1063–1084. [Google Scholar]
- Vignais, P.M.; Billoud, B. Occurrence, classification, and biological function of hydrogenases: An overview. Chem. Rev. 2007, 107, 4206–4272. [Google Scholar]
- Vignais, P.M.; Billoud, B.; Meyer, J. Classification and phylogeny of hydrogenases. FEMS Microbiol. Rev. 2001, 25, 455–501. [Google Scholar]
- Conrad, R. Soil microorganisms as controllers of atmospheric trace gases (H2, CO, CH4, OCS, N2O, and NO). Microbiol. Rev. 1996, 60, 609–640. [Google Scholar]
- Robson, R.L.; Postgate, J.R. Oxygen and hydrogen in biological nitrogen fixation. Annu. Rev. Microbiol. 1980, 34, 183–207. [Google Scholar]
- Cammack, R. Bioinorganic chemistry: Hydrogenase sophistication. Nature 1999, 397, 214–215. [Google Scholar]
- Mulder, D.W.; Boyd, E.S.; Sarma, R.; Lange, R.K.; Endrizzi, J.A.; Broderick, J.B.; Peters, J. W. Stepwise [FeFe]-hydrogenase H-cluster assembly revealed in the structure of HydAΔEFG. Nature 2010, 465, 248–251. [Google Scholar]
- Peters, J.W.; Lanzilotta, W.N.; Lemon, B.J.; Seefeldt, L.C. X-ray crystal structure of the Fe-only hydrogenase (CpI) from Clostridium pasteurianum to 1.8 angstrom resolution. Science 1998, 282, 1853–1858. [Google Scholar]
- Posewitz, M.C.; Mulder, D.W.; Peters, J.W. New frontiers in hydrogenase structure and biosynthesis. Curr. Chem. Biol. 2008, 2, 178–199. [Google Scholar]
- Stapleton, J.A.; Swartz, J.R. A cell-free microtiter plate screen for improved [FeFe] hydrogenases. PLoS One 2010, 5, e10554. [Google Scholar]
- Stapleton, J.A.; Swartz, J.R. Development of an in vitro compartmentalization screen for high-throughput directed evolution of [FeFe] hydrogenases. PLoS One 2010, 5, e15275. [Google Scholar] [PubMed]
- Happe, T.; Mosler, B.; Naber, J.D. Induction, localization and metal content of hydrogenase in the green alga Chlamydomonas reinhardtii. Eur. J. Biochem. 1994, 222, 769–774. [Google Scholar]
- Ghirardi, M.L.; Posewitz, M.C.; Maness, P.C.; Dubini, A.; Yu, J.; Seibert, M. Hydrogenases and hydrogen photoproduction in oxygenic photosynthetic organisms. Annu. Rev. Plant Biol. 2007, 58, 71–91. [Google Scholar]
- Forestier, M.; King, P.; Zhang, L.; Posewitz, M.; Schwarzer, S.; Happe, T.; Ghirardi, M.L.; Seibert, M. Expression of two [Fe]-hydrogenases in Chlamydomonas reinhardtii under anaerobic conditions. Eur. J. Biochem. 2003, 270, 2750–2758. [Google Scholar]
- Adams, M.W.W. The structure and mechanism of iron-hydrogenases. Biochim. Biophys. Acta 1990, 1020, 115–145. [Google Scholar]
- Chen, J.S.; Mortenson, L.E. Purification and properties of hydrogenase from Clostridium pasteurianum W5. Biochim. Biophys. Acta 1974, 371, 283–298. [Google Scholar]
- Calusinska, M.; Happe, T.; Joris, B.; Wilmotte, A. The surprising diversity of clostridial hydrogenases: A comparative genomic perspective. Microbiology 2010, 156, 1575–1588. [Google Scholar]
- Malki, S.; Saimmaime, I.; de Luca, G.; Rousset, M.; Dermoun, Z.; Belaich, J.P. Characterization of an operon encoding an NADP-reducing hydrogenase in Desulfovibrio fructosovorans. J. Bacteriol. 1995, 177, 2628–2636. [Google Scholar]
- Soboh, B.; Linder, D.; Hedderich, R. A multisubunit membrane-bound [NiFe] hydrogenaseand an NADH-dependent Fe-only hydrogenase in the fermenting bacterium Thermoanaerobacter tengcongensis. Microbiology 2004, 150, 2451–2463. [Google Scholar]
