1. Introduction
Hepatocellular carcinoma (HCC) ranks among the most common malignancies worldwide with a poor prognosis. Although there have been significant developments in diagnosis, surgical treatment and adjuvant therapies for HCC, metastasis is the dominating cause of death in the majority of patients, the overall five-year survival rate of which is limited to 5%–6% [
1,
2]. Surgery is the main treatment for HCC; however, the recurrence rate after curative treatment is still disappointingly high. Given the grim outlook of HCC, novel therapeutic targets of genes and new modalities of effective chemoprevention and treatment are highly awaited.
Chemokines are a family of low molecular weight (8–10 kDa) pro-inflammatory cytokines, which bind to chemokine receptors and sustain the migration of neutrophils, lymphocytes, monocytes, DC cells and so on. Recent studies [
3,
4] suggested that chemokine receptors expressed highly on cancer cells might play a role in tumor progression and metastasis. The actin polymerization and pseudopodia formation within the cells may participate in this process, and the combination of chemokines and their receptors plays a very important role in determining the metastatic destination of tumor cells. Support for this theory comes from the overexpression of chemokine receptors CCR
7 and CXCR
4 in breast cancer cells, and the expression levels of their ligands (SDF-1 and 6) are much higher in the usual metastatic destination (lymph nodes, lung and liver) than the rare one (small intestine, skin, brain tissue and skeletal muscle) [
5]. Lu [
6] found that the expression level of CCR2 is much higher in cell stains that have stronger invasiveness, by the detection of CCR2 mRNA and protein expression levels in prostate cancer cells. As for providing a new research direction in anti-tumor therapy, organ selective transfer has become a hot spot in the field of tumor metastasis.
Chemokine receptor 6 (CCR6) is a member of the CC chemokine receptor family, whose main expression is observed in the spleen, lymph nodes, thymus and fetal liver tissue [
7]. CCL20, a ligand of CCR6, is also known as a liver and activation-regulated chemokine and as MIP-3α. Immunohistochemical analysis of 64 colorectal cancer cases [
8] found that the expression of CCR6 is closely associated with the hepatic metastasis of colorectal cancer, prompting that CCR6 and its ligands are involved in this process. Moreover, it is in thymus and ovarian cancer [
9]. It is speculated that CCR6-mediated chemotaxis may be a common mechanism of malignant tumor metastasis, suggesting that CCR6 signaling pathway suppression may prevent the risk of liver metastasis.
Hepatic metastase, a highly organized and non-random organ-specific process, includes several sequential steps. It may develop not only as a result of intrahepatic metastasis, but also hematogenous metastasis, within which pulmonary distant metastasis is common. Speculation is reasonable that CCR6 and its ligand CCL20 may be determinant in intrahepatic metastasis [
10] and distant migration of lungs based on the constitutive expression of CCL20 and CCR6 in the liver, lungs and other organs.
Based on many previous reports [
11–
13], we suppose CCR6 may play major roles in hepatocellular carcinoma metastasis and participate in regulating the migration and invasion of HCC, and CCR6 expression would have some correlation with metastasis of HCC. In the present study, we examined the relationship of CCR6/CCL20 and the biological behavior of HCC, which would help us understand the mechanism of the invasion and metastasis of HCC.
3. Experimental Section
3.1. Cell Lines and Cell Culture
Normal liver cell line L02 and different invasive potential HCC cell lines, SMMC-7721, MHCC-97L, MHCC-97H, HCCLM3 and HCCLM6, were used. All kinds of cells were maintained in DMEM with high sugar (Gibco, Darmstadt, Germany) containing 10% fetal bovine serum (Gibco), at 37 °C in a 5% CO2 humidified atmosphere.
