TGF-β1 Protection against Aβ1–42-Induced Neuroinflammation and Neurodegeneration in Rats

Abstract
:1. Introduction
2. Results
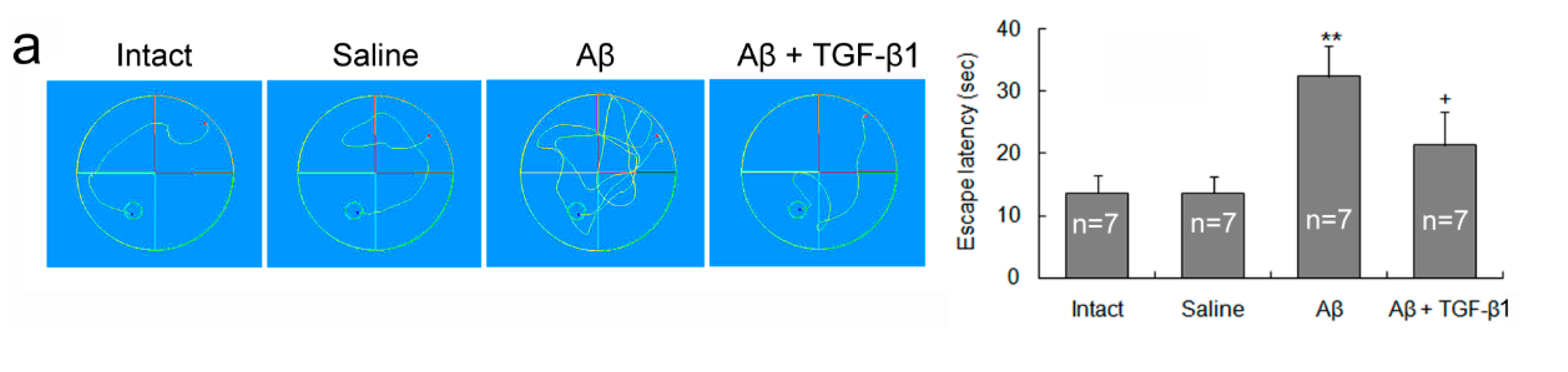
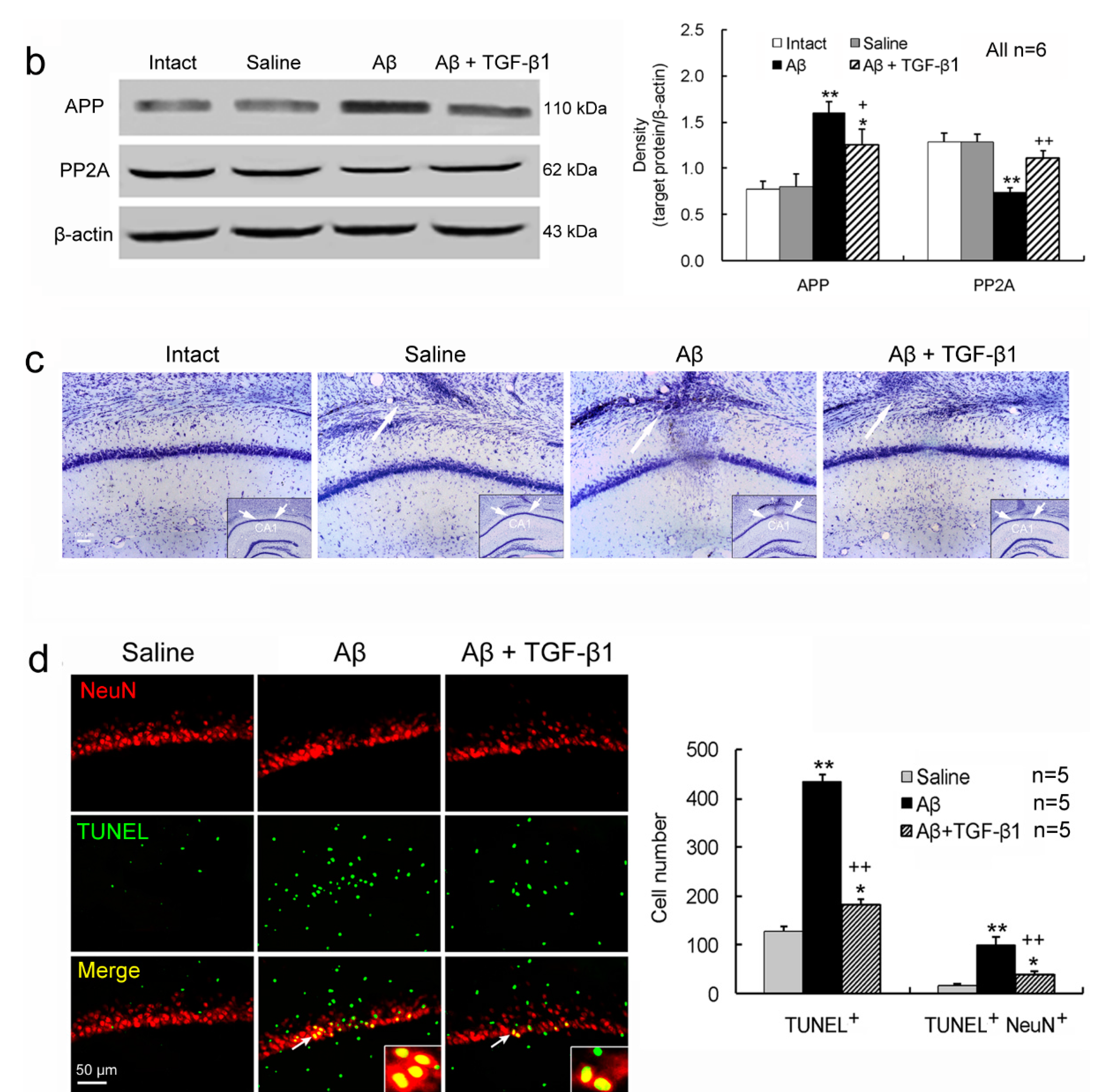
2.1. TGF-β1 Ameliorates Aβ1–42-Induced Impairments of Cognition and Neurons


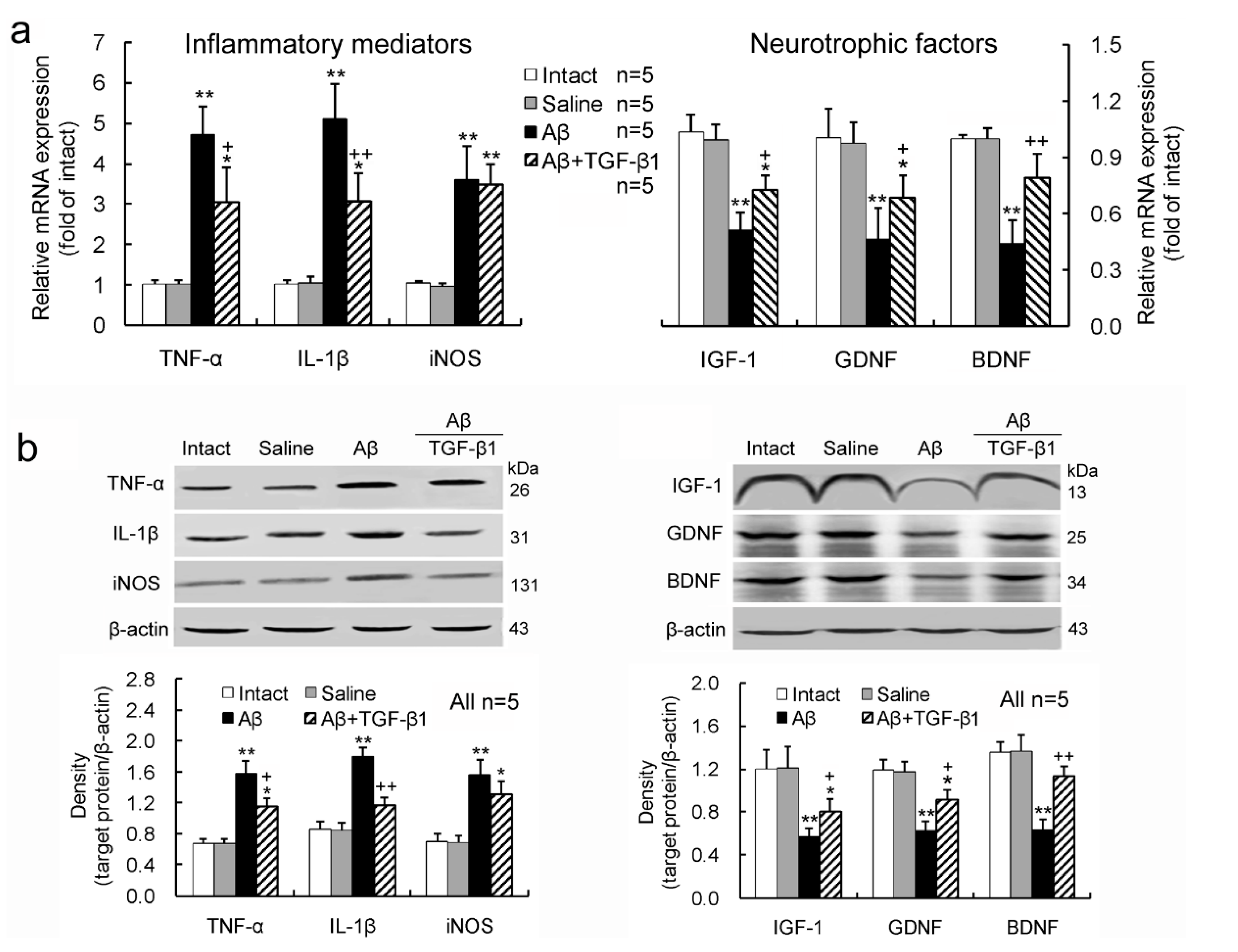
2.2. TGF-β1 Reduces the Up-Regulation of Inflammatory Mediators and Down-Regulation of Neurotrophic Factors Induced by Aβ1–42 in the Hypothalamus

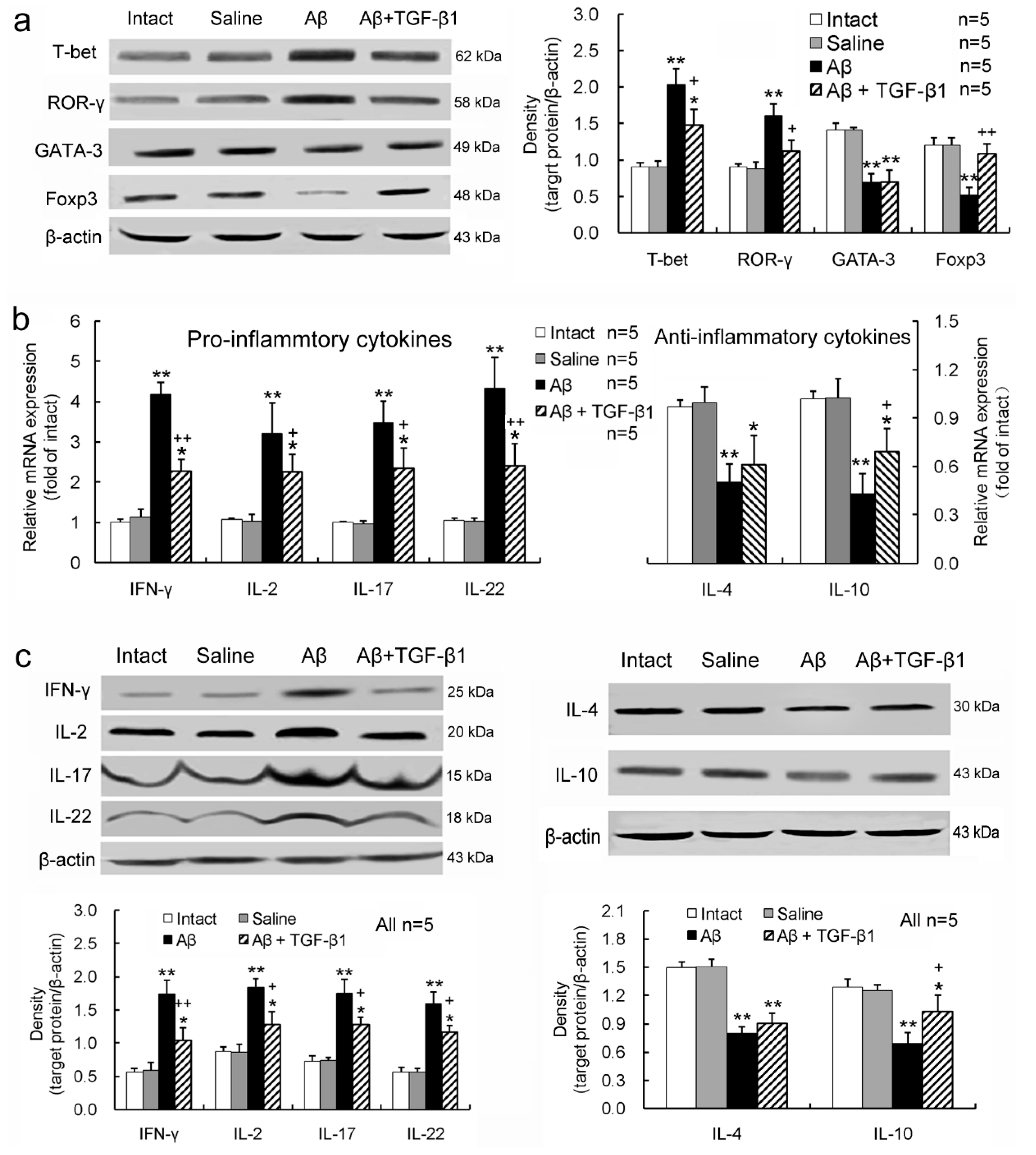
2.3. TGF-β1 Alleviates Pro-Inflammatory Enhancement and Anti-Inflammatory Attenuation of T-Lymphocytes Induced by Aβ1–42 in the Hippocampus
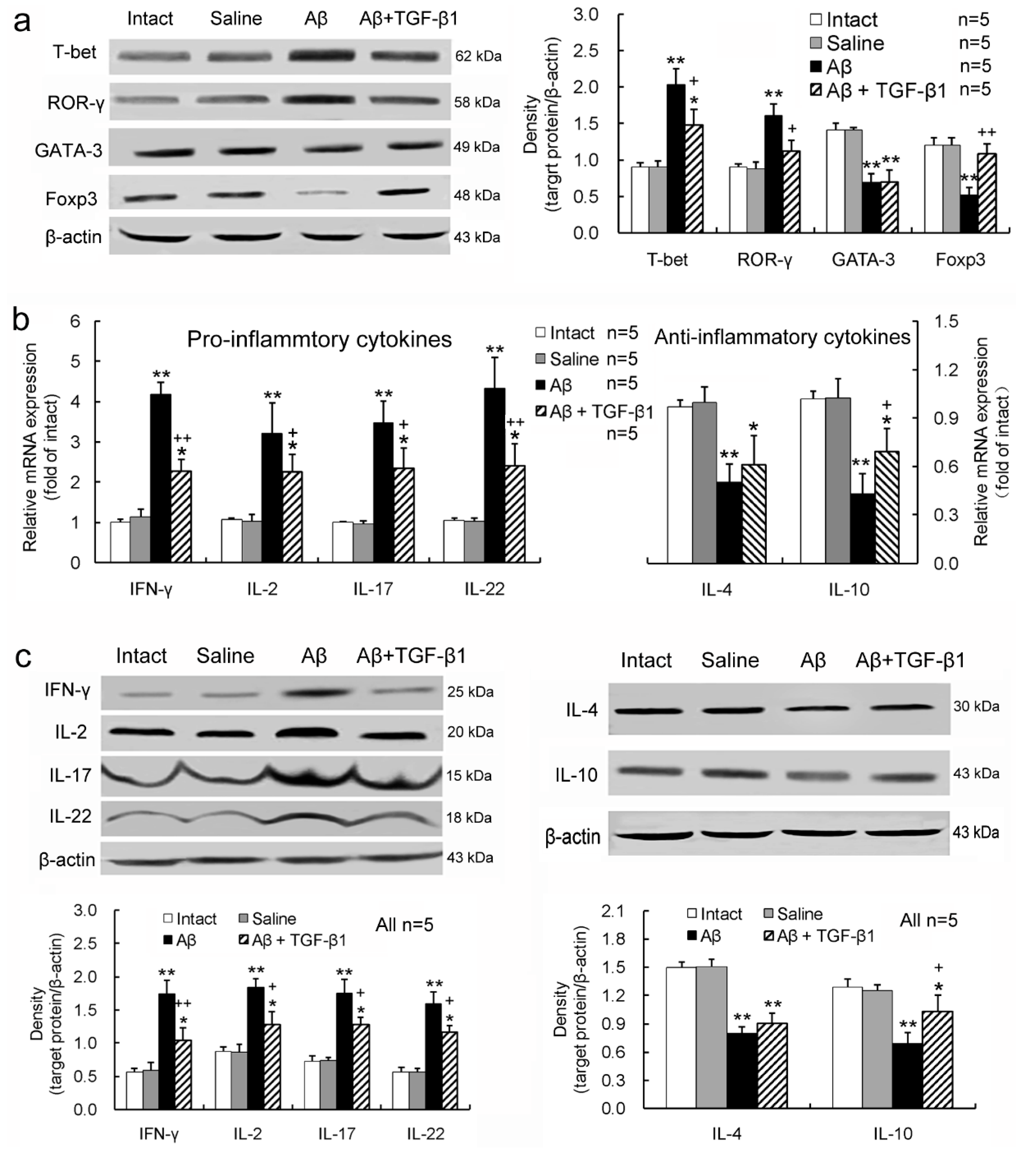
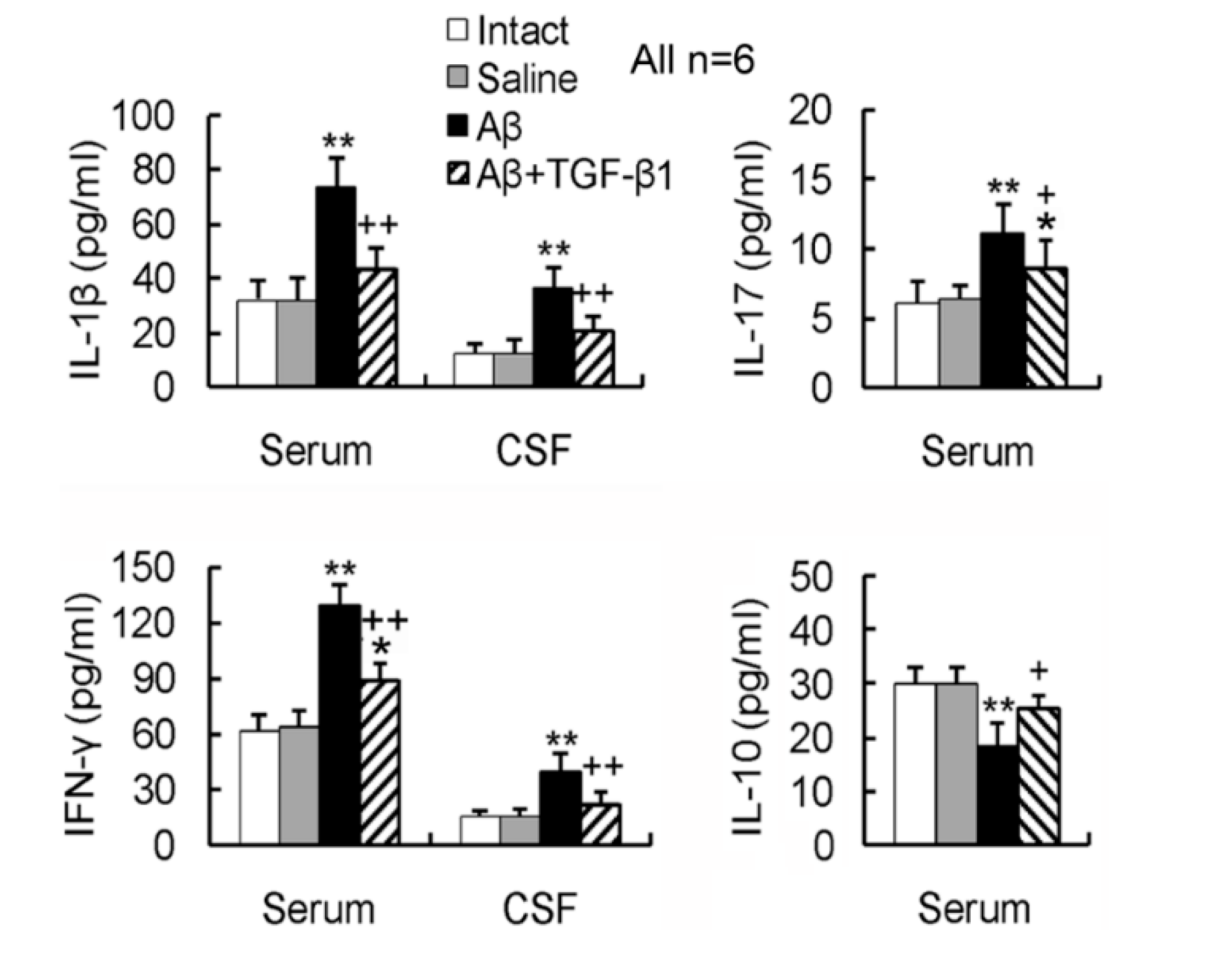
2.4. TGF-β1 Reverses the Aβ1–42-Induced Elevation of Pro-Inflammatory Cytokines and Reduction of Anti-Inflammatory Cytokine in Serum or CSF

3. Discussion
4. Experimental Section
4.1. Animals
4.2. Preparation of Aβ1–42-Induced AD Rat Model
4.3. TGF-β1 Treatment
4.4. Morris Water Maze
4.5. Nissl Staining
4.6. TUNEL Staining
4.7. Western Blot Analysis
4.8. Real-Time PCR Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sense Primer | Antisense Primer |
|---|---|---|
| TNF-α | 5'-CCACCACGCTCTTCTGTCTAC-3' | 5'-ATCTGAGTGTGGGGTCTGG-3' |
| IL-1β | 5'-CTTCCTTGTGCAAGTGTCTG-3' | 5'-CAGGTCATTCTCATCACTGTC-3' |
| iNOS | 5'-CAGCTGGGCTGTACAAACCTT-3' | 5'-CATTGGAAGTGAAGCGTTTCG-3' |
| IGF-1 | 5'-TTTTACTTCAACAAGCCCACA-3' | 5'-CATCCACAATGCCCGTCT-3' |
| GDNF | 5'-ATTCAAGCCACCATCAAAAG-3' | 5'-TCAGTTCCTCCTTGGTTTCG-3' |