- Verhagen, M.F.; Adams, M.W. Fe-only hydrogenase from Thermotoga maritima. Methods Enzymol. 2001, 331, 216–226. [Google Scholar]
- Schut, G.J.; Adams, M.W. The iron-hydrogenase of Thermotoga maritima utilizes ferredoxin and NADH synergistically: A new perspective on anaerobic hydrogen production. J. Bacteriol. 2009, 191, 4451–4457. [Google Scholar]
- Herrmann, G.; Jayamani, E.; Mai, G.; Buckel, W. Energy conservation via electron-transferring flavoprotein in anaerobic bacteria. J. Bacteriol. 2008, 190, 784–791. [Google Scholar]
- Buckel, W.; Thauer, R.K. Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochim. Biophys. Acta 2013, 1827, 94–113. [Google Scholar]
- Wang, S.; Huang, H.; Kahnt, J.; Mueller, A.P.; Kopke, M.; Thauer, R.K. NADP-specific electron-bifurcating [FeFe]-hydrogenase in a functional complex with formate dehydrogenase in Clostridium autoethanogenum grown on CO. J. Bacteriol. 2013, 195, 4373–4386. [Google Scholar]
- Wang, S.; Huang, H.; Kahnt, J.; Thauer, R.K. A reversible electron-bifurcating ferredoxin- and NAD-dependent [FeFe]-hydrogenase (HydABC) in Moorella thermoacetica. J Bacteriol. 2013, 195, 1267–1275. [Google Scholar]
- Stripp, S.T.; Goldet, G.; Brandmayr, C.; Sanganas, O.; Vincent, K.A.; Haumann, M.; Armstrong, F.A.; Happe, T. How oxygen attacks [FeFe] hydrogenases from photosynthetic organisms. Proc. Natl. Acad. Sci. USA 2009, 106, 17331–17336. [Google Scholar]
- Lambertz, C.; Leidel, N.; Havelius, K.G.V.; Noth, J.; Chernev, P.; Winkler, M.; Happe, T.; Haumann, M. O2-reactions at the six-iron activesite (H-cluster) in [FeFe]-hydrogenase. J. Biol. Chem. 2011, 286, 40614–40623. [Google Scholar]
- Stripp, S.; Sanganas, O.; Happe, T.; Haumann, M. The structure of the active site H-cluster of [FeFe] hydrogenase from the green alga Chlamydomonas reinhardtii studied by X-ray absorption spectroscopy. Biochemistry 2009, 48, 5042–5049. [Google Scholar]
- Juszczak, A.; Aono, S.; Adams, M.W. The extremely thermophilic eubacterium, Thermotoga maritima, contains a novel iron-hydrogenase whose cellular activity is dependent upon tungsten. J. Biol. Chem. 1991, 266, 13834–13841. [Google Scholar]
- Baffert, C.; Demuez, M.; Cournac, L.; Burlat, B.; Guigliarelli, B.; Bertrand, P.; Girbal, L.; Léger, C. Hydrogen-activating enzymes: Activity does not correlate with oxygen sensitivity. Angew. Chem. (Int. Ed. Engl.) 2008, 47, 2052–2054. [Google Scholar]
- Bruska, M.K.; Stiebritz, M.T.; Reiher, M. Regioselectivity of H-cluster oxidation. J. Am. Chem. Soc. 2011, 133, 20588–20603. [Google Scholar]
- Stiebritz, M.T.; Reiher, M. Hydrogenases and oxygen. Chem. Sci. 2012, 3, 1739–1751. [Google Scholar]
- Kubas, A.; de Sancho, D.; Best, R.B.; Blumberger, J. Aerobic damage to [FeFe]-hydrogenases: Activation barriers for the chemical attachment of O2. Angew. Chem. (Int. Ed. Engl.) 2014, 126, 4165–4168. [Google Scholar]
- Fourmond, V.; Greco, C.; Sybirna, K.; Baffert, C.; Wang, P.H.; Ezanno, P.; Montefiori, M.; Bruschi, M.; Meynial-Salles, I.; Soucaille, P.; et al. The oxidative inactivation of FeFe hydrogenase reveals the flexibility of the H-cluster. Nat. Chem. 2014, 6, 336–342. [Google Scholar]