3.2. RNA Oligos and Transfection
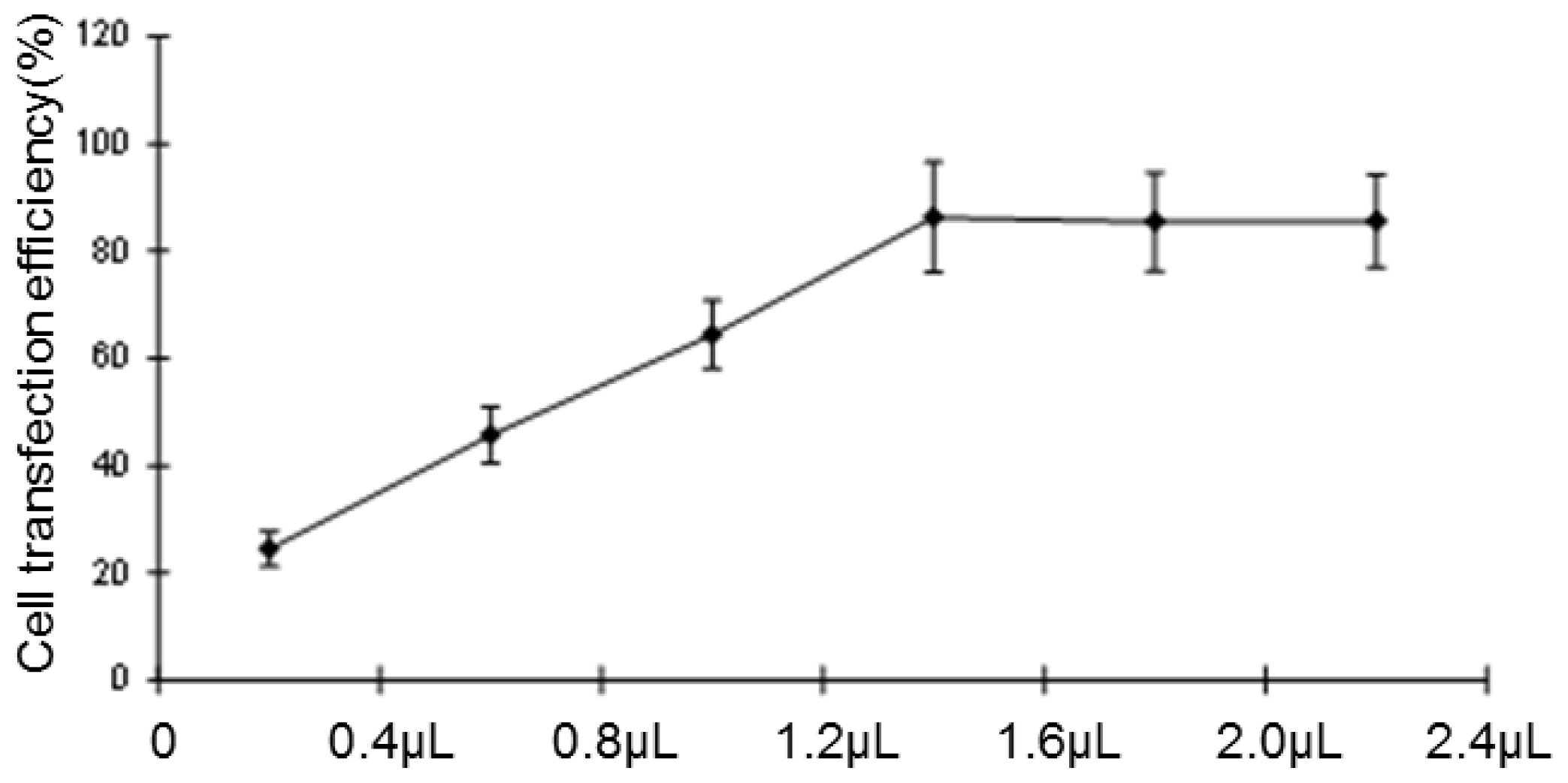
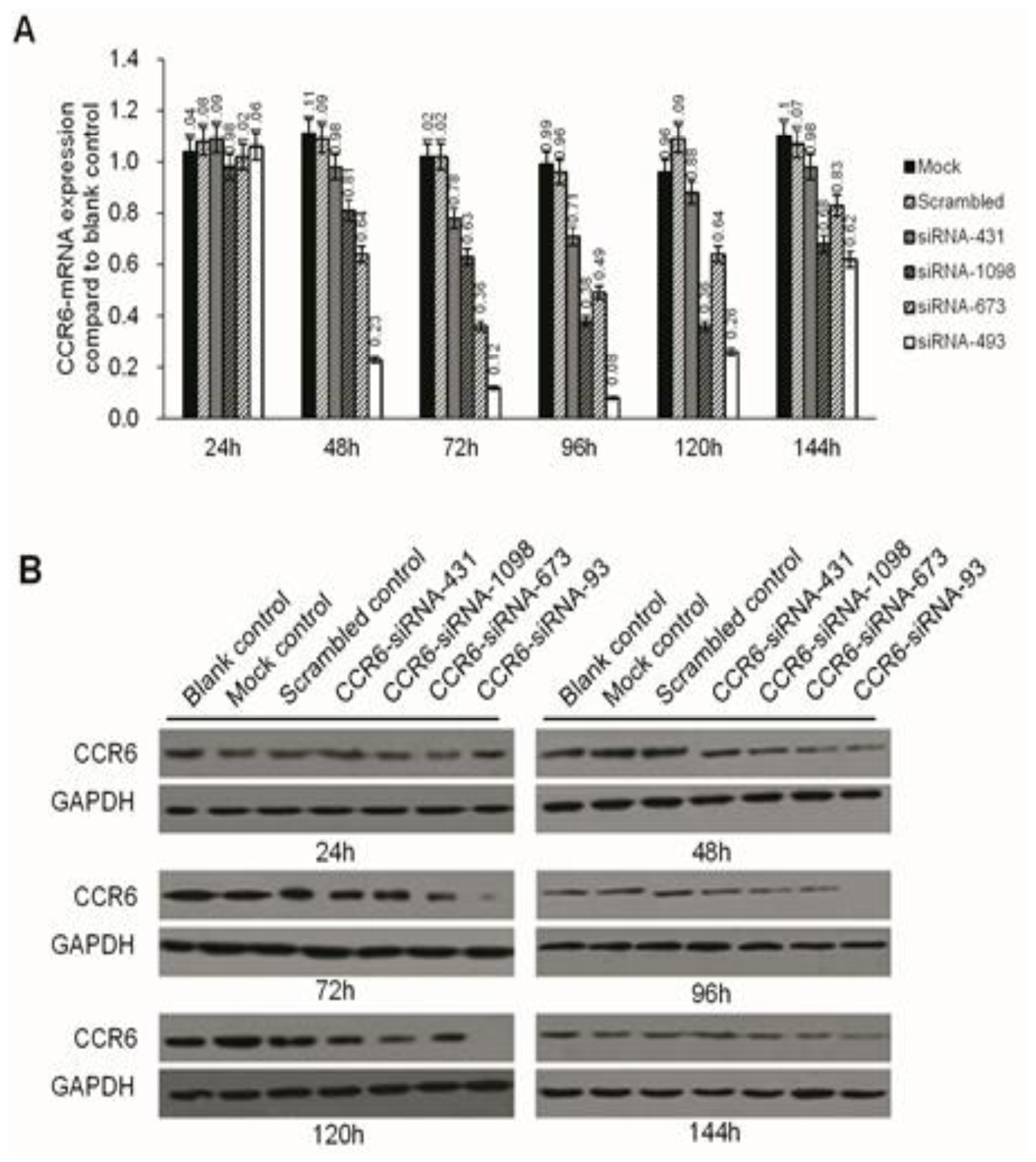
Negative miRNA control (miR-NC), HCCLM6-mock (liposomes only), HCCLM6-scrambled control (irrelevant sequence with the same base arrangement as siRNA) and siRNA of CCR6 (pGPH1/GFP/Neo-CCR6-siRNA-431, -1098, -673, -493) were purchased from Gene Pharma (Shanghai, China). About 12–24 h before transfection, the cells were seeded in 24-well culture plates (0.5 × 105/well). At the time of transfection, cells were 80% confluent. The complete medium was replaced by serum-free medium and incubated at 37 °C with 100 μL of siRNA complex solutions at the indicated concentration. After 24 h, the medium was removed, and the cells were cultured for 24 h in complete culture medium (with 10% FBS) without any transfection reagents. The transfection efficiency was evaluated by the number of cells expressing green fluorescent protein with a fluorescence microscope. Due to the carrying of the Neo gene in the pGPH1/GFP/Neo plasmid, which endowed G418 resistance to the successful transfected cells, G418 (Gibco) was used to screen for stable, transfected strains. The expression of CCR6 in the level of gene and protein were then screened for the most effective siRNA sequence.