| BDNF | 5'-ATCCCATGGGTTACACGAAGGAAG-3' | 5'-AGTAAGGGCCCGAACATACGATTG-3' |
| IFN-γ | 5'-GCCCTCTCTGGCTGTTACTG-3' | 5'-TACCGTCCTTTTGCCAGTTC-3' |
| IL-2 | 5'-CCATGATGCTCACGTTTAAATTTT-3' | 5'-CATTTTCCAGGCACTGAAGATG-3' |
| IL-17 | 5'-TGGACTCTGAGCCGCAATG-3' | 5'-GGCGGACAATAGAGGAAACG-3' |
| IL-22 | 5'-AGCGGTGATGACCAGAACA-3' | 5'-CTCAGGGACATAAACAGCAGA-3' |
| IL-4 | 5'-ACCTTGCTGTCACCCTGTTCT-3' | 5'-CTCTCTCAGAGGGCTGTCGTTA-3' |
| IL-10 | 5'-TGGCAACCCAAGTAACCCT-3' | 5'-CACCCACTTCCCAGTCAGC-3' |
| β-actin | 5'-CGTTGACATCCGTAAAGACC-3' | 5'-TAGAGCCACCAATCCACAC-3' |
4.9. Enzyme-Linked Immunosorbent Assay (ELISA)
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pál, G.; Vincze, C.; Renner, É.; Wappler, E.A.; Nagy, Z.; Lovas, G.; Dobolyi, A. Time course, distribution and cell types of induction of transforming growth factor betas following middle cerebral artery occlusion in the rat brain. PLoS One 2012, 7, e46731. [Google Scholar]
- Wyss-Coray, T.; Lin, C.; Yan, F.; Yu, G.Q.; Rohde, M.; McConlogue, L.; Masliah, E.; Mucke, L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat. Med. 2001, 7, 612–618. [Google Scholar]
- Vincze, C.; Pál, G.; Wappler, E.A.; Szabó, E.R.; Nagy, Z.G.; Lovas, G.; Dobolyi, A. Distribution of mRNAs encoding transforming growth factors-beta1, -2, and -3 in the intact rat brain and after experimentally induced focal ischemia. J. Comp. Neurol. 2010, 518, 3752–3770. [Google Scholar]