- Liebgott, P.P.; Leroux, F.; Burlat, B.; Dementin, S.; Baffert, C.; Lautier, T.; Fourmond, V.; Ceccaldi, P.; Cavazza, C.; Meynial-Salles, I.; et al. Relating diffusion along the substrate tunnel and oxygen sensitivity in hydrogenase. Nat. Chem. Biol. 2010, 6, 63–70. [Google Scholar]
- Nagy, L.; Meuser, J.; Plummer, S.; Seibert, M.; Ghirardi, M.; King, P.; Ahmann, D.; Posewitz, M. Application of gene-shuffling for the rapid generation of novel [FeFe]-hydrogenase libraries. Biotechnol. Lett. 2007, 29, 421–430. [Google Scholar]
- Bingham, A.S.; Smith, P.R.; Swartz, J.R. Evolution of an [FeFe] hydrogenase with decreased oxygen sensitivity. Int. J. Hydrogen. Energy 2012, 37, 2965–2976. [Google Scholar]
- Boyd, E.S.; Hamilton, T.L.; Spear, J.R.; Lavin, M.; Peters, J.W. [FeFe]-hydrogenase in Yellowstone National Park: Evidence for dispersal limitation and phylogenetic niche conservatism. ISME J. 2010, 4, 1485–1495. [Google Scholar]
- Boyd, E.S.; Spear, J.R.; Peters, J.W. [FeFe]-hydrogenase genetic diversity provides insight into molecular adaptation in a saline microbial mat community. Appl. Environ. Microbiol. 2009, 75, 4620–4623. [Google Scholar]
- Schmidt, O.; Drake, H.L.; Horn, M.A. Hitherto unknown [FeFe]-hydrogenase gene diversity in anaerobes and anoxic enrichments from a moderately acidic fen. Appl. Environ. Microbiol. 2010, 76, 2027–2031. [Google Scholar]
- Chivian, D.; Brodie, E.L.; Alm, E.J.; Culley, D.E.; Dehal, P.S.; de Santis, T.Z.; Gihring, T.M.; Lapidus, A.; Lin, L.H.; Lowry, S.R.; et al. Environmental genomics reveals a single-species ecosystem deep within Earth. Science 2008, 322, 275–278. [Google Scholar]
- Warnecke, F.; Luginbuhl, P.; Ivanova, N.; Ghassemian, M.; Richardson, T.H.; Stege, J.T.; Cayouette, M.; McHardy, A.C.; Djordjevic, G.; Aboushadi, N.; et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 2007, 450, 560–565. [Google Scholar]
- Cohen, J.; Kim, K.; King, P.; Seibert, M.; Schulten, K. Finding gas diffusion pathways in proteins: Application to O2 and H2 transport in CpI [FeFe]-hydrogenase and the role of packing defects. Structure 2005, 13, 1321–1329. [Google Scholar]
- Goldet, G.; Brandmayr, C.; Stripp, S.T.; Happe, T.; Cavazza, C.; Fontecilla-Camps, J.C.; Armstrong, F.A.J. Electrochemical kinetic investigations of the reactions of [FeFe]-hydrogenases with carbon monoxide and oxygen: Comparing the importance of gas tunnels and active-site electronic/redox effects. J. Am. Chem. Soc. 2009, 131, 14979–14989. [Google Scholar]
- King, P.W.; Svedruzic, D.; Cohen, J.; Schulten, K.; Seibert, M.; Ghirardi, M.L. Structural and functional investigations of biological catalysts for optimization of solar-driven, H2 production systems. In Proceedings of the SPIE Optics and Photonics, San Diego, CA, USA, 13–17 August 2006; p. 9.
- Lautier, T.; Ezanno, P.; Baffert, C.; Fourmond, V.; Cournac, L.; Fontecilla-Camps, J.C.; Soucaille, P.; Bertrand, P.; Meynial-Salles, I.; Leger, C. The quest for a functional substrate access tunnel in FeFe hydrogenase. Faraday Discuss. 2011, 148, 385–407. [Google Scholar]
- Ghirardi, M.L.; Cohen, J.; King, P.; Schulten, K.; Kim, K.; Seibert, M. [FeFe]-hydrogenases and photobiological hydrogen production. In Proceedings of the SPIE Optics and Photonics, San Diego, CA, USA, 13–17 August 2006; p. 6.