3.3. Real-Time PCR
RNA was extracted using the RNeasy Mini kit and synthesized for cDNA. Quantitative real-time PCR was conducted in an SYBR Green PCR Kit using the ABI Prism 7500 Real-time PCR System, and each sample was run in triplicate.
3.4. Western Blotting
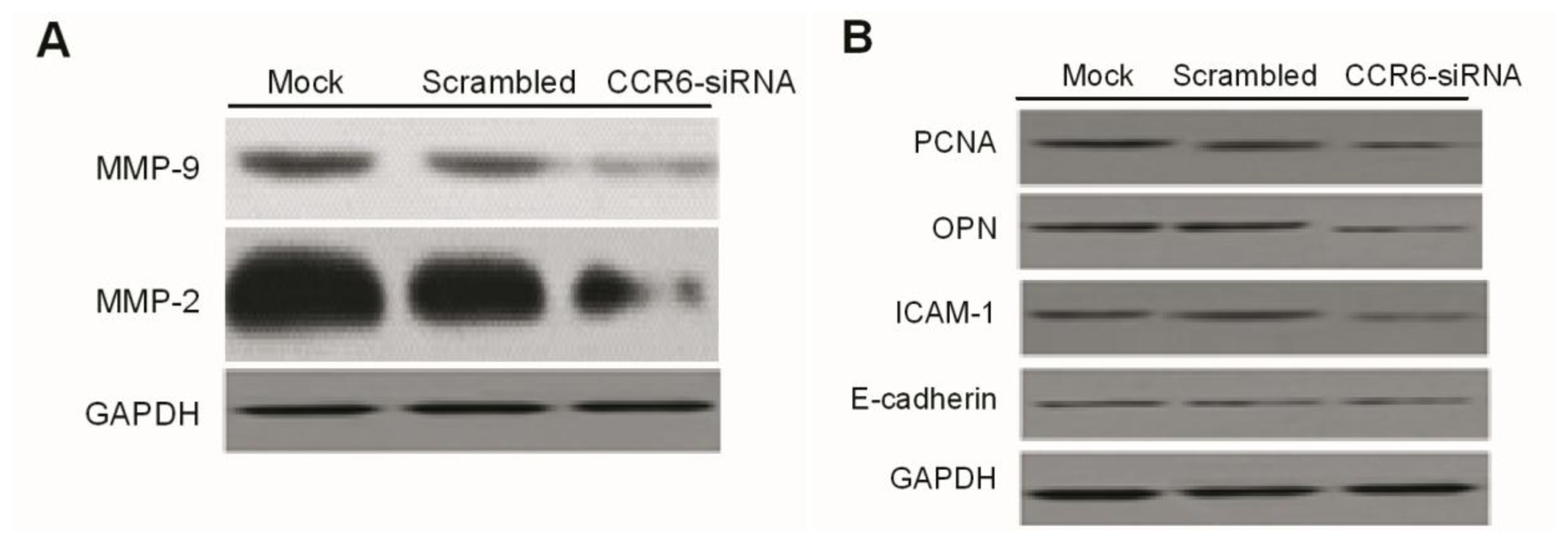
Western blotting was done as previously described. Briefly, at the end of the designated experiments, cell lysates were separated with 10% SDS-PAGE and clear protein extracts were obtained by centrifugation for 30 min at 4 °C. Twenty to forty milligrams of protein mixed with loading buffer were loaded per lane, separated by 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to PVDF membrane filters. After each membrane was blocked with 5% milk for 1 h at room temperature (RT), it was probed with specific monoclonal antibodies (Abcam, Cambridge, MA, USA) overnight at 4 °C. After washing with phosphate-buffered saline (PBS), the membranes were incubated with corresponding secondary antibodies (Abcam) TBST-5% nonfat milk for 1 h at RT. The immuno-reactive bands were visualized by enhanced chemiluminescence reagent, and GAPDH served as the loading control.