- De Groot, C.J.; Montagne, L.; Barten, A.D.; Sminia, P.; van der Valk, P. Expression of transforming growth factor (TGF)-beta1, -beta2, and -beta3 isoforms and TGF-beta type I and type II receptors in multiple sclerosis lesions and human adult astrocyte cultures. J. Neuropathol. Exp. Neurol 1999, 58, 174–187. [Google Scholar]
- De Sampaio e Spohr, T.C.; Martinez, R.; da Silva, E.F.; Neto, V.M.; Gomes, F.C. Neuro-glia interaction effects on GFAP gene: A novel role for transforming growth factor-beta1. Eur. J. Neurosci. 2002, 16, 2059–2069. [Google Scholar]
- Da Cunha, A.; Jefferson, J.A.; Jackson, R.W.; Vitković, L. Glial cell-specific mechanisms of TGF-beta 1 induction by IL-1 in cerebral cortex. J. Neuroimmunol. 1993, 42, 71–85. [Google Scholar]
- Ata, A.K.; Funa, K.; Olsson, Y. Expression of various TGF-beta isoforms and type I receptor in necrotizing human brain lesions. Acta Neuropathol. 1997, 93, 326–333. [Google Scholar]
- Vivien, D.; Bernaudin, M.; Buisson, A.; Divoux, D.; MacKenzie, E.T.; Nouvelot, A. Evidence of type I and type II transforming growth factor-beta receptors in central nervous tissues: Changes induced by focal cerebral ischemia. J. Neurochem. 1998, 70, 2296–2304. [Google Scholar]
- Ata, K.A.; Lennmyr, F.; Funa, K.; Olsson, Y.; Terént, A. Expression of transforming growth factor-beta1, 2, 3 isoforms and type I and II receptors in acute focal cerebral ischemia: An immunohistochemical study in rat after transient and permanent occlusion of middle cerebral artery. Acta Neuropathol. 1999, 97, 447–455. [Google Scholar]
- Van der Wal, E.A.; Gomez–Pinilla, F.; Cotman, C.W. Transforming growth factor-beta 1 is in plaques in Alzheimer and Down pathologies. Neuroreport 1993, 4, 69–72. [Google Scholar]