- Meuser, J.E.; Baxter, B.K.; Spear, J.R.; Peters, J.W.; Posewitz, M.C.; Boyd, E.S. Contrasting patterns of community assembly in the stratified water column of Great Salt Lake, Utah. Microb. Ecol. 2013, 66, 268–280. [Google Scholar]
- Beer, L.L.; Boyd, E.S.; Peters, J.W.; Posewitz, M.C. Engineering algae for biohydrogen and biofuel production. Curr. Opin. Biotechnol. 2009, 20, 1–8. [Google Scholar]
- Schuchmann, K.; Müller, V. A bacterial electron-bifurcating hydrogenase. J. Biol. Chem. 2012, 287, 31165–31171. [Google Scholar]
- Shaw, A.J.; Hogsett, D.A.; Lynd, L.R. Identification of the [FeFe]-hydrogenase responsible for hydrogen generation in Thermoanaerobacterium saccharolyticum and demonstration of increased ethanol yield via hydrogenase knockout. J. Bacteriol. 2009, 191, 6457–6464. [Google Scholar]
- Lanyi, J.K. Salt-dependent properties of proteins from extremely halophilic bacteria. Bacteriol. Rev. 1974, 38, 272–290. [Google Scholar]
- Dennis, P.P.; Shimmin, L.C. Evolutionary divergence and salinity-mediated selection in halophilic archaea. Microbiol. Mol. Biol. Rev. 1997, 61, 90–104. [Google Scholar]
- Reistad, R. On the composition and nature of the bulk protein of the extremely halophilic bacteria. Arch. Mikrobiol. 1970, 71, 353–360. [Google Scholar]
- Spahr, P.F. Amino acid composition of ribosomes from Escherichia coli. J. Mol. Biol. 1962, 4, 395–406. [Google Scholar]
- Visentin, L.P.; Chow, C.; Matheson, A.T.; Yaguchi, M.; Rollin, F. Halobacterium cutirubrum ribosomes: Properties of the ribosomal proteins and ribonucleic acid. Biochem. J. 1972, 130, 103–110. [Google Scholar]
- Hutcheon, G.W.; Vasisht, N.; Bolhuis, A. Characterization of a highly stable alpha-amylase from the halophilic archaeon Haloarcula hispanica. Extremophiles 2005, 9, 487–495. [Google Scholar]
- Joo, W.A.; Kim, C.W. Proteomics of halophilic archaea. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2005, 815, 237–250. [Google Scholar]
- Paul, S.; Bag, S.; Das, S.; Harvill, E.; Dutta, C. Molecular signature of hypersaline adaptation: Insights from genome and proteome composition of halophilic prokaryotes. Genome Biol. 2008, 9, R70. [Google Scholar]
- Lange, R.; Staaland, H.; Mostad, A. The effect of salinity and temperature on solubility of oxygen and respiratory rate in oxygen-dependent marine invertebrates. J. Exp. Mar. Biol. Ecol. 1972, 9, 217–229. [Google Scholar]
- Nicolet, Y.; Lemon, B.J.; Fontecilla-Camps, J.C.; Peters, J.W. A novel FeS cluster in Fe-only hydrogenases. Trends Biochem. Sci. 2000, 25, 138–143. [Google Scholar]
- Nicolet, Y.; Piras, C.; Legrand, P.; Hatchikian, C.E.; Fontecilla-Camps, J.C. Desulfovibrio desulfuricans iron hydrogenase: The structure shows unusual coordination to an active site Fe binuclear center. Struct. Fold. Des. 1999, 7, 13–23. [Google Scholar]
- Rees, D.C. Great Metalloclusters in Enzymology. Annu. Rev. Biochem. 2002, 71, 221–246. [Google Scholar]
- Meyer, J. Iron-sulfur protein folds, iron-sulfur chemistry, and evolution. J. Biol. Inorg. Chem. 2008, 13, 157–170. [Google Scholar]
- Boyd, E.S.; Jackson, R.A.; Encarnacion, G.; Zahn, J.A.; Beard, T.; Leavitt, W.D.; Pi, Y.; Zhang, C.L.; Pearson, A.; Geesey, G.G. Isolation, characterization, and ecology of sulfur-respiring Crenarchaea inhabiting acid-sulfate-chloride geothermal springs in Yellowstone National Park. Appl. Environ. Microbiol. 2007, 73, 6669–6677. [Google Scholar]