3.5. MTT Assay
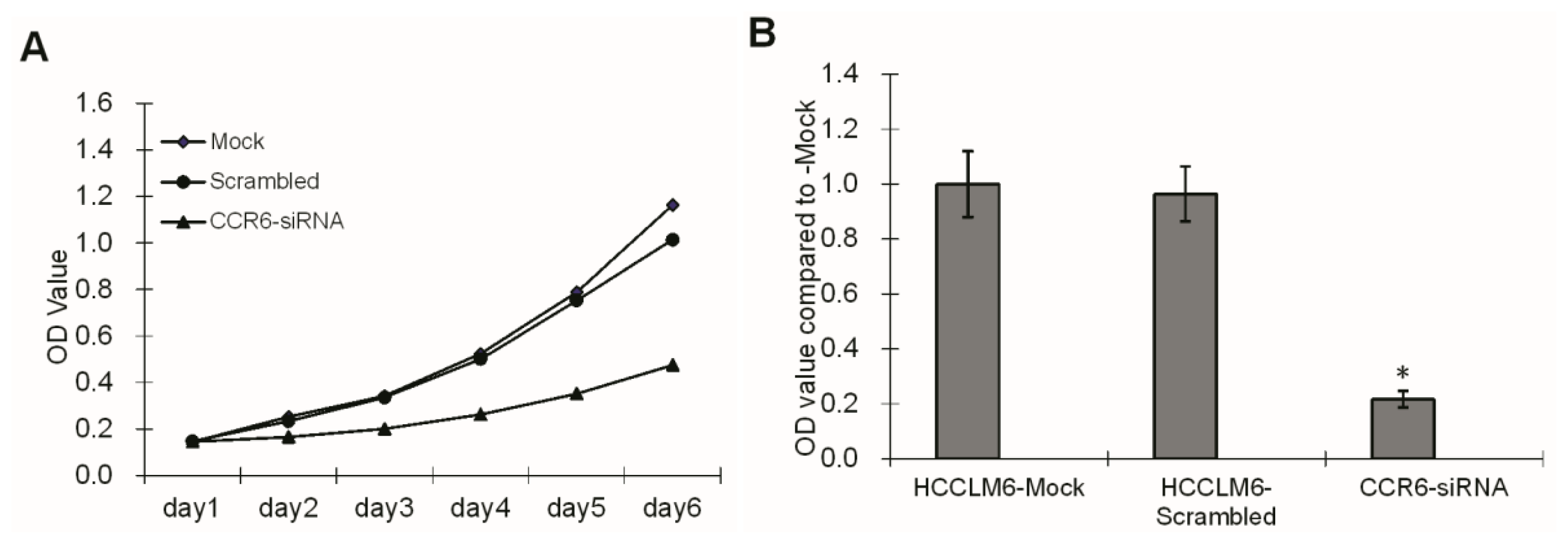
HCCLM6 cell proliferation was tested using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. For the assay, three groups of cells, HCCLM6-mock, HCCLM6-scrambled and CCR6-siRNA, were freshly isolated and plated in 96-well flat bottom cell culture plates at a concentration of 1 × 105 cells/well containing 100 μL of DMEM (supplemented with 10% FBS) culture medium. At 2, 3, 4, 5 and 6 d of culture, 20 μL of MTT solution (Sigma, St. Louis, MO, USA) in a concentration of 5 mg/mL was added per well. After 4 h of incubation, the culture medium was dissolved with 150 μL DMSO (Sigma, St. Louis, MO, USA). Plates were kept on an orbital shaker for 10 min and the optical density (OD) was read on a multiwell scanning spectrophotometer at 570 nm.
3.6. Adhesion Assay
Logarithmic growth of cells in three groups, HCCLM6-mock, HCCLM6-scrambled and CCR6-siRNA, were cultured in DMEM (free of serum) for 24 h and resuspended by serum-containing medium. Suspensions of cells were seeded in 96-well plates at a density of 4 × 104/well, which were coated with Matrigel (Invitrogen, Carlsbad, CA, USA) and incubated in 10% FCS for 1h. After being cultured at 37 °C in 5% CO2 humidified atmosphere for another 1 h and being washed twice with PBS, each well of the 96-well plates was added to 20 μL fresh MTT solution in a concentration of 5 mg/mL. Then, the medium was replaced by 150 μL DMSO after 2 h. Plates were shaken in a dark place for 10 min, and the optical density (OD) was recorded at 570 nm.