- Chao, C.C.; Hu, S.; Frey, W.H.; Ala, T.A.; Tourtellotte, W.W.; Peterson, P.K. Transforming growth factor beta in Alzheimer’s disease. Clin. Diagn. Lab. Immunol. 1994, 1, 109–110. [Google Scholar]
- Zetterberg, H.; Andreasen, N.; Blennow, K. Increased cerebrospinal fluid levels of transforming growth factor-beta1 in Alzheimer’s disease. Neurosci. Lett. 2004, 367, 194–196. [Google Scholar]
- Chao, C.C.; Ala, T.A.; Hu, S.; Crossley, K.B.; Sherman, R.E.; Peterson, P.K.; Frey, W.H., 2nd. Serum cytokine levels in patients with Alzheimer’s disease. Clin. Diagn. Lab. Immunol. 1994, 1, 443–446. [Google Scholar]
- Wyss-Coray, T.; Masliah, E.; Mallory, M.; McConlogue, L.; Johnson-Wood, K.; Lin, C.; Mucke, L. Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer’s disease. Nature 1997, 389, 603–606. [Google Scholar]
- Wyss-Coray, T.; Lacombe, P.; Lin, C.; Sanan, D.A.; Mucke, L.; Masliah, E. Alzheimer’s disease-like cerebrovascular pathology in transforming growth factor-β1 transgenic mice and functional metabolic correlates. Ann. N. Y. Acad. Sci. 2000, 903, 317–323. [Google Scholar]
- Salins, P.; He, Y.; Olson, K.; Glazner, G.; Kashour, T.; Amara, F. TGF-beta1 is increased in a transgenic mouse model of familial Alzheimer’s disease and causes neuronal apoptosis. Neurosci. Lett. 2008, 430, 81–86. [Google Scholar]
- Brionne, T.C.; Tesseur, I.; Masliah, E.; Wyss-Coray, T. Loss of TGF-beta1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron 2003, 40, 1133–1145. [Google Scholar]