- Boyd, E.S.; Leavitt, W.D.; Geesey, G.G. CO2 uptake and fixation by a thermoacidophilic microbial community attached to precipitated sulfur in a geothermal spring. Appl. Environ. Microbiol. 2009, 75, 4289–4296. [Google Scholar]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar]
- Schloss, P.D.; Handelsman, J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 2005, 71, 1501–1506. [Google Scholar]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: New York, NY, USA, 2005; pp. 571–607. [Google Scholar]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar]
- Marchler-Bauer, A.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; de Weese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; et al. CDD: Specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009, 37, D205–D210. [Google Scholar]
- Boyd, E.S.; Lange, R.K.; Mitchell, A.C.; Havig, J.R.; Lafrenière, M.J.; Hamilton, T.L.; Shock, E.L.; Peters, J.W.; Skidmore, M. Diversity, abundance, and potential activity of nitrifying and nitrate-reducing microbial assemblages in a subglacial ecosystem. Appl. Environ. Microbiol. 2011, 77, 4778–4787. [Google Scholar]
- Anisimova, M.; Gil, M.; Dufayard, J.F.; Dessimoz, C.; Gascuel, O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst. Biol. 2011, 60, 685–699. [Google Scholar]
- Posada, D. ModelTest Server: A web-based tool for the statistical selection of models of nucleotide substitution online. Nucleic Acids Res. 2006, 34, W700–W703. [Google Scholar]
- Swofford, D.L. PAUP *: Kembel, S.W. Phylocom: Software for the Analysis of Phylogenetic Community Structure and Character Evolution. Phylogenetic Analysis Using Parsimony (* and Other Methods). version 4, Sinauer Associate: Sunderland, MA, USA, 2001. [Google Scholar]
- Webb, C.O.; Ackerly, D.D.; Kembel, S.W. Phylocom: Software for the analysis of phylogenetic community structure and character evolution. Bioinformatics 2008, 24, 2098–2100. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boyd, E.S.; Hamilton, T.L.; Swanson, K.D.; Howells, A.E.; Baxter, B.K.; Meuser, J.E.; Posewitz, M.C.; Peters, J.W. [FeFe]-Hydrogenase Abundance and Diversity along a Vertical Redox Gradient in Great Salt Lake, USA. Int. J. Mol. Sci. 2014, 15, 21947-21966. https://doi.org/10.3390/ijms151221947
Boyd ES, Hamilton TL, Swanson KD, Howells AE, Baxter BK, Meuser JE, Posewitz MC, Peters JW. [FeFe]-Hydrogenase Abundance and Diversity along a Vertical Redox Gradient in Great Salt Lake, USA. International Journal of Molecular Sciences. 2014; 15(12):21947-21966. https://doi.org/10.3390/ijms151221947
Chicago/Turabian StyleBoyd, Eric S., Trinity L. Hamilton, Kevin D. Swanson, Alta E. Howells, Bonnie K. Baxter, Jonathan E. Meuser, Matthew C. Posewitz, and John W. Peters. 2014. "[FeFe]-Hydrogenase Abundance and Diversity along a Vertical Redox Gradient in Great Salt Lake, USA" International Journal of Molecular Sciences 15, no. 12: 21947-21966. https://doi.org/10.3390/ijms151221947
APA StyleBoyd, E. S., Hamilton, T. L., Swanson, K. D., Howells, A. E., Baxter, B. K., Meuser, J. E., Posewitz, M. C., & Peters, J. W. (2014). [FeFe]-Hydrogenase Abundance and Diversity along a Vertical Redox Gradient in Great Salt Lake, USA. International Journal of Molecular Sciences, 15(12), 21947-21966. https://doi.org/10.3390/ijms151221947