3.7. Fibronectin Adhesion Assay
In this assay, 96-well plates were coated with 100 μL fibronectin (Sigma, St. Louis, MO, USA) overnight at 4 °C. As a positive control, holes were coated with 1% BSA, and for negative control, holes were coated with 100 μL/mL polylysine. The optimal concentrations of BSA and polylysine were chosen empirically by titration. Then, the plates were blocked for 30 min at 37 °C with 200 μL of 1% BSA and washed with PBS. After being subsequently immersed in cell culture media with serum-free high glucose DMEM, the wells were seeded by the suspension of HCCLM6 cells with 5.0 × 104 cells/well and then maintained at 37 °C in a humidified atmosphere containing 5% CO2 for 30 min. After removal of the culture medium, the samples were fixed with ethanol for 10 min and washed with PBS. Crystal violet hydrate solution was transferred to the 96-well plates for 25 min. After being rinsed with water, each well was added to 200 μL of solubilization solution containing 0.5% TrtionX-100 and shaken overnight. The absorbance in 570 nm of the samples were evaluated by an enzyme mark instrument.
3.8. Wound Healing Assay
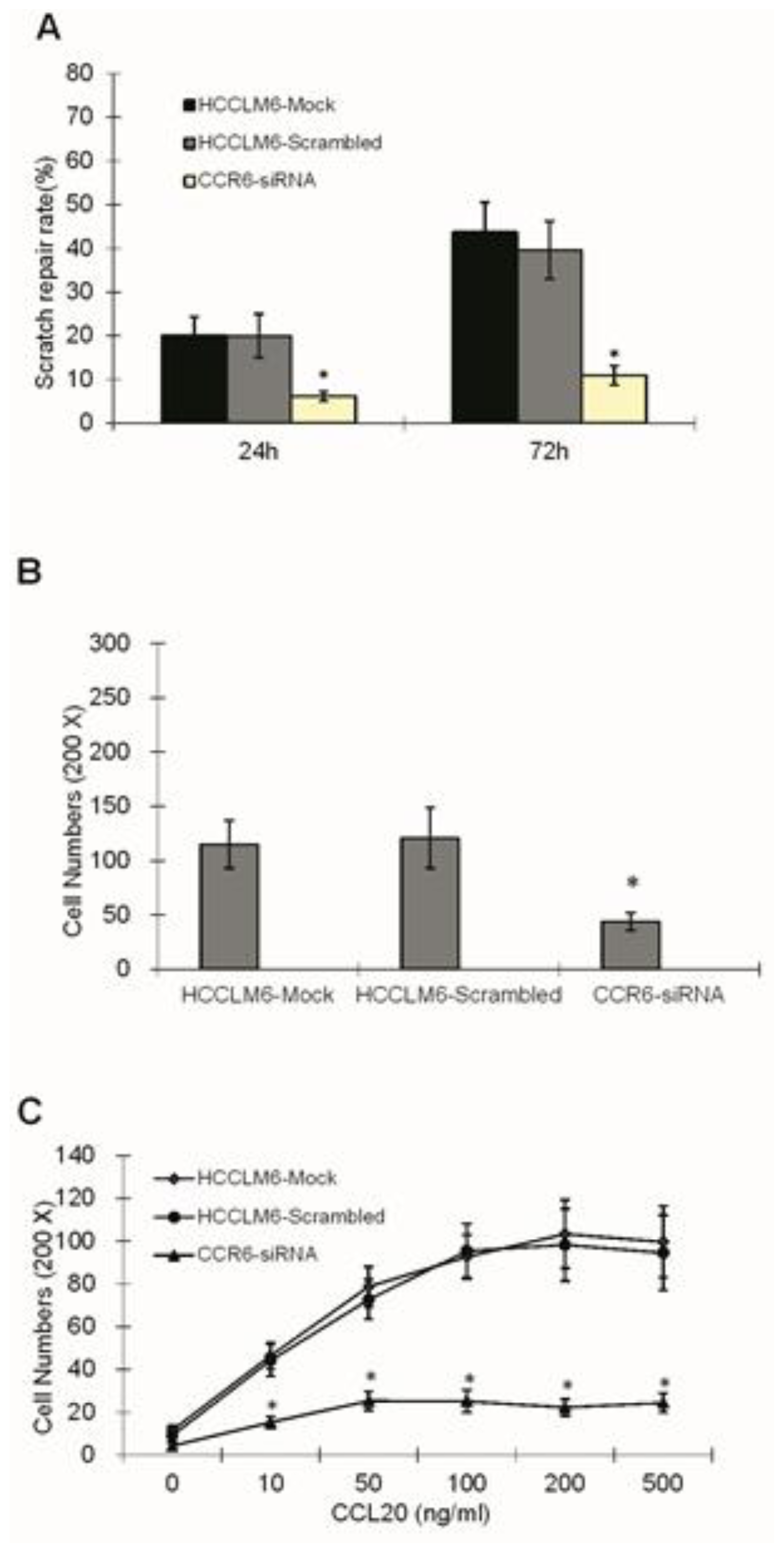
Suspension of cells in three groups (HCCLM6-mock, HCCLM6-scrambled and CCR6-siRNA) were plated on 6-well plates with a concentration of 5 × 105/mL, the plates of which had been smeared by Matrigel glue and incubated in 10% FBS for 1 h. Until confluence with a monolayer of cells, each well was infused with fresh DMEM (free of FBS) for 24 h and scratched in a reproducible way using a sterile 200 μL pipette tip. The same wound area was photographed at once under a Zeiss Axioskop microscope and was also done after being cultured in DMEM (supplemented with 10% FBS) for 24 and 72 h. The distance between the edges of the scratch defect were measured and averaged from four separate experiments.
3.9. Matrigel Invasion Assay
A manmade Matrigel invasion chamber (Corning Corporation, Midland, MI, USA) was used according to the manufacturer’s instruction. The upper portion of the transwell chamber was coated with 100 μL Matrigel with 2.0 mg/mL (dissolved with serum-free DMEM) and incubated at 37 °C for 3 h. Then, 1 × 106/mL cells (previously starved in serum-free DMEM for 24 h) in the groups of HCCLM6-mock, HCCLM6-scrambled and CCR6-siRNA were trypsinized, washed, resuspended and added to the upper chamber in serum-free DMEM, and migration at 37 °C towards 15% FBS containing growth media was determined after 24 h. Cells invading through the Matrigel and migrating to the bottom chamber were fixed with 4% paraformaldehyde, stained with 1% hematoxylin-eosin staining (HE), photographed and counted.