- Lesné, S.; Docagne, F.; Gabriel, C.; Liot, G.; Lahiri, D.K.; Buée, L.; Plawinski, L.; Delacourte, A.; MacKenzie, E.T.; Buisson, A.; et al. Transforming growth factor-beta1 potentiates amyloid-beta generation in astrocytes and in transgenic mice. J. Biol. Chem. 2003, 278, 18408–18418. [Google Scholar]
- Zhang, J.; Ke, K.F.; Liu, Z.; Qiu, Y.H.; Peng, Y.P. Th17 cell-mediated neuroinflammation is involved in neurodegeneration of aβ1–42-induced Alzheimer’s disease model rats. PLoS One 2013, 8, e75786. [Google Scholar]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar]
- Passos, G.F.; Figueiredo, C.P.; Prediger, R.D.; Silva, K.A.; Siqueira, J.M.; Duarte, F.S.; Leal, P.C.; Medeiros, R.; Calixto, J.B. Involvement of phosphoinositide 3-kinase gamma in the neuro-inflammatory response and cognitive impairments induced by beta-amyloid 1–40 peptide in mice. Brain Behav. Immun. 2010, 24, 493–501. [Google Scholar]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar]
- Gandy, S.; Greengard, P. Processing of Alzheimer A beta-amyloid precursor protein: Cell biology, regulation, and role in Alzheimer disease. Int. Rev. Neurobiol. 1994, 36, 29–50. [Google Scholar]
- McLarnon, J.G.; Ryu, J.K. Relevance of Aβ1−42 intrahippocampal injection as an animal model of inflamed Alzheimer’s disease brain. Curr. Alzheimer Res. 2008, 5, 475–480. [Google Scholar]
- Burton, T.; Liang, B.; Dibrov, A.; Amara, F. Transforming growth factor-beta-induced transcription of the Alzheimer beta-amyloid precursor protein gene involves interaction between the CTCF-complex and Smads. Biochem. Biophys. Res. Commun. 2002, 295, 713–723. [Google Scholar]
- Tichauer, J.E.; von Bernhardi, R. Transforming growth factor-β stimulates β amyloid uptake by microglia through Smad3-dependent mechanisms. J. Neurosci. Res. 2012, 90, 1970–1980. [Google Scholar]
- Tesseur, I.; Zou, K.; Esposito, L.; Bard, F.; Berber, E.; Can, J.V.; Lin, A.H.; Crews, L.; Tremblay, P.; Mathews, P.; et al. Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer’s pathology. J. Clin. Investig. 2006, 116, 3060–3069. [Google Scholar]
- Caraci, F.; Spampinato, S.; Sortino, M.A.; Bosco, P.; Battaglia, G.; Bruno, V.; Drago, F.; Nicoletti, F.; Copani, A. Dysfunction of TGF-β1 signaling in Alzheimer’s disease: Perspectives for neuroprotection. Cell Tissue Res. 2012, 347, 291–301. [Google Scholar]
- Bosco, P.; Ferri, R.; Salluzzo, M.G.; Castellano, S.; Signorelli, M.; Nicoletti, F.; Nuovo, S.D.; Drago, F.; Caraci, F. Role of the transforming-growth-factor-β1 gene in late-onset Alzheimer’s disease: Implications for the treatment. Curr. Genomics 2013, 14, 147–156. [Google Scholar]
- Flores, B.; von Bernhardi, R. Transforming growth factor β1 modulates amyloid β-induced glial activation through the Smad3-dependent induction of MAPK phosphatase-1. J. Alzheimers Dis. 2012, 32, 417–429. [Google Scholar]
- Huang, W.C.; Yen, F.C.; Shie, F.S.; Pan, C.M.; Shiao, Y.J.; Yang, C.N.; Huang, F.L.; Sung, Y.J.; Tsay, H.J. TGF-beta1 blockade of microglial chemotaxis toward Abeta aggregates involves SMAD signaling and down-regulation of CCL5. J. Neuroinflamm. 2010, 7, 28. [Google Scholar]