3.10. Chemotaxis Assay
Cells resuspended by DMEM (free of serum) to a concentration of 1 × 106/mL were seeded in the upper chamber. The lower chamber was covered with chemoattractant CCL20 (R&D Systems, Minneapolis, MN, USA) diluted by serum-free DMEM, whose concentration gradient was 0, 10, 50, 100, 200 and 500 ng/mL. After incubation for 24 h at 37 °C with 5% CO2, cells on the lower surface of the membrane were stained and counted under a light microscope (×200). Assays were performed in triplicate.
3.11. Gelatin Zymography
To evaluate the nature of the enzymes responsible for the observed gelatinolytic activity, 2 × 105/mL cells in the groups of HCCLM6-mock, HCCLM6-scrambled and CCR6-siRNA were vaccinated in 6-well plates. After being cultivated in serum-free DMEM for 24 h, proteins were then extracted and the concentration adjusted to 2.0 mg/mL. SDS-PAGE substrate zymography was carried out with separating gels containing 1 mg/mL of gelatin. Ten microliters of extract were mixed with an equal volume of 2× loading buffer, and then, the mixture was applied to each well of the gel. After electrophoresis was complete, which was then run at 20 mA at 4 °C, the gel was washed twice in washing buffer for 30 min and incubated in developing buffer for 18 h at 37 °C. Gels were stained with 0.25% Coomassie brilliant blue solution and then destained with 30% methanol and 10% acetic acid. Gelatinolytic activities were detected as unstained bands against the background of Coomassie blue-stained gelatin.
3.12. Statistical Analysis
By using the software SPSS16.0 (Chicago, IL, USA), data were presented as the mean ± SEM, and statistical differences were evaluated by one-way analysis of variance, comparison of positive difference with chi-square test (continuous correction). Image analysis of graphics was measured by Image Pro-Plus 6.0 software. For all analyses, we considered p < 0.05 to be statistically significant.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