- Browne, T.C.; McQuillan, K.; McManus, R.M.; O’Reilly, J.A.; Mills, K.H.; Lynch, M.A. IFN-γ Production by amyloid β-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer’s disease. J. Immunol. 2013, 190, 2241–2251. [Google Scholar]
- Li, M.; Shang, D.S.; Zhao, W.D.; Tian, L.; Li, B.; Fang, W.G.; Zhu, L.; Man, S.M.; Chen, Y.H. Amyloid beta interaction with receptor for advanced glycation end products up-regulates brain endothelial CCR5 expression and promotes T-cells crossing the blood-brain barrier. J. Immunol. 2009, 182, 5778–5788. [Google Scholar]
- Forlenza, O.V.; Diniz, B.S.; Talib, L.L.; Mendonça, V.A.; Ojopi, E.B.; Gattaz, W.F.; Teixeira, A.L. Increased serum IL-1beta level in Alzheimer’s disease and mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 2009, 28, 507–512. [Google Scholar]
- Khemka, V.K.; Ganguly, A.; Bagchi, D.; Ghosh, A.; Bir, A.; Biswas, A.; Chattopadhyay, S.; Chakrabarti, S. Raised serum proinflammatory cytokines in Alzheimer’s disease with depression. Aging Dis. 2014, 5, 170–176. [Google Scholar]
- Pike, C.J.; Burdick, D.; Walencewicz, A.J.; Glabe, C.G.; Cotman, C.W. Neurodegeneration induced by beta-amyloid peptides in vitro: The role of peptide assembly state. J. Neurosci. 1993, 13, 1676–1687. [Google Scholar]
- Giuffrida, M.L.; Grasso, G.; Ruvo, M.; Pedone, C.; Saporito, A.; Marasco, D.; Pignataro, B.; Cascio, C.; Copani, A.; Rizzarelli, E. Abeta (25–35) and its C-and/or N-blocked derivatives: Copper driven structural features and neurotoxicity. J. Neurosci. Res. 2007, 85, 623–633. [Google Scholar]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 5th ed.; Elsevier Inc.: Burlington, VT, USA, 2005. [Google Scholar]
- Henrich-Noack, P.; Prehn, J.H.; Krieglstein, J. TGF-beta 1 protects hippocampal neurons against degeneration caused by transient global ischemia. Dose-response relationship and potential neuroprotective mechanisms. Stroke 1996, 27, 1609–1615. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, W.-X.; Chen, J.-H.; Lu, J.-H.; Peng, Y.-P.; Qiu, Y.-H. TGF-β1 Protection against Aβ1–42-Induced Neuroinflammation and Neurodegeneration in Rats. Int. J. Mol. Sci. 2014, 15, 22092-22108. https://doi.org/10.3390/ijms151222092
Shen W-X, Chen J-H, Lu J-H, Peng Y-P, Qiu Y-H. TGF-β1 Protection against Aβ1–42-Induced Neuroinflammation and Neurodegeneration in Rats. International Journal of Molecular Sciences. 2014; 15(12):22092-22108. https://doi.org/10.3390/ijms151222092
Chicago/Turabian StyleShen, Wei-Xing, Jia-Hui Chen, Jian-Hua Lu, Yu-Ping Peng, and Yi-Hua Qiu. 2014. "TGF-β1 Protection against Aβ1–42-Induced Neuroinflammation and Neurodegeneration in Rats" International Journal of Molecular Sciences 15, no. 12: 22092-22108. https://doi.org/10.3390/ijms151222092
APA StyleShen, W. -X., Chen, J. -H., Lu, J. -H., Peng, Y. -P., & Qiu, Y. -H. (2014). TGF-β1 Protection against Aβ1–42-Induced Neuroinflammation and Neurodegeneration in Rats. International Journal of Molecular Sciences, 15(12), 22092-22108. https://doi.org/10.3390/ijms151222